Running Loose or Getting Lost: How HIV-1 Counters and Capitalizes on APOBEC3-Induced Mutagenesis through Its Vif Protein

 ,
, 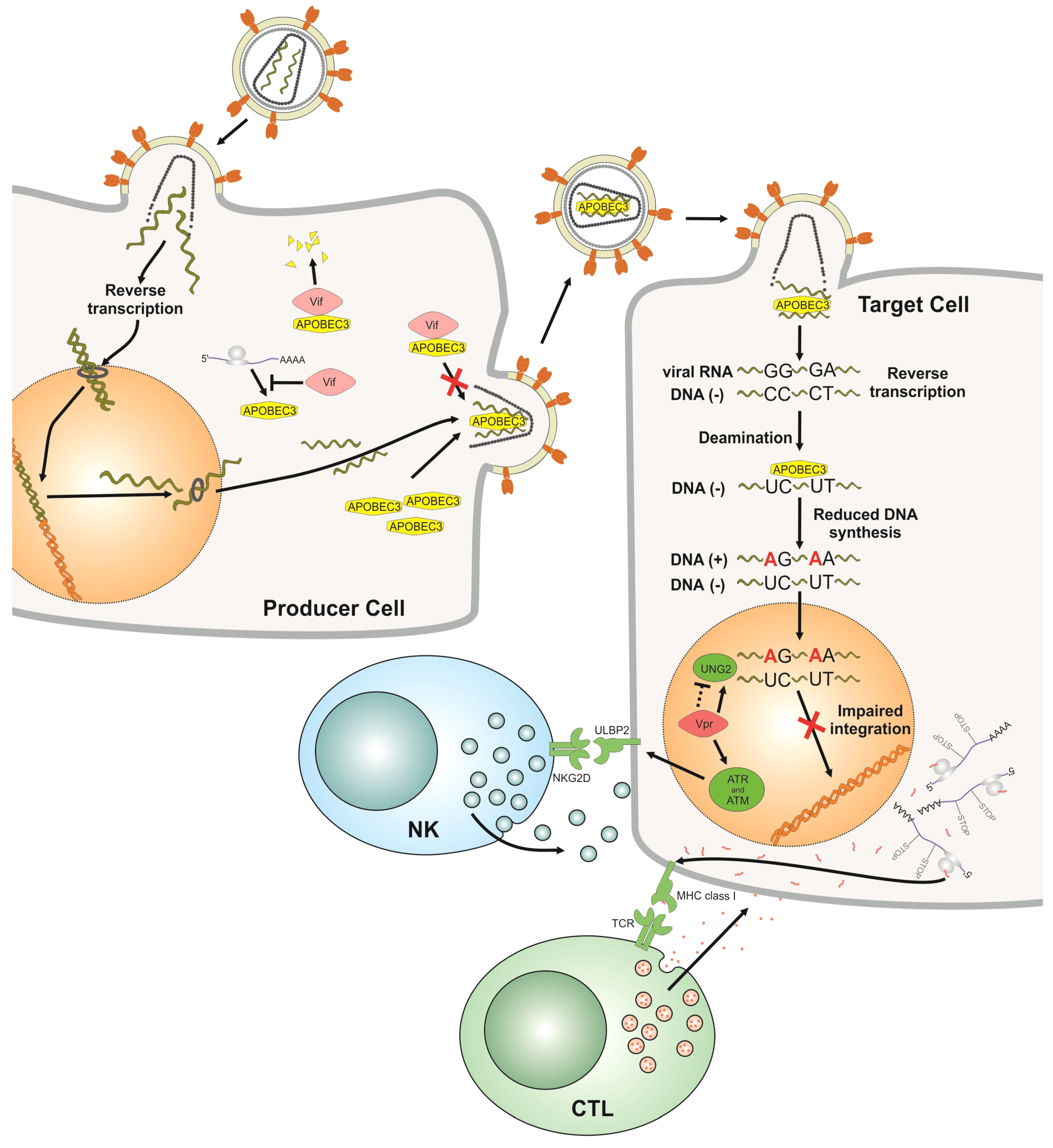{kind=link}
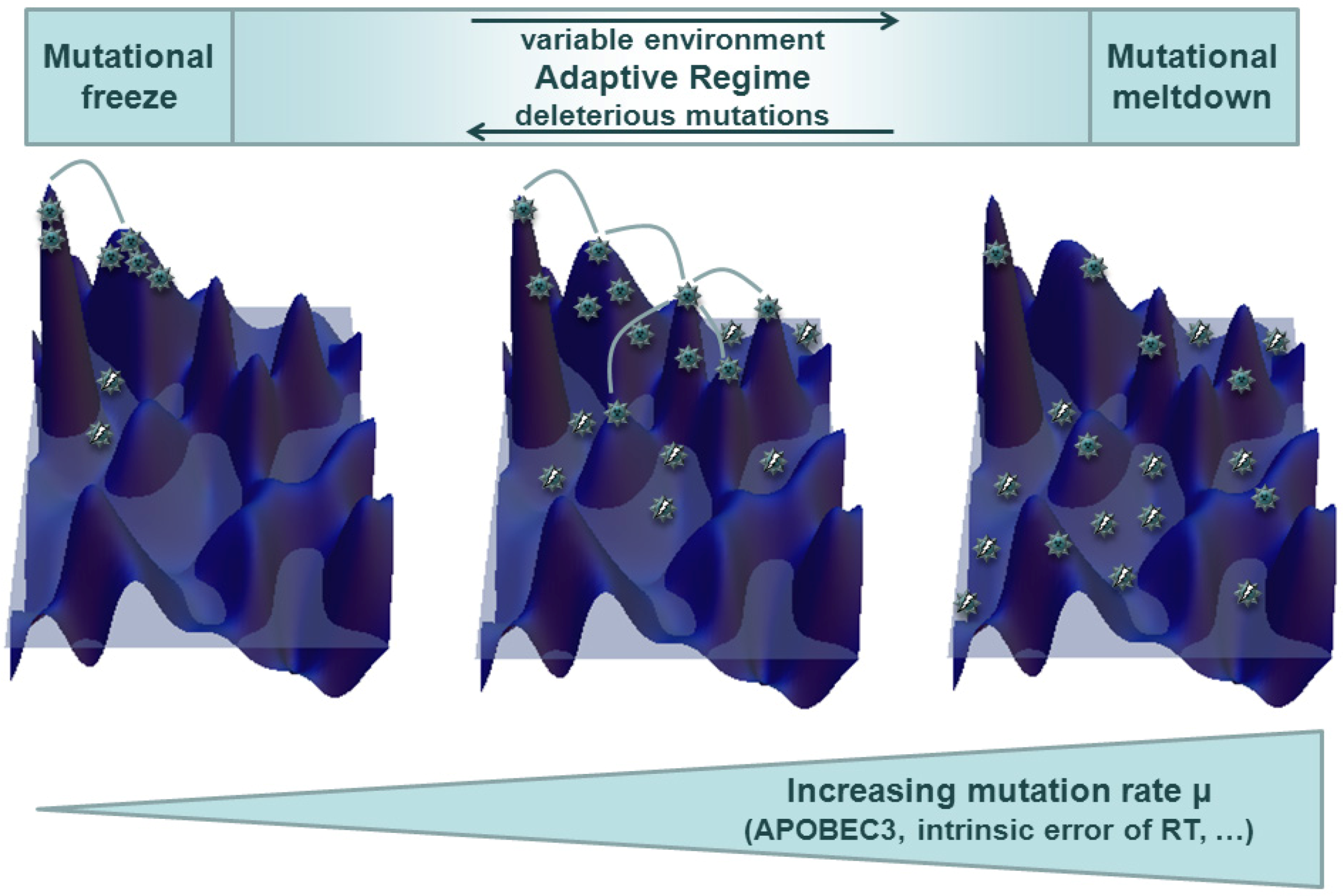{kind=link}
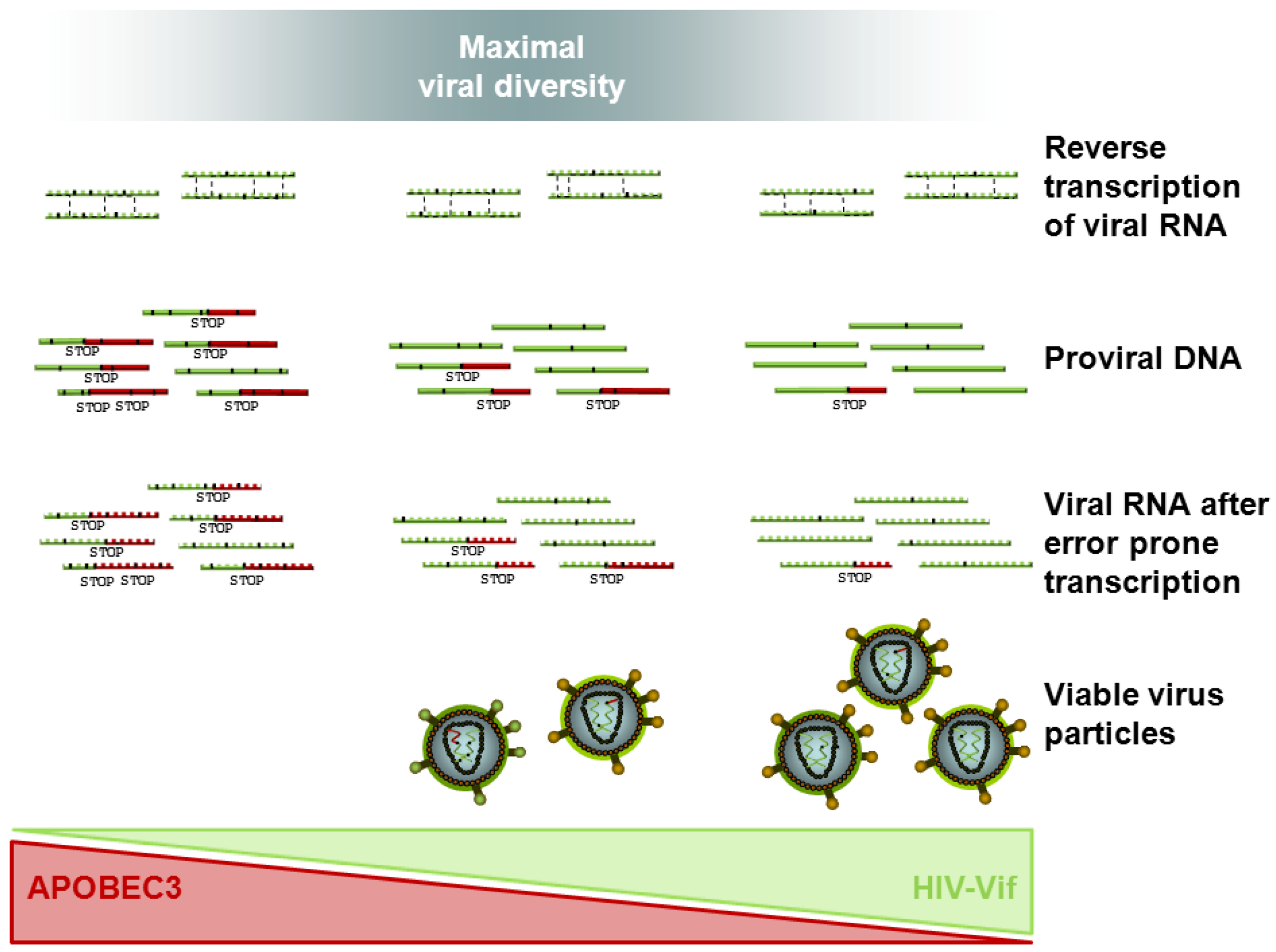{kind=link}
Abstract
:1. Introduction
2. The Interplay between Host Restriction Factors from the APOBEC3 Protein Family and HIV-1 Vif in the Viral Replication Cycle
2.1. APOBEC3
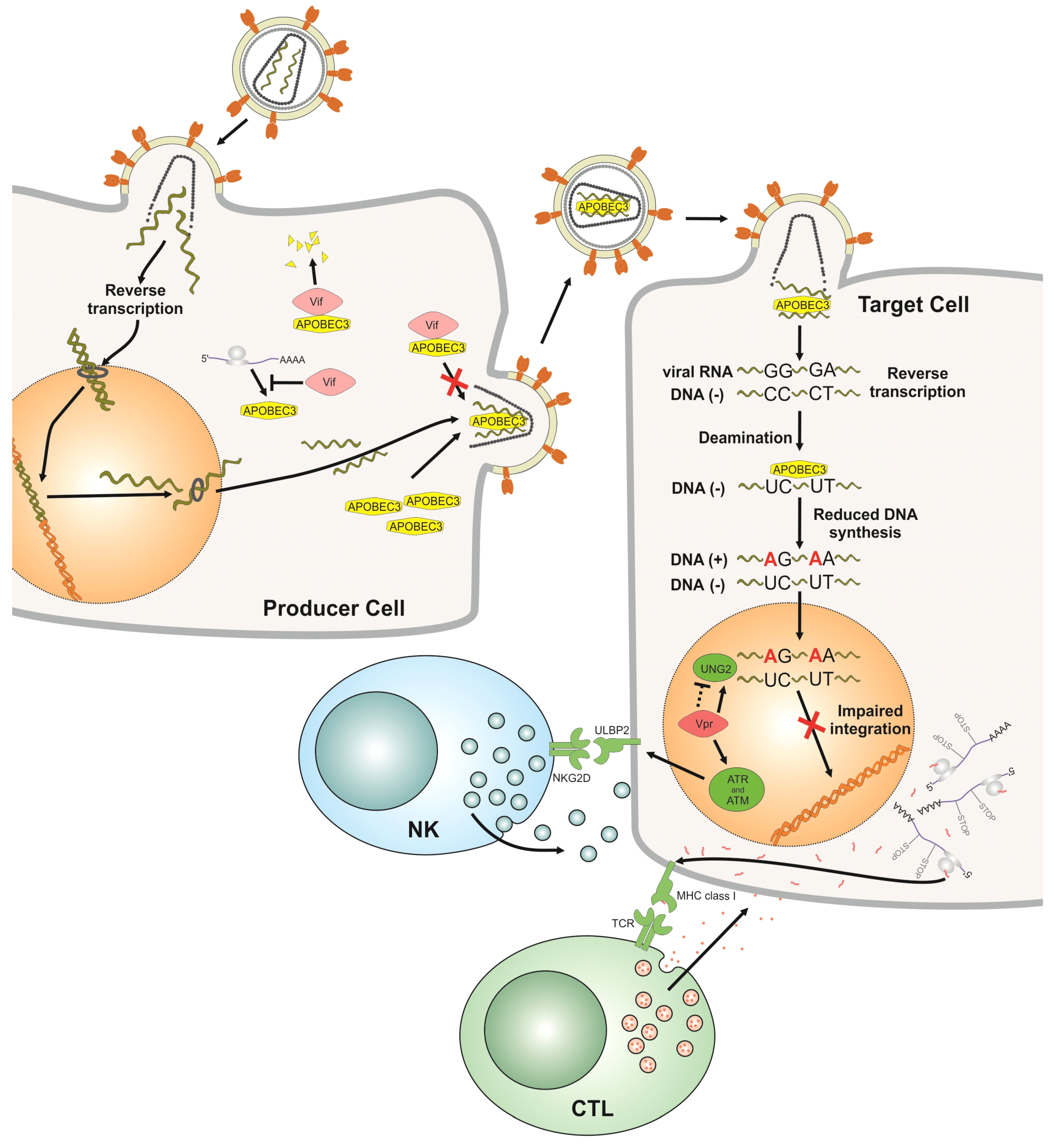
2.2. Vif
3. Models for Viral Evolution and Host Pathogen Co-Evolution/Interactions
3.1. Quasispecies in Static Environments
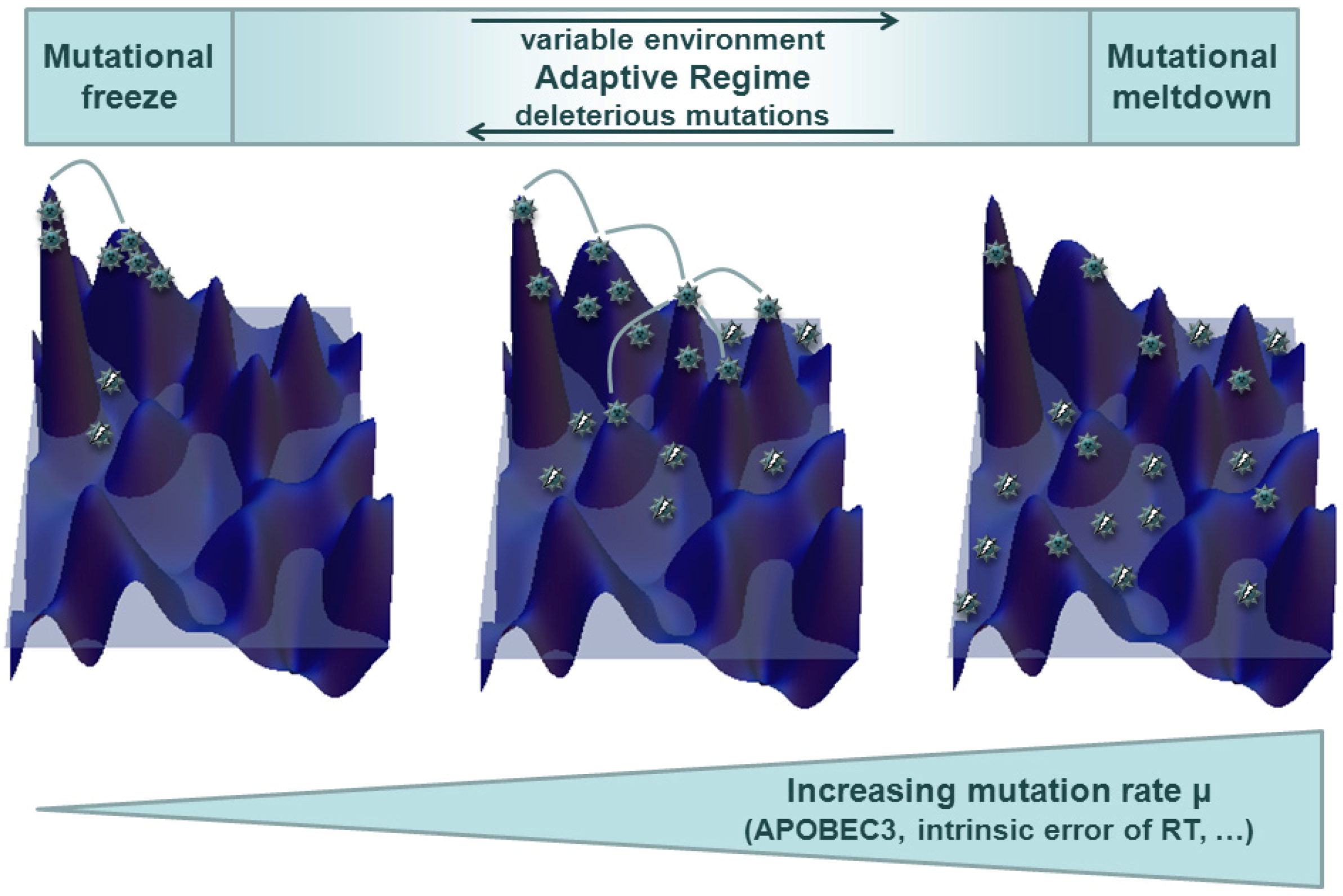
3.2. Quasispecies in Dynamic Environments and Co-Evolution in the Host Context
4. Coevolution of HIV Vif and APOBEC3 — Impact on Viral Escape and Drug Resistance
4.1. In Vitro Evidence of HIV Vif APOBEC3 Coevolution
4.2. In Vivo Clues of HIV Vif APOBEC3 Co-Evolution: Clinical Data on HIV Dynamics in Patients
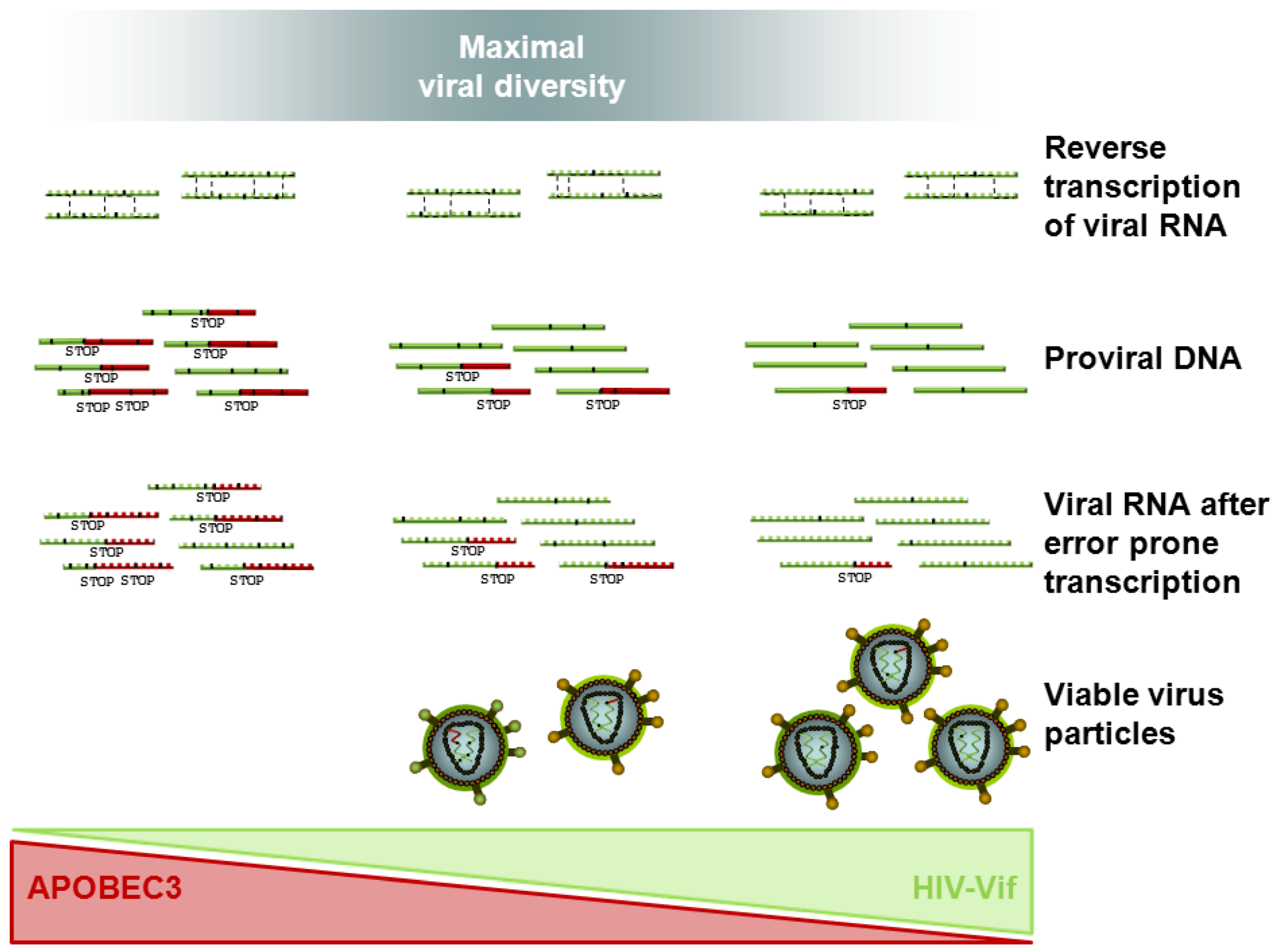
4.3. Tracing HIV Vif-APOBEC3 Coevolution in the Genome — Towards an Interpretation and Extrapolation of in Vitro and in Vivo Data through Modeling Studies
5. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- De, C.E. Perspectives for the chemotherapy of AIDS. Anticancer Res. 1987, 7, 1023–1038. [Google Scholar]
- Oette, M.; Reuter, S.; Kaiser, R.; Lengauer, T.; Fatkenheuer, G.; Knechten, H.; Hower, M.; Pfister, H.; Haussinger, D. Epidemiology of transmitted drug resistance in chronically HIV-infected patients in Germany: The RESINA study 2001-2009. Intervirology 2012, 55, 154–159. [Google Scholar] [CrossRef]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar]
- Sanjuan, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar]
- Rosenbloom, D.I.; Hill, A.L.; Rabi, S.A.; Siliciano, R.F.; Nowak, M.A. Antiretroviral dynamics determines HIV evolution and predicts therapy outcome. Nat. Med. 2012, 18, 1378–1385. [Google Scholar]
- Hill, A.L.; Rosenbloom, D.I.; Nowak, M.A. Evolutionary dynamics of HIV at multiple spatial and temporal scales. J. Mol. Med. (Berl) 2012, 90, 543–561. [Google Scholar] [CrossRef]
- Yebra, G.; Holguin, A. Mutation Vif-22H, which allows HIV-1 to use the APOBEC3G hypermutation to develop resistance, could appear more quickly in certain non-B variants. J. Antimicrob. Chemother. 2011, 66, 941–942. [Google Scholar] [CrossRef]
- Simon, V.; Zennou, V.; Murray, D.; Huang, Y.; Ho, D.D.; Bieniasz, P.D. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog. 2005, 1, e6. [Google Scholar] [CrossRef]
- Iwabu, Y.; Kinomoto, M.; Tatsumi, M.; Fujita, H.; Shimura, M.; Tanaka, Y.; Ishizaka, Y.; Nolan, D.; Mallal, S.; Sata, T.; et al. Differential anti-APOBEC3G activity of HIV-1 Vif proteins derived from different subtypes. J. Biol. Chem. 2010, 285, 35350–35358. [Google Scholar]
- Compton, A.A.; Hirsch, V.M.; Emerman, M. The host restriction factor APOBEC3G and retroviral Vif protein coevolve due to ongoing genetic conflict. Cell Host Microbe 2012, 11, 91–98. [Google Scholar] [CrossRef]
- Pillai, S.K.; Wong, J.K.; Barbour, J.D. Turning up the volume on mutational pressure: Is more of a good thing always better? (A case study of HIV-1 Vif and APOBEC3). Retrovirology 2008, 5, 26. [Google Scholar] [CrossRef]
- Korber, B.; Muldoon, M.; Theiler, J.; Gao, F.; Gupta, R.; Lapedes, A.; Hahn, B.H.; Wolinsky, S.; Bhattacharya, T. Timing the ancestor of the HIV-1 pandemic strains. Science 2000, 288, 1789–1796. [Google Scholar] [CrossRef]
- Malim, M.H.; Bieniasz, P.D. HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar]
- Schaller, T.; Goujon, C.; Malim, M.H. AIDS/HIV. HIV interplay with SAMHD1. Science 2012, 335, 1313–1314. [Google Scholar]
- Berger, A.; Sommer, A.F.; Zwarg, J.; Hamdorf, M.; Welzel, K.; Esly, N.; Panitz, S.; Reuter, A.; Ramos, I.; Jatiani, A.; et al. SAMHD1-deficient CD14+ cells from individuals with Aicardi-Goutieres syndrome are highly susceptible to HIV-1 infection. PLoS Pathog. 2011, 7, e1002425. [Google Scholar]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar]
- Smith, H.C.; Bennett, R.P.; Kizilyer, A.; McDougall, W.M.; Prohaska, K.M. Functions and regulation of the APOBEC family of proteins. Semin. Cell Dev. Biol. 2012, 23, 258–268. [Google Scholar]
- LaRue, R.S.; Jonsson, S.R.; Silverstein, K.A.; Lajoie, M.; Bertrand, D.; El-Mabrouk, N.; Hotzel, I.; Andresdottir, V.; Smith, T.P.; Harris, R.S. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol. Biol. 2008, 9, 104. [Google Scholar] [CrossRef]
- Münk, C.; Willemsen, A.; Bravo, I.G. An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals. BMC Evol. Biol 2012, 12, 71. [Google Scholar]
- Hultquist, J.F.; Harris, R.S. Leveraging APOBEC3 proteins to alter the HIV mutation rate and combat AIDS. Future Virol. 2009, 4, 605. [Google Scholar] [CrossRef]
- Refsland, E.W.; Stenglein, M.D.; Shindo, K.; Albin, J.S.; Brown, W.L.; Harris, R.S. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: Implications for HIV-1 restriction. Nucleic Acids Res. 2010, 38, 4274–4284. [Google Scholar] [CrossRef]
- Koning, F.A.; Newman, E.N.; Kim, E.Y.; Kunstman, K.J.; Wolinsky, S.M.; Malim, M.H. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 2009, 83, 9474–9485. [Google Scholar]
- Soros, V.B.; Yonemoto, W.; Greene, W.C. Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog. 2007, 3, e15. [Google Scholar] [CrossRef]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar]
- Mariani, R.; Chen, D.; Schröfelbauer, B.; Navarro, F.; König, R.; Bollman, B.; Münk, C.; Nymark-McMahon, H.; Landau, N.R. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 2003, 114, 21–31. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar]
- Knoepfel, S.A.; Salisch, N.C.; Huelsmann, P.M.; Rauch, P.; Walter, H.; Metzner, K.J. Comparison of G-to-A mutation frequencies induced by APOBEC3 proteins in H9 cells and peripheral blood mononuclear cells in the context of impaired processivities of drug-resistant human immunodeficiency virus type 1 reverse transcriptase variants. J. Virol. 2008, 82, 6536–6545. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Chen, K.; Zhang, C.; Huang, S.; Zhang, H. Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of APOBEC3G-edited nascent HIV-1 DNA. J. Biol. Chem. 2007, 282, 11667–11675. [Google Scholar]
- Schrofelbauer, B.; Yu, Q.; Zeitlin, S.G.; Landau, N.R. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J. Virol. 2005, 79, 10978–10987. [Google Scholar]
- Kaiser, S.M.; Emerman, M. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase Apobec3G. J. Virol. 2006, 80, 875–882. [Google Scholar] [CrossRef]
- Langlois, M.A.; Neuberger, M.S. Human APOBEC3G can restrict retroviral infection in avian cells and acts independently of both UNG and SMUG1. J. Virol. 2008, 82, 4660–4664. [Google Scholar]
- Mbisa, J.L.; Barr, R.; Thomas, J.A.; Vandegraaff, N.; Dorweiler, I.J.; Svarovskaia, E.S.; Brown, W.L.; Mansky, L.M.; Gorelick, R.J.; Harris, R.S.; et al. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 2007, 81, 7099–7110. [Google Scholar]
- Yan, N.; O'Day, E.; Wheeler, L.A.; Engelman, A.; Lieberman, J. HIV DNA is heavily uracilated, which protects it from autointegration. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 9244–9249. [Google Scholar]
- Norman, J.M.; Mashiba, M.; McNamara, L.A.; Onafuwa-Nuga, A.; Chiari-Fort, E.; Shen, W.; Collins, K.L. The antiviral factor APOBEC3G enhances the recognition of HIV-infected primary T cells by natural killer cells. Nat. Immunol. 2011, 12, 975–983. [Google Scholar]
- Luo, K.; Wang, T.; Liu, B.; Tian, C.; Xiao, Z.; Kappes, J.; Yu, X.F. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J. Virol. 2007, 81, 7238–7248. [Google Scholar]
- Mbisa, J.L.; Bu, W.; Pathak, V.K. APOBEC3F and APOBEC3G inhibit HIV-1 DNA integration by different mechanisms. J. Virol. 2010, 84, 5250–5259. [Google Scholar] [CrossRef]
- Guo, F.; Cen, S.; Niu, M.; Yang, Y.; Gorelick, R.J.; Kleiman, L. The interaction of APOBEC3G with human immunodeficiency virus type 1 nucleocapsid inhibits tRNA3Lys annealing to viral RNA. J. Virol. 2007, 81, 11322–11331. [Google Scholar]
- Guo, F.; Cen, S.; Niu, M.; Saadatmand, J.; Kleiman, L. Inhibition of formula-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 2006, 80, 11710–11722. [Google Scholar] [CrossRef]
- Li, X.Y.; Guo, F.; Zhang, L.; Kleiman, L.; Cen, S. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J. Biol. Chem. 2007, 282, 32065–32074. [Google Scholar]
- Bishop, K.N.; Verma, M.; Kim, E.Y.; Wolinsky, S.M.; Malim, M.H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008, 4, e1000231. [Google Scholar]
- Wang, X.; Ao, Z.; Chen, L.; Kobinger, G.; Peng, J.; Yao, X. The cellular antiviral protein APOBEC3G interacts with HIV-1 reverse transcriptase and inhibits its function during viral replication. J. Virol. 2012, 86, 3777–3786. [Google Scholar] [CrossRef]
- Holmes, R.K.; Koning, F.A.; Bishop, K.N.; Malim, M.H. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 2007, 282, 2587–2595. [Google Scholar]
- Bishop, K.N.; Holmes, R.K.; Malim, M.H. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 2006, 80, 8450–8458. [Google Scholar] [CrossRef]
- Newman, E.N.; Holmes, R.K.; Craig, H.M.; Klein, K.C.; Lingappa, J.R.; Malim, M.H.; Sheehy, A.M. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 2005, 15, 166–170. [Google Scholar] [CrossRef]
- Iwatani, Y.; Chan, D.S.; Wang, F.; Maynard, K.S.; Sugiura, W.; Gronenborn, A.M.; Rouzina, I.; Williams, M.C.; Musier-Forsyth, K.; Levin, J.G. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007, 35, 7096–7108. [Google Scholar] [CrossRef]
- Russell, R.A.; Moore, M.D.; Hu, W.S.; Pathak, V.K. APOBEC3G induces a hypermutation gradient: purifying selection at multiple steps during HIV-1 replication results in levels of G-to-A mutations that are high in DNA, intermediate in cellular viral RNA, and low in virion RNA. Retrovirology 2009, 6, 16. [Google Scholar] [CrossRef]
- Casartelli, N.; Guivel-Benhassine, F.; Bouziat, R.; Brandler, S.; Schwartz, O.; Moris, A. The antiviral factor APOBEC3G improves CTL recognition of cultured HIV-infected T cells. J. Exp. Med. 2010, 207, 39–49. [Google Scholar] [CrossRef]
- Courcoul, M.; Patience, C.; Rey, F.; Blanc, D.; Harmache, A.; Sire, J.; Vigne, R.; Spire, B. Peripheral blood mononuclear cells produce normal amounts of defective Vif- human immunodeficiency virus type 1 particles which are restricted for the preretrotranscription steps. J. Virol. 1995, 69, 2068–2074. [Google Scholar]
- Fisher, A.G.; Ensoli, B.; Ivanoff, L.; Chamberlain, M.; Petteway, S.; Ratner, L.; Gallo, R.C.; Wong-Staal, F. The sor gene of HIV-1 is required for efficient virus transmission in vitro. Science 1987, 237, 888–893. [Google Scholar]
- Gabuzda, D.H.; Lawrence, K.; Langhoff, E.; Terwilliger, E.; Dorfman, T.; Haseltine, W.A.; Sodroski, J. Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J. Virol. 1992, 66, 6489–6495. [Google Scholar]
- Strebel, K.; Daugherty, D.; Clouse, K.; Cohen, D.; Folks, T.; Martin, M.A. The HIV 'A' (sor) gene product is essential for virus infectivity. Nature 1987, 328, 728–730. [Google Scholar]
- von Schwedler, U.; Song, J.; Aiken, C.; Trono, D. Vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J. Virol. 1993, 67, 4945–4955. [Google Scholar]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.F. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003, 302, 1056–1060. [Google Scholar]
- Yu, Y.; Xiao, Z.; Ehrlich, E.S.; Yu, X.; Yu, X.F. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 2004, 18, 2867–2872. [Google Scholar] [CrossRef]
- Mehle, A.; Goncalves, J.; Santa-Marta, M.; McPike, M.; Gabuzda, D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004, 18, 2861–2866. [Google Scholar] [CrossRef]
- Marin, M.; Rose, K.M.; Kozak, S.L.; Kabat, D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 2003, 9, 1398–1403. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Malim, M.H. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 2003, 9, 1404–1407. [Google Scholar] [CrossRef]
- Kobayashi, M.; Takaori-Kondo, A.; Miyauchi, Y.; Iwai, K.; Uchiyama, T. Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J Biol Chem. 2005, 280, 18573–18578. [Google Scholar]
- Liu, B.; Sarkis, P.T.; Luo, K.; Yu, Y.; Yu, X.F. Regulation of Apobec3F and human immunodeficiency virus type 1 Vif by Vif-Cul5-ElonB/C E3 ubiquitin ligase. J. Virol. 2005, 79, 9579–9587. [Google Scholar] [CrossRef]
- Bergeron, J.R.; Huthoff, H.; Veselkov, D.A.; Beavil, R.L.; Simpson, P.J.; Matthews, S.J.; Malim, M.H.; Sanderson, M.R. The SOCS-box of HIV-1 Vif interacts with ElonginBC by induced-folding to recruit its Cul5-containing ubiquitin ligase complex. PLoS Pathog. 2010, 6, e1000925. [Google Scholar]
- Jager, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 2012, 481, 371–375. [Google Scholar]
- Zhang, W.; Du, J.; Evans, S.L.; Yu, Y.; Yu, X.F. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature 2012, 481, 376–379. [Google Scholar]
- Jager, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global landscape of HIV-human protein complexes. Nature 2012, 481, 365–370. [Google Scholar]
- Kopietz, F.; Jaguva Vasudevan, AA.; Krämer, M.; Muckenfuss, H.; Sanzenbacher, R.; Cichutek, K.; Flory, E.; Münk, C. Interaction of Vif with APOBEC3G is not dependent on serine/threonine phosphorylation status. J. Gen. Virol. 2012, 93, 2425–2430. [Google Scholar]
- Dang, Y.; Wang, X.; York, I.A.; Zheng, Y.H. Identification of a critical T(Q/D/E)x5ADx2(I/L) motif from primate lentivirus Vif proteins that regulate APOBEC3G and APOBEC3F neutralizing activity. J. Virol. 2010, 84, 8561–8570. [Google Scholar] [CrossRef]
- Pery, E.; Rajendran, K.S.; Brazier, A.J.; Gabuzda, D. Regulation of APOBEC3 proteins by a novel YXXL motif in human immunodeficiency virus type 1 Vif and simian immunodeficiency virus SIVagm Vif. J. Virol. 2009, 83, 2374–2381. [Google Scholar] [CrossRef]
- He, Z.; Zhang, W.; Chen, G.; Xu, R.; Yu, X.F. Characterization of conserved motifs in HIV-1 Vif required for APOBEC3G and APOBEC3F interaction. J. Mol. Biol. 2008, 381, 1000–1011. [Google Scholar] [CrossRef]
- Russell, R.A.; Pathak, V.K. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J. Virol. 2007, 81, 8201–8210. [Google Scholar] [CrossRef]
- Dang, Y.; Wang, X.; Zhou, T.; York, I.A.; Zheng, Y.H. Identification of a novel WxSLVK motif in the N terminus of human immunodeficiency virus and simian immunodeficiency virus Vif that is critical for APOBEC3G and APOBEC3F neutralization. J. Virol. 2009, 83, 8544–8552. [Google Scholar]
- Dang, Y.; Davis, R.W.; York, I.A.; Zheng, Y.H. Identification of 81LGxGxxIxW89 and 171EDRW174 domains from human immunodeficiency virus type 1 Vif that regulate APOBEC3G and APOBEC3F neutralizing activity. J. Virol. 2010, 84, 5741–5750. [Google Scholar] [CrossRef]
- Kao, S.; Miyagi, E.; Khan, M.A.; Takeuchi, H.; Opi, S.; Goila-Gaur, R.; Strebel, K. Production of infectious human immunodeficiency virus type 1 does not require depletion of APOBEC3G from virus-producing cells. Retrovirology 2004, 1, 27. [Google Scholar] [CrossRef] [Green Version]
- Kao, S.; Goila-Gaur, R.; Miyagi, E.; Khan, M.A.; Opi, S.; Takeuchi, H.; Strebel, K. Production of infectious virus and degradation of APOBEC3G are separable functional properties of human immunodeficiency virus type 1 Vif. Virology 2007, 369, 329–339. [Google Scholar] [CrossRef]
- Opi, S.; Kao, S.; Goila-Gaur, R.; Khan, M.A.; Miyagi, E.; Takeuchi, H.; Strebel, K. Human immunodeficiency virus type 1 Vif inhibits packaging and antiviral activity of a degradation-resistant APOBEC3G variant. J. Virol. 2007, 81, 8236–8246. [Google Scholar] [CrossRef]
- Santa-Marta, M.; da Silva, F.A.; Fonseca, A.M.; Goncalves, J. HIV-1 Vif can directly inhibit apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G-mediated cytidine deamination by using a single amino acid interaction and without protein degradation. J. Biol. Chem. 2005, 280, 8765–8775. [Google Scholar]
- Goila-Gaur, R.; Khan, M.A.; Miyagi, E.; Kao, S.; Opi, S.; Takeuchi, H.; Strebel, K. HIV-1 Vif promotes the formation of high molecular mass APOBEC3G complexes. Virology 2008, 372, 136–146. [Google Scholar] [CrossRef]
- Britan-Rosich, E.; Nowarski, R.; Kotler, M. Multifaceted counter-APOBEC3G mechanisms employed by HIV-1 Vif. J. Mol. Biol. 2011, 410, 1065–1076. [Google Scholar] [CrossRef]
- Kao, S.; Khan, M.A.; Miyagi, E.; Plishka, R.; Buckler-White, A.; Strebel, K. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J. Virol. 2003, 77, 11398–11407. [Google Scholar] [CrossRef]
- Stopak, K.; de, N.C.; Yonemoto, W.; Greene, W.C. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 2003, 12, 591–601. [Google Scholar] [CrossRef]
- Felsenstein, J. Inferring Phylogenies; Sinauer Associates: Sunderland, Massachusetts, USA, 2004. [Google Scholar]
- Quasispecies: Concepts and Implications for Virology; Domingo, E. (Ed.) Springer: Berlin, Germany, 2006.
- Wilke, C.O. Quasispecies theory in the context of population genetics. BMC Evol. Biol. 2005, 5, 44. [Google Scholar] [CrossRef] [Green Version]
- Eigen, M. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften 1971, 58, 465–523. [Google Scholar] [CrossRef]
- Eigen, M.; Schuster, P. The hypercycle. A principle of natural self-organization. Part A: Emergence of the hypercycle. Naturwissenschaften 1977, 64, 541–565. [Google Scholar] [CrossRef]
- Domingo, E.; Biebricher, C.K.; Eigen, M.; Holland, J.J. Quasispecies and RNA Virus Evolution; Landes Bioscience: Georgetown, TA, USA, 2000. [Google Scholar]
- Bull, J.J.; Meyers, L.A.; Lachmann, M. Quasispecies made simple. PLoS Comput. Biol. 2005, 1, e61. [Google Scholar] [CrossRef]
- Wagner, G.P.; Krall, P. What is the difference between models of error thresholds and Muller's ratchet? J. Math. Biol. 1993, 32, 33–44. [Google Scholar] [CrossRef]
- Schuster, P.; Swetina, J. Stationary mutant distributions and evolutionary optimization. Bull. Math. Biol. 1988, 50, 635–660. [Google Scholar]
- Wilke, C.O.; Wang, J.L.; Ofria, C.; Lenski, R.E.; Adami, C. Evolution of digital organisms at high mutation rates leads to survival of the flattest. Nature 2001, 412, 331–333. [Google Scholar] [CrossRef]
- Baake, E.; Gabriel, W. Biological evolution through mutation, selection, and drift: An introductory review. In Annual Reviews of Computational Physics; World Scientific Publishing: London, UK, 2000; Volume 7, pp. 203–264. [Google Scholar]
- Nowak, M. Evolutionary Dynamics; Harvard University Press: Cambridge, Massachusetts, USA, 2006. [Google Scholar]
- Park, J.M.; Munoz, E.; Deem, M.W. Quasispecies theory for finite populations. Phys. Rev. E. Stat. Nonlin. Soft. Matter Phys. 2010, 81, 011902. [Google Scholar] [CrossRef]
- Muller, H.J. The relation of recombination to mutational advance. Mutat. Res. Fund. Mol. Mech. Mutagen. 1964, 1, 2–9. [Google Scholar] [CrossRef]
- Felsenstein, J. The evolutionary advantage of recombination. Genetics 1974, 78, 737–756. [Google Scholar]
- Barton, N.H.; Charlesworth, B. Why sex and recombination? Science 1998, 281, 1986–1990. [Google Scholar]
- Drossel, B. Biological evolution and statistical physics. Adv. Phys. 2001, 50, 209–295. [Google Scholar] [CrossRef]
- Kouyos, R.D.; Salathe, M.; Otto, S.P.; Bonhoeffer, S. The role of epistasis on the evolution of recombination in host-parasite coevolution. Theor. Popul. Biol. 2009, 75, 1–13. [Google Scholar]
- Kouyos, R.D.; Silander, O.K.; Bonhoeffer, S. Epistasis between deleterious mutations and the evolution of recombination. Trends Ecol. Evol. 2007, 22, 308–315. [Google Scholar] [CrossRef]
- Salathe, M.; Kouyos, R.D.; Bonhoeffer, S. The state of affairs in the kingdom of the Red Queen. Trends Ecol. Evol. 2008, 23, 439–445. [Google Scholar] [CrossRef]
- Salathe, M.; Kouyos, R.D.; Regoes, R.R.; Bonhoeffer, S. Rapid parasite adaptation drives selection for high recombination rates. Evolution 2008, 62, 295–300. [Google Scholar] [CrossRef]
- Smith, R.A.; Loeb, L.A.; Preston, B.D. Lethal mutagenesis of HIV. Virus Res. 2005, 107, 215–228. [Google Scholar] [CrossRef]
- Dapp, M.J.; Holtz, C.M.; Mansky, L.M. Concomitant lethal mutagenesis of human immunodeficiency virus type 1. J. Mol. Biol. 2012, 419, 158–170. [Google Scholar] [CrossRef]
- Sadler, H.A.; Stenglein, M.D.; Harris, R.S.; Mansky, L.M. APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis. J. Virol. 2010, 84, 7396–7404. [Google Scholar]
- Vijay, N.N.; Vasantika; Ajmani, R.; Perelson, A.S.; Dixit, N.M. ecombination increases human immunodeficiency virus fitness, but not necessarily diversity. J. Gen. Virol. 2008, 89, 1467–1477. [Google Scholar] [CrossRef]
- Althaus, C.L.; Bonhoeffer, S. Stochastic interplay between mutation and recombination during the acquisition of drug resistance mutations in human immunodeficiency virus type 1. J. Virol. 2005, 79, 13572–13578. [Google Scholar] [CrossRef]
- Cobey, S.; Koelle, K. Capturing escape in infectious disease dynamics. Trends Ecol. Evol. 2008, 23, 572–577. [Google Scholar] [CrossRef]
- Nilsson, M.; Snoad, N. Error thresholds for quasispecies on dynamic fitness landscapes. Phys. Rev. Lett. 2000, 84, 191–194. [Google Scholar] [CrossRef]
- Wilke, C.O.; Ronnewinkel, C.; Martinetz, T. Dynamic fitness landscapes in molecular evolution. Phys. Rep. 2001, 349, 395–446. [Google Scholar]
- Wilke, C.O. Evolution in time-dependent fitness landscapes. arXiv 1998. arXiv:physics/9811021. [Google Scholar]
- Orr, H.A. The rate of adaptation in asexuals. Genetics 2000, 155, 961–968. [Google Scholar]
- Kaplan, R.W. Evolutionary adjustment of spontaneous mutation rates. Humangenetik 1972, 16, 39–42. [Google Scholar] [CrossRef]
- Kimura, M. On the evolutionary adjustment of spontaneous mutation rates. Genet. Res. 1967, 9, 23–34. [Google Scholar] [CrossRef]
- Nilsson, M.; Snoad, N. Optimal mutation rates in dynamic environments. Bull. Math. Biol. 2002, 64, 1033–1043. [Google Scholar] [CrossRef]
- Kamp, C. A quasispecies approach to viral evolution in the context of an adaptive immune system. Microb. Infect. 2003, 5, 1397–1405. [Google Scholar] [CrossRef]
- Kamp, C.; Wilke, C.O.; Adami, C.; Bornholdt, S. Viral evolution under the pressure of an adaptive immune system: optimal mutation rates for viral escape. Complexity 2002, 8, 28–33. [Google Scholar] [CrossRef]
- Bonhoeffer, S.; Sniegowski, P. Virus evolution: The importance of being erroneous. Nature 2002, 420, 367–369. [Google Scholar] [CrossRef]
- Kamp, C.; Bornholdt, S. Coevolution of quasispecies: B-cell mutation rates maximize viral error catastrophes. Phys. Rev. Lett. 2002, 88, 068104. [Google Scholar] [CrossRef]
- Brumer, Y.; Shakhnovich, E.I. Host-parasite coevolution and optimal mutation rates for semiconservative quasispecies. Phys. Rev. E Stat. Nonlin. Soft. Matter Phys. 2004, 69, 061909. [Google Scholar] [CrossRef]
- Bianconi, G.; Fichera, D.; Franz, S.; Peliti, L. Modeling microevolution in a changing environment: The evolving quasispecies and the diluted champion process. J. Stat. Mech. Theor. Exp. 2011, 2011, 08022. [Google Scholar] [CrossRef]
- Kim, E.Y.; Bhattacharya, T.; Kunstman, K.; Swantek, P.; Koning, F.A.; Malim, M.H.; Wolinsky, S.M. Human APOBEC3G-mediated editing can promote HIV-1 sequence diversification and accelerate adaptation to selective pressure. J. Virol. 2010, 84, 10402–10405. [Google Scholar] [CrossRef]
- Mulder, L.C.; Harari, A.; Simon, V. Cytidine deamination induced HIV-1 drug resistance. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 5501–5506. [Google Scholar] [CrossRef]
- Fourati, S.; Malet, I.; Lambert, S.; Soulie, C.; Wirden, M.; Flandre, P.; Fofana, D.B.; Sayon, S.; Simon, A.; Katlama, C.; et al. E138K and M184I mutations in HIV-1 reverse transcriptase co-emerge as a result of APOBEC3 editing in the absence of drug exposure. AIDS 2012, 26, 1619–1624. [Google Scholar]
- Vartanian, J.P.; Meyerhans, A.; Asjo, B.; Wain-Hobson, S. Selection, recombination, and G----A hypermutation of human immunodeficiency virus type 1 genomes. J. Virol. 1991, 65, 1779–1788. [Google Scholar]
- Fourati, S.; Malet, I.; Binka, M.; Boukobza, S.; Wirden, M.; Sayon, S.; Simon, A.; Katlama, C.; Simon, V.; Calvez, V.; et al. Partially active HIV-1 Vif alleles facilitate viral escape from specific antiretrovirals. AIDS 2010, 24, 2313–2321. [Google Scholar]
- Zielonka, J.; Bravo, I.G.; Marino, D.; Conrad, E.; Perkovic, M.; Battenberg, M.; Cichutek, K.; Münk, C. Restriction of equine infectious anemia virus by equine APOBEC3 cytidine deaminases. J. Virol. 2009, 83, 7547–7559. [Google Scholar]
- Shen, X.; Leutenegger, C.M.; Stefano, C.K.; Pedersen, N.C.; Sparger, E.E. A feline immunodeficiency virus vif-deletion mutant remains attenuated upon infection of newborn kittens. J. Gen. Virol. 2007, 88, 2793–2799. [Google Scholar]
- Daniel, M.D.; Kirchhoff, F.; Czajak, S.C.; Sehgal, P.K.; Desrosiers, R.C. Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science 1992, 258, 1938–1941. [Google Scholar]
- Desrosiers, R.C.; Lifson, J.D.; Gibbs, J.S.; Czajak, S.C.; Howe, A.Y.; Arthur, L.O.; Johnson, R.P. Identification of highly attenuated mutants of simian immunodeficiency virus. J. Virol. 1998, 72, 1431–1437. [Google Scholar]
- Zielonka, J.; Marino, D.; Hofmann, H.; Yuhki, N.; Löchelt, M.; Münk, C. Vif of Feline Immunodeficiency Virus from Domestic Cats Protects against APOBEC3 Restriction Factors from Many Felids. J. Virol. 2010, 84, 7312–7324. [Google Scholar] [CrossRef]
- Mussil, B.; Sauermann, U.; Motzkus, D.; Stahl-Hennig, C.; Sopper, S. Increased APOBEC3G and APOBEC3F expression is associated with low viral load and prolonged survival in simian immunodeficiency virus infected rhesus monkeys. Retrovirology 2011, 8, 77. [Google Scholar] [CrossRef]
- Pace, C.; Keller, J.; Nolan, D.; James, I.; Gaudieri, S.; Moore, C.; Mallal, S. Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with APOBEC3G and vif genetic variation. J. Virol. 2006, 80, 9259–9269. [Google Scholar] [CrossRef]
- Kieffer, T.L.; Kwon, P.; Nettles, R.E.; Han, Y.; Ray, S.C.; Siliciano, R.F. G-->A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. J. Virol. 2005, 79, 1975–1980. [Google Scholar]
- Janini, M.; Rogers, M.; Birx, D.R.; McCutchan, F.E. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4(+) T cells. J. Virol. 2001, 75, 7973–7986. [Google Scholar]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 7552–7557. [Google Scholar]
- Rose, P.P.; Korber, B.T. Detecting hypermutations in viral sequences with an emphasis on G --> A hypermutation. Bioinformatics 2000, 16, 400–401. [Google Scholar] [CrossRef]
- Wood, N.; Bhattacharya, T.; Keele, B.F.; Giorgi, E.; Liu, M.; Gaschen, B.; Daniels, M.; Ferrari, G.; Haynes, B.F.; McMichael, A.; et al. HIV evolution in early infection: Selection pressures, patterns of insertion and deletion, and the impact of APOBEC. PLoS Pathog. 2009, 5, e1000414. [Google Scholar] [CrossRef] [Green Version]
- Kijak, G.H.; Janini, L.M.; Tovanabutra, S.; Sanders-Buell, E.; Arroyo, M.A.; Robb, M.L.; Michael, N.L.; Birx, D.L.; McCutchan, F.E. Variable contexts and levels of hypermutation in HIV-1 proviral genomes recovered from primary peripheral blood mononuclear cells. Virology 2008, 376, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Koulinska, I.N.; Chaplin, B.; Mwakagile, D.; Essex, M.; Renjifo, B. Hypermutation of HIV type 1 genomes isolated from infants soon after vertical infection. AIDS Res. Hum. Retrovir. 2003, 19, 1115–1123. [Google Scholar]
- Amoedo, N.D.; Afonso, A.O.; Cunha, S.M.; Oliveira, R.H.; Machado, E.S.; Soares, M.A. Expression of APOBEC3G/3F and G-to-A hypermutation levels in HIV-1-infected children with different profiles of disease progression. PLoS One 2011, 6, e24118. [Google Scholar]
- De Maio, F.A.; Rocco, C.A.; Aulicino, P.C.; Bologna, R.; Mangano, A.; Sen, L. APOBEC3-Mediated Editing in HIV Type 1 from Pediatric Patients and Its Association with APOBEC3G/CUL5 Polymorphisms and Vif Variability. AIDS Res. Hum. Retrovir. 2012, 28, 619–627. [Google Scholar] [CrossRef]
- Wei, M.; Xing, H.; Hong, K.; Huang, H.; Tang, H.; Qin, G.; Shao, Y. Biased G-to-A hypermutation in HIV-1 proviral DNA from a long-term non-progressor. AIDS 2004, 18, 1863–1865. [Google Scholar]
- Gandhi, S.K.; Siliciano, J.D.; Bailey, J.R.; Siliciano, R.F.; Blankson, J.N. Role of APOBEC3G/F-mediated hypermutation in the control of human immunodeficiency virus type 1 in elite suppressors. J. Virol. 2008, 82, 3125–3130. [Google Scholar] [CrossRef]
- Kourteva, Y.; De, P.M.; Allos, T.; McMunn, C.; D'Aquila, R.T. APOBEC3G expression and hypermutation are inversely associated with human immunodeficiency virus type 1 (HIV-1) burden in vivo. Virology 2012, 430, 1–9. [Google Scholar] [CrossRef]
- Land, A.M.; Ball, T.B.; Luo, M.; Pilon, R.; Sandstrom, P.; Embree, J.E.; Wachihi, C.; Kimani, J.; Plummer, F.A. Human immunodeficiency virus (HIV) type 1 proviral hypermutation correlates with CD4 count in HIV-infected women from Kenya. J. Virol. 2008, 82, 8172–8182. [Google Scholar] [CrossRef]
- Piantadosi, A.; Humes, D.; Chohan, B.; McClelland, R.S.; Overbaugh, J. Analysis of the percentage of human immunodeficiency virus type 1 sequences that are hypermutated and markers of disease progression in a longitudinal cohort, including one individual with a partially defective Vif. J. Virol. 2009, 83, 7805–7814. [Google Scholar] [CrossRef]
- Ulenga, N.K.; Sarr, A.D.; Hamel, D.; Sankale, J.L.; Mboup, S.; Kanki, P.J. The level of APOBEC3G (hA3G)-related G-to-A mutations does not correlate with viral load in HIV type 1-infected individuals. AIDS Res. Hum. Retrovir. 2008, 24, 1285–1290. [Google Scholar] [CrossRef]
- Jin, X.; Brooks, A.; Chen, H.; Bennett, R.; Reichman, R.; Smith, H. APOBEC3G/CEM15 (hA3G) mRNA levels associate inversely with human immunodeficiency virus viremia. J. Virol. 2005, 79, 11513–11516. [Google Scholar]
- Ulenga, N.K.; Sarr, A.D.; Thakore-Meloni, S.; Sankale, J.L.; Eisen, G.; Kanki, P.J. Relationship between human immunodeficiency type 1 infection and expression of human APOBEC3G and APOBEC3F. J Infect. Dis. 2008, 198, 486–492. [Google Scholar] [CrossRef]
- Vazquez-Perez, J.A.; Ormsby, C.E.; Hernandez-Juan, R.; Torres, K.J.; Reyes-Teran, G. APOBEC3G mRNA expression in exposed seronegative and early stage HIV infected individuals decreases with removal of exposure and with disease progression. Retrovirology 2009, 6, 23. [Google Scholar] [CrossRef]
- Cho, S.J.; Drechsler, H.; Burke, R.C.; Arens, M.Q.; Powderly, W.; Davidson, N.O. APOBEC3F and APOBEC3G mRNA levels do not correlate with human immunodeficiency virus type 1 plasma viremia or CD4+ T-cell count. J. Virol. 2006, 80, 2069–2072. [Google Scholar] [CrossRef]
- Reddy, K.; Winkler, C.A.; Werner, L.; Mlisana, K.; Abdool Karim, S.S.; Ndung'u, T. APOBEC3G expression is dysregulated in primary HIV-1 infection and polymorphic variants influence CD4+ T-cell counts and plasma viral load. AIDS 2010, 24, 195–204. [Google Scholar] [CrossRef]
- Mous, K.; Jennes, W.; Camara, M.; Seydi, M.; Daneau, G.; Mboup, S.; Kestens, L.; Van, O. X Expression analysis of LEDGF/p75, APOBEC3G, TRIM5alpha, and tetherin in a Senegalese cohort of HIV-1-exposed seronegative individuals. PLoS One 2012, 7, e33934. [Google Scholar]
- Bell, C.M.; Connell, B.J.; Capovilla, A.; Venter, W.D.; Stevens, W.S.; Papathanasopoulos, M.A. Molecular characterization of the HIV type 1 subtype C accessory genes vif, vpr, and vpu. AIDS Res. Hum. Retrovir. 2007, 23, 322–330. [Google Scholar] [CrossRef]
- Stephens, E.B.; Singh, D.K.; Pacyniak, E.; McCormick, C. Comparison of Vif sequences from diverse geographical isolates of HIV type 1 and SIV(cpz) identifies substitutions common to subtype C isolates and extensive variation in a proposed nuclear transport inhibition signal. AIDS Res. Hum. Retrovir. 2001, 17, 169–177. [Google Scholar] [CrossRef]
- Jacobs, G.B.; Nistal, M.; Laten, A.; van Rensburg, E.J.; Rethwilm, A.; Preiser, W.; Bodem, J.; Engelbrecht, S. Molecular analysis of HIV type 1 vif sequences from Cape Town, South Africa. AIDS Res. Hum. Retrovir. 2008, 24, 991–994. [Google Scholar] [CrossRef]
- Gupta, N.; Banerjea, A.C. C-terminal half of HIV-1 Vif C possesses major determinant for APOBEC3G degradation. AIDS 2009, 23, 141–143. [Google Scholar] [CrossRef]
- Adekale, M.A.; Cane, P.A.; McCrae, M.A. Changes in the Vif protein of HIV-1 associated with the development of resistance to inhibitors of viral protease. J. Med. Virol. 2005, 75, 195–201. [Google Scholar] [CrossRef]
- Saurya, S.; Lichtenstein, Z.; Karpas, A. Characterization of pol, vif, vpr, and vpu genes of HIV type 1 in AIDS patients with high viral load and stable CD4+ T cell counts on combination therapy. AIDS Res. Hum. Retrovir. 2002, 18, 1151–1155. [Google Scholar] [CrossRef]
- Wieland, U.; Hartmann, J.; Suhr, H.; Salzberger, B.; Eggers, H.J.; Kuhn, J.E. In vivo genetic variability of the HIV-1 vif gene. Virology 1994, 203, 43–51. [Google Scholar] [CrossRef]
- An, P.; Duggal, P.; Wang, L.H.; O'Brien, S.J.; Donfield, S.; Goedert, J.J.; Phair, J.; Buchbinder, S.; Kirk, G.D.; Winkler, C.A. Polymorphisms of CUL5 are associated with CD4+ T cell loss in HIV-1 infected individuals. PLoS Genet. 2007, 3, e19. [Google Scholar] [CrossRef]
- Binka, M.; Ooms, M.; Steward, M.; Simon, V. The activity spectrum of Vif from multiple HIV-1 subtypes against APOBEC3G, APOBEC3F, and APOBEC3H. J. Virol. 2012, 86, 49–59. [Google Scholar] [CrossRef]
- OhAinle, M.; Kerns, J.A.; Li, M.M.; Malik, H.S.; Emerman, M. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 2008, 4, 249–259. [Google Scholar] [CrossRef]
- Wang, X.; Abudu, A.; Son, S.; Dang, Y.; Venta, P.J.; Zheng, Y.H. Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. J. Virol. 2011, 85, 3142–3152. [Google Scholar] [CrossRef]
- Hassaine, G.; Agostini, I.; Candotti, D.; Bessou, G.; Caballero, M.; Agut, H.; Autran, B.; Barthalay, Y.; Vigne, R. Characterization of human immunodeficiency virus type 1 vif gene in long-term asymptomatic individuals. Virology 2000, 276, 169–180. [Google Scholar] [CrossRef]
- Sakurai, A.; Jere, A.; Yoshida, A.; Yamada, T.; Iwamoto, A.; Adachi, A.; Fujita, M. Functional analysis of HIV-1 vif genes derived from Japanese long-term nonprogressors and progressors for AIDS. Microbes. Infect. 2004, 6, 799–805. [Google Scholar]
- Zhang, L.; Huang, Y.; Yuan, H.; Tuttleton, S.; Ho, D.D. Genetic characterization of vif, vpr, and vpu sequences from long-term survivors of human immunodeficiency virus type 1 infection. Virology 1997, 228, 340–349. [Google Scholar] [CrossRef]
- Alexander, L.; quino-DeJesus, M.J.; Chan, M.; Andiman, W.A. Inhibition of human immunodeficiency virus type 1 (HIV-1) replication by a two-amino-acid insertion in HIV-1 Vif from a nonprogressing mother and child. J. Virol. 2002, 76, 10533–10539. [Google Scholar]
- Farrow, M.A.; Somasundaran, M.; Zhang, C.; Gabuzda, D.; Sullivan, J.L.; Greenough, T.C. Nuclear localization of HIV type 1 Vif isolated from a long-term asymptomatic individual and potential role in virus attenuation. AIDS Res. Hum. Retrovir. 2005, 21, 565–574. [Google Scholar]
- De Maio, F.A.; Rocco, C.A.; Aulicino, P.C.; Bologna, R.; Mangano, A.; Sen, L. Effect of HIV-1 Vif variability on progression to pediatric AIDS and its association with APOBEC3G and CUL5 polymorphisms. Infect. Genet. Evol. 2011, 11, 1256–1262. [Google Scholar] [CrossRef]
- Gourraud, P.A.; Karaouni, A.; Woo, J.M.; Schmidt, T.; Oksenberg, J.R.; Hecht, F.M.; Liegler, T.J.; Barbour, J.D. APOBEC3H haplotypes and HIV-1 pro-viral vif DNA sequence diversity in early untreated human immunodeficiency virus-1 infection. Hum. Immunol. 2011, 72, 207–212. [Google Scholar] [CrossRef]
- Valcke, H.S.; Bernard, N.F.; Bruneau, J.; Alary, M.; Tsoukas, C.M.; Roger, M. APOBEC3G genetic variants and their association with risk of HIV infection in highly exposed Caucasians. AIDS 2006, 20, 1984–1986. [Google Scholar] [CrossRef]
- Do, H.; Vasilescu, A.; Diop, G.; Hirtzig, T.; Heath, S.C.; Coulonges, C.; Rappaport, J.; Therwath, A.; Lathrop, M.; Matsuda, F.; et al. Exhaustive genotyping of the CEM15 (APOBEC3G) gene and absence of association with AIDS progression in a French cohort. J. Infect. Dis. 2005, 191, 159–163. [Google Scholar]
- An, P.; Bleiber, G.; Duggal, P.; Nelson, G.; May, M.; Mangeat, B.; Alobwede, I.; Trono, D.; Vlahov, D.; Donfield, S.; et al. APOBEC3G genetic variants and their influence on the progression to AIDS. J. Virol. 2004, 78, 11070–11076. [Google Scholar] [CrossRef]
- Bizinoto, M.C.; Leal, E.; Diaz, R.S.; Janini, L.M. Loci polymorphisms of the APOBEC3G gene in HIV type 1-infected Brazilians. AIDS Res. Hum. Retrovir. 2011, 27, 137–141. [Google Scholar] [CrossRef]
- Harari, A.; Ooms, M.; Mulder, L.C.; Simon, V. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J. Virol. 2009, 83, 295–303. [Google Scholar] [CrossRef]
- Sadler, H.A.; Stenglein, M.D.; Harris, R.S.; Mansky, L.M. APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis. J. Virol. 2010, 84, 7396–7404. [Google Scholar]
- Heger, E.; Thielen, A.; Gilles, R.; Obermeier, M.; Lengauer, T.; Kaiser, R.; Trapp, S. APOBEC3G/F as one possible driving force for co-receptor switch of the human immunodeficiency virus-1. Med. Microbiol. Immunol. 2012, 201, 7–16. [Google Scholar] [CrossRef]
- Iyengar, S.; Schwartz, D.H. Acquisition of CD4-dependence by CD4-independent SIV passaged in human peripheral blood mononuclear cells. Retrovirology 2012, 9, 61. [Google Scholar] [CrossRef]
- Pillai, S.K.; Wong, J.K.; Barbour, J.D. Turning up the volume on mutational pressure: Is more of a good thing always better? (A case study of HIV-1 Vif and APOBEC3). Retrovirology 2008, 5, 26. [Google Scholar] [CrossRef]
- Muller, V.; Bonhoeffer, S. Guanine-adenine bias: A general property of retroid viruses that is unrelated to host-induced hypermutation. Trends Genet. 2005, 21, 264–268. [Google Scholar] [CrossRef]
- Jern, P.; Russell, R.A.; Pathak, V.K.; Coffin, J.M. Likely role of APOBEC3G-mediated G-to-A mutations in HIV-1 evolution and drug resistance. PLoS Pathog. 2009, 5, e1000367. [Google Scholar]
- Ebrahimi, D.; Anwar, F.; Davenport, M.P. APOBEC3 has not left an evolutionary footprint on the HIV-1 genome. J. Virol. 2011, 85, 9139–9146. [Google Scholar] [CrossRef]
- Nowarski, R.; Britan-Rosich, E.; Shiloach, T.; Kotler, M. Hypermutation by intersegmental transfer of APOBEC3G cytidine deaminase. Nat. Struct. Mol. Biol. 2008, 15, 1059–1066. [Google Scholar] [CrossRef]
- Xu, H.; Chertova, E.; Chen, J.; Ott, D.E.; Roser, J.D.; Hu, W.S.; Pathak, V.K. Stoichiometry of the antiviral protein APOBEC3G in HIV-1 virions. Virology 2007, 360, 247–256. [Google Scholar] [CrossRef]
- Browne, E.P.; Allers, C.; Landau, N.R. Restriction of HIV-1 by APOBEC3G is cytidine deaminase-dependent. Virology 2009, 387, 313–321. [Google Scholar] [CrossRef]
- Armitage, A.E.; Deforche, K.; Chang, C.H.; Wee, E.; Kramer, B.; Welch, J.J.; Gerstoft, J.; Fugger, L.; McMichael, A.; Rambaut, A.; et al. APOBEC3G-induced hypermutation of human immunodeficiency virus type-1 is typically a discrete "All or Nothing" phenomenon. PLoS Genet. 2012, 8, e1002550. [Google Scholar] [CrossRef]
- Smyth, R.P.; Davenport, M.P.; Mak, J. The origin of genetic diversity in HIV-1. Virus Res. 2012, 169, 415–424. [Google Scholar] [CrossRef]
- Neher, R.A.; Leitner, T. Recombination rate and selection strength in HIV intra-patient evolution. PLoS Comput. Biol. 2010, 6, e1000660. [Google Scholar] [CrossRef]
- Zhuang, J.; Jetzt, A.E.; Sun, G.; Yu, H.; Klarmann, G.; Ron, Y.; Preston, B.D.; Dougherty, J.P. Human immunodeficiency virus type 1 recombination: Rate, fidelity, and putative hot spots. J. Virol. 2002, 76, 11273–11282. [Google Scholar] [CrossRef]
- Nora, T.; Charpentier, C.; Tenaillon, O.; Hoede, C.; Clavel, F.; Hance, A.J. Contribution of recombination to the evolution of human immunodeficiency viruses expressing resistance to antiretroviral treatment. J. Virol. 2007, 81, 7620–7628. [Google Scholar]
- Bretscher, M.T.; Althaus, C.L.; Muller, V.; Bonhoeffer, S. Recombination in HIV and the evolution of drug resistance: For better or for worse? Bioessays 2004, 26, 180–188. [Google Scholar] [CrossRef]
- Althaus, C.L.; Bonhoeffer, S. Stochastic interplay between mutation and recombination during the acquisition of drug resistance mutations in human immunodeficiency virus type 1. J. Virol. 2005, 79, 13572–13578. [Google Scholar]
- Rouzine, I.M.; Coffin, J.M. Evolution of human immunodeficiency virus under selection and weak recombination. Genetics 2005, 170, 7–18. [Google Scholar] [CrossRef]
- Perales, C.; Iranzo, J.; Manrubia, S.C.; Domingo, E. The impact of quasispecies dynamics on the use of therapeutics. Trends Microbiol. 2012. [Google Scholar] [CrossRef]
- Iranzo, J.; Perales, C.; Domingo, E.; Manrubia, S.C. Tempo and mode of inhibitor-mutagen antiviral therapies: A multidisciplinary approach. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 16008–16013. [Google Scholar]
- Hosseini, I.; Gabhann, F.M. Multi-scale modeling of HIV infection in vitro and APOBEC3G-based anti-retroviral therapy. PLoS Comput. Biol. 2012, 8, e1002371. [Google Scholar] [CrossRef]
- Lobkovsky, A.E.; Wolf, Y.I.; Koonin, E.V. Predictability of evolutionary trajectories in fitness landscapes. PLoS Comput. Biol. 2011, 7, e1002302. [Google Scholar] [CrossRef]
- Franke, J.; Klozer, A.; de Visser, J.A.; Krug, J. Evolutionary accessibility of mutational pathways. PLoS Comput. Biol. 2011, 7, e1002134. [Google Scholar] [CrossRef]
- da, S.J.; Coetzer, M.; Nedellec, R.; Pastore, C.; Mosier, D.E. Fitness epistasis and constraints on adaptation in a human immunodeficiency virus type 1 protein region. Genetics 2010, 185, 293–303. [Google Scholar] [CrossRef]
- Kouyos, R.D.; von Wyl, V.; Hinkley, T.; Petropoulos, C.J.; Haddad, M.; Whitcomb, J.M.; Boni, J.; Yerly, S.; Cellerai, C.; Klimkait, T.; et al. Assessing predicted HIV-1 replicative capacity in a clinical setting. PLoS Pathog. 2011, 7, e1002321. [Google Scholar]
- Kouyos, R.D.; Leventhal, G.E.; Hinkley, T.; Haddad, M.; Whitcomb, J.M.; Petropoulos, C.J.; Bonhoeffer, S. Exploring the complexity of the HIV-1 fitness landscape. PLoS Genet. 2012, 8, e1002551. [Google Scholar]
- Hinkley, T.; Martins, J.; Chappey, C.; Haddad, M.; Stawiski, E.; Whitcomb, J.M.; Petropoulos, C.J.; Bonhoeffer, S. A systems analysis of mutational effects in HIV-1 protease and reverse transcriptase. Nat. Genet. 2011, 43, 487–489. [Google Scholar] [CrossRef]
- Ali, A.; Wang, J.; Nathans, R.S.; Cao, H.; Sharova, N.; Stevenson, M.; Rana, T.M. Synthesis and structure-activity relationship studies of HIV-1 virion infectivity factor (Vif) inhibitors that block viral replication. ChemMedChem 2012, 7, 1217–1229. [Google Scholar]
- Cen, S.; Peng, Z.G.; Li, X.Y.; Li, Z.R.; Ma, J.; Wang, Y.M.; Fan, B.; You, X.F.; Wang, Y.P.; Liu, F.; et al. Small molecular compounds inhibit HIV-1 replication through specifically stabilizing APOBEC3G. J. Biol. Chem. 2010, 285, 16546–16552. [Google Scholar]
- Nathans, R.; Cao, H.; Sharova, N.; Ali, A.; Sharkey, M.; Stranska, R.; Stevenson, M.; Rana, T.M. Small-molecule inhibition of HIV-1 Vif. Nat. Biotechnol. 2008, 26, 1187–1192. [Google Scholar]
- Zuo, T.; Liu, D.; Lv, W.; Wang, X.; Wang, J.; Lv, M.; Huang, W.; Wu, J.; Zhang, H.; Jin, H.; et al. Small-molecule inhibition of human immunodeficiency virus type 1 replication by targeting the interaction between Vif and ElonginC. J. Virol. 2012, 86, 5497–5507. [Google Scholar]
- Ao, Z.; Wang, X.; Bello, A.; Jayappa, K.D.; Yu, Z.; Fowke, K.; He, X.; Chen, X.; Li, J.; Kobinger, G.; et al. Characterization of anti-HIV activity mediated by R88-APOBEC3G mutant fusion proteins in CD4+ T cells, peripheral blood mononuclear cells, and macrophages. Hum. Gene Ther. 2011, 22, 1225–1237. [Google Scholar]
- Ao, Z.; Yu, Z.; Wang, L.; Zheng, Y.; Yao, X. Vpr14-88-Apobec3G fusion protein is efficiently incorporated into Vif-positive HIV-1 particles and inhibits viral infection. PLoS One 2008, 3, e1995. [Google Scholar] [CrossRef]
- Green, L.A.; Liu, Y.; He, J.J. Inhibition of HIV-1 infection and replication by enhancing viral incorporation of innate anti-HIV-1 protein A3G: A non-pathogenic Nef mutant-based anti-HIV strategy. J. Biol. Chem. 2009, 284, 13363–13372. [Google Scholar] [CrossRef]
- Goila-Gaur, R.; Khan, M.A.; Miyagi, E.; Kao, S.; Strebel, K. Targeting APOBEC3A to the viral nucleoprotein complex confers antiviral activity. Retrovirology 2007, 4, 61. [Google Scholar] [CrossRef]
- Aguiar, R.S.; Lovsin, N.; Tanuri, A.; Peterlin, B.M. Vpr.A3A chimera inhibits HIV replication. J. Biol. Chem. 2008, 283, 2518–2525. [Google Scholar]
- Li, M.; Shandilya, S.M.; Carpenter, M.A.; Rathore, A.; Brown, W.L.; Perkins, A.L.; Harki, D.A.; Solberg, J.; Hook, D.J.; Pandey, K.K.; et al. First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem. Biol. 2012, 7, 506–517. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Münk, C.; Jensen, B.-E.O.; Zielonka, J.; Häussinger, D.; Kamp, C. Running Loose or Getting Lost: How HIV-1 Counters and Capitalizes on APOBEC3-Induced Mutagenesis through Its Vif Protein. Viruses 2012, 4, 3132-3161. https://doi.org/10.3390/v4113132
Münk C, Jensen B-EO, Zielonka J, Häussinger D, Kamp C. Running Loose or Getting Lost: How HIV-1 Counters and Capitalizes on APOBEC3-Induced Mutagenesis through Its Vif Protein. Viruses. 2012; 4(11):3132-3161. https://doi.org/10.3390/v4113132
Chicago/Turabian StyleMünk, Carsten, Björn-Erik O. Jensen, Jörg Zielonka, Dieter Häussinger, and Christel Kamp. 2012. "Running Loose or Getting Lost: How HIV-1 Counters and Capitalizes on APOBEC3-Induced Mutagenesis through Its Vif Protein" Viruses 4, no. 11: 3132-3161. https://doi.org/10.3390/v4113132