Cellular Aspects of Prion Replication In Vitro

Abstract
:1. Introduction
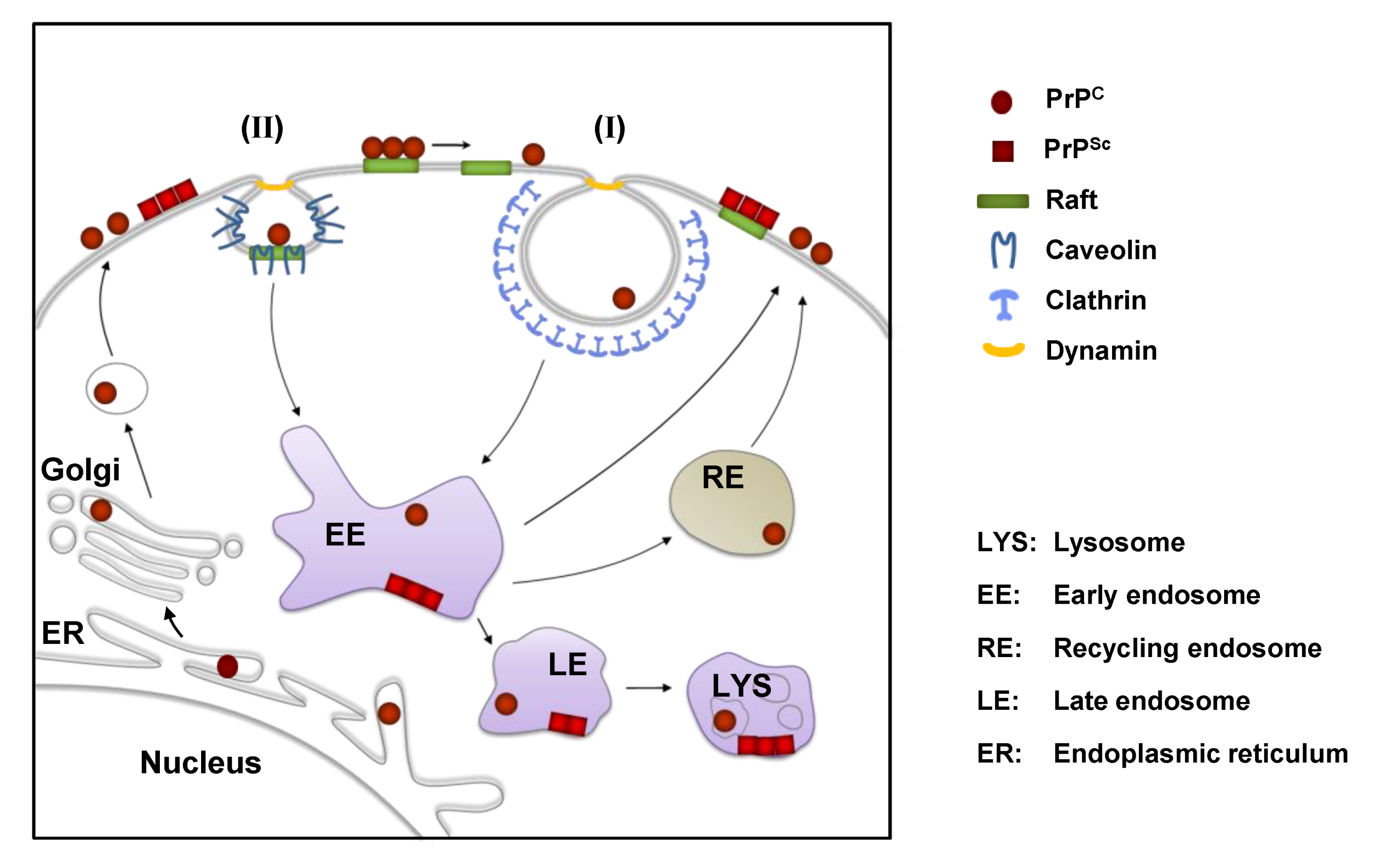
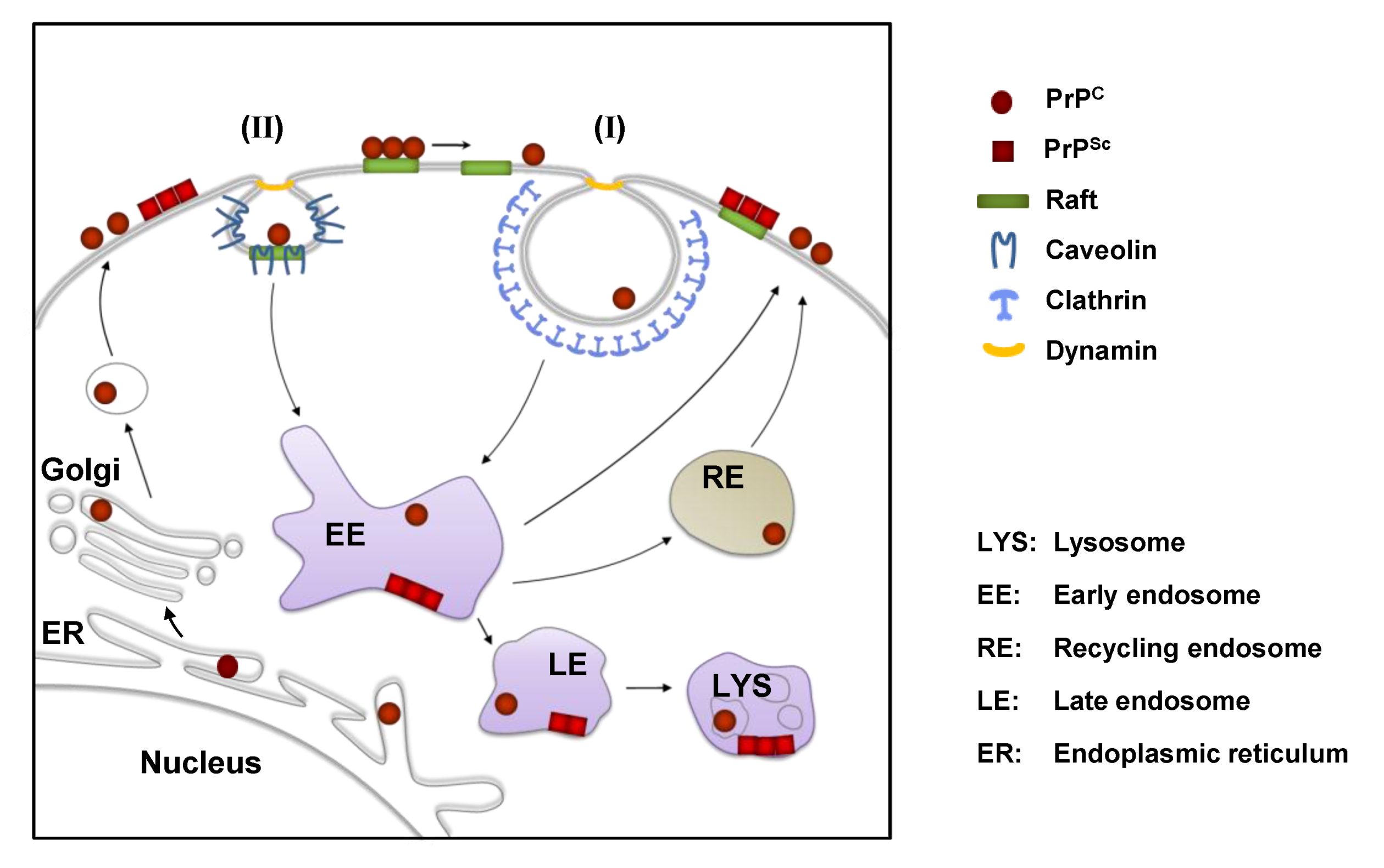
2. The Cellular Prion Protein PrPC: Structure, Biosynthesis and Intracellular Trafficking
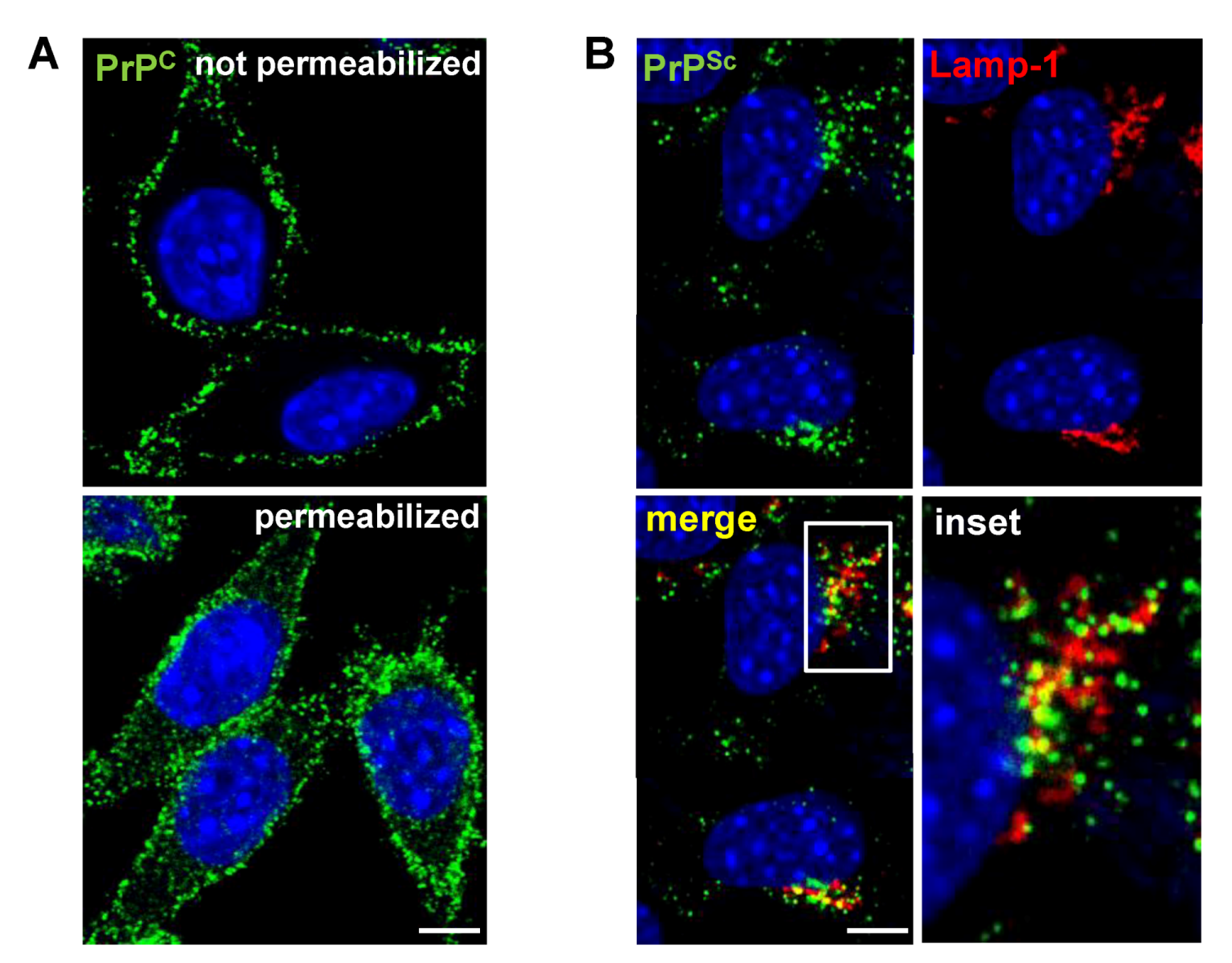
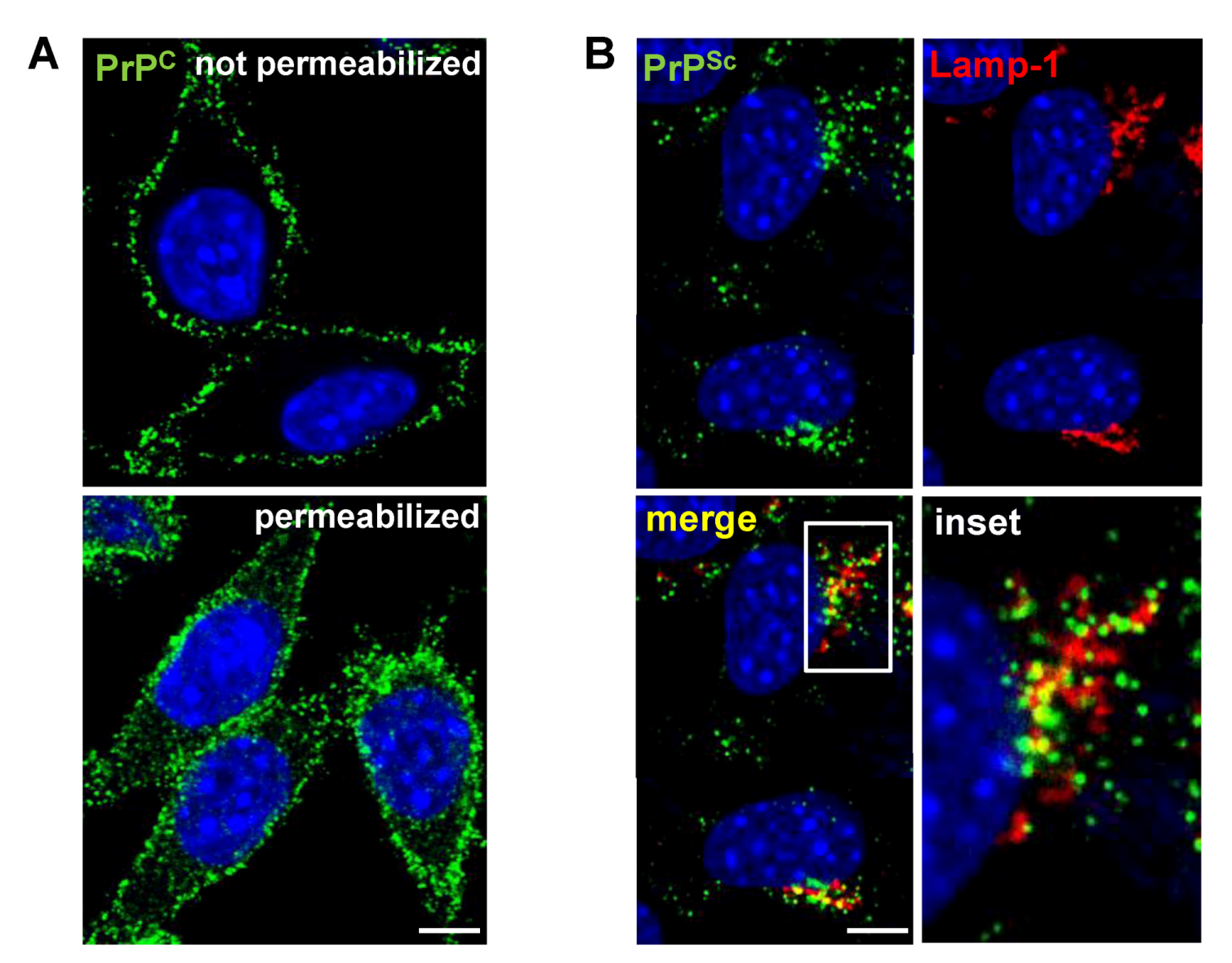
3. Cell Surface Receptors for PrPC
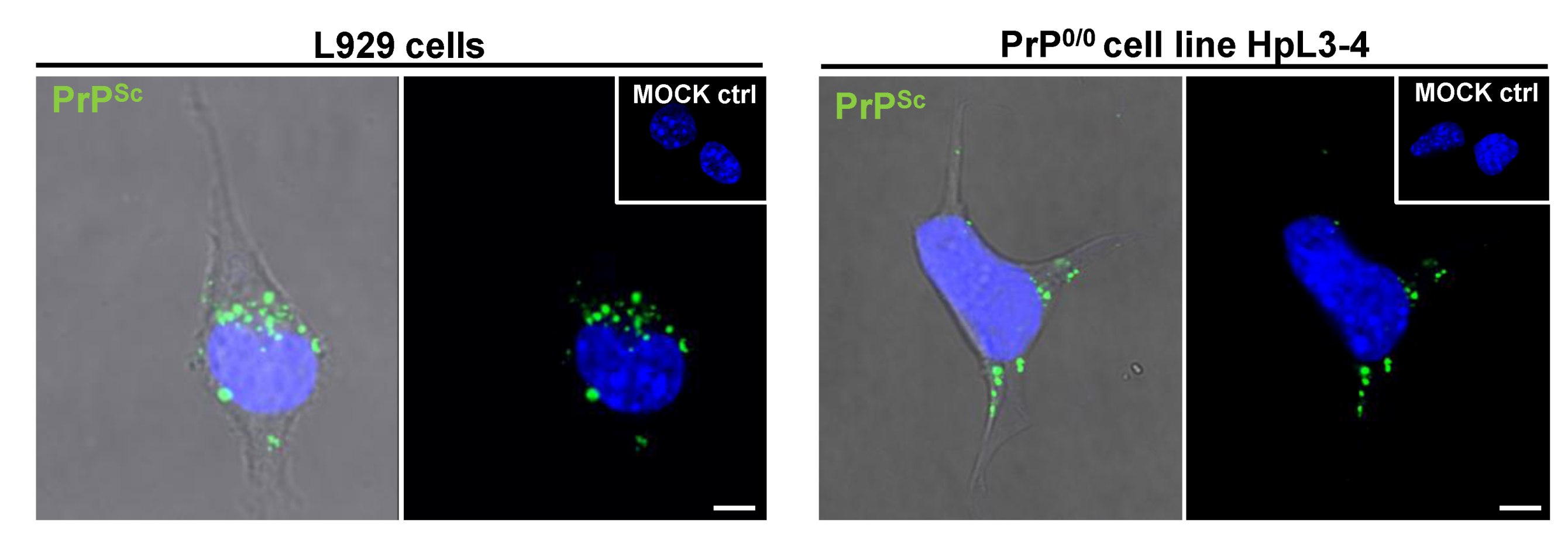
4. Cellular Models for Studying PrPSc Formation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell designation | Tissue of origin or cell type | Species of origin | Prion strain | References |
|---|---|---|---|---|
| 1. Neuronal or brain-derived cells | ||||
| N2a | neuroblastoma cell line* | mouse | Chandler, RML, 139A, 22L, C506, Fukuoka-1, FU CJD | [95,96,127,139,140,141,142,143,144] |
| GT1 | hypothalamic cell line | mouse | Chandler, RML, 139A, 22L, kCJD, FU CJD , M1000 | [96,97,99,120,139,145] |
| SN56 | cholinergic septal cell line | mouse | Chandler, ME7, 22L | [146] |
| HpL3-4 | hippocampal PrP-deficient cell line, upon ectopic expression of moPrP* | mouse | 22L | [121,147] |
| CF10 | brain derived PrP-deficient cell line, upon ectopic expression of moPrP | mouse | 22L | [122] |
| SMB | prion-infected brain cell | mouse | Chandler, 139A, 22F, 79A | [93,148,149] |
| CAD | catecholaminergic cell line | mouse | RML, 22L, 22F, 79A, 139A, ME7 | [125,150,151,152] |
| MG20 | microglial cell line overexpressing PrPC | tg20 mouse | Chandler, ME7, Obihiro, mouse-adapted BSE | [98] |
| PC12 | pheochromocytoma cell line | rat | 139A, ME7 | [153,154,155] |
| HaB | brain-derived cell line | hamster | Sc237 | [131] |
| SH-SY5Y | neuroblastoma cell line | human | sCJD brain material | [119] |
| MDB | primary brain cells, SV40 transformed | mule deer | CWD | [129] |
| 2. Primary neuronal or brain-derived cells | ||||
| CGN | cerebellar granule neurons overexpressing ovine PrPC | tgov mouse | mo 127S | [111] |
| CAS | cerebellar astrocytes overexpressing ovine PrPC | tgov mouse | mo 127S | [111] |
| NSC | neural stem cells | mouse | 22L, RML | [112,113,115] |
| 3. Non-neuronal cells | ||||
| C2C12 | skeletal myoblast cell line | mouse | 22L | [108] |
| L fibroblasts | fibroblast cell line | mouse | ME7, Chandler | [106] |
| L929 | fibroblast cell line | mouse | 22L, RML, ME7 | [107] |
| NIH/3T3 | fibroblast cell line | mouse | 22L | [107] |
| MSC-80 | Schwann cell line | mouse | Chandler | [156] |
| MovS | Schwann cell-like from dorsal root ganglia | tgov mouse | PG127, SSBP/1, scrapie field isolates | [104,157] |
| moRK13 | epithelial cell line expressing mouse PrPC | rabbit | Fukuoka-1, 22L, Chandler, M1000, mo sCJD | [99,100,101,120] |
| voRK13 | epithelial cell line expressing vole PrPC | rabbit | vo BSE | [100] |
| ovRK13/ RoV9 | epithelial cell line expressing ovine PrPC | rabbit | PG127, LA404, SSBP/1, scrapie field isolates | [102,103,104] |
| elkRK13 | epithelial cell line expressing elk PrPC | rabbit | CWD | [105,109] |
| 4. Primary non-neuronal cells | ||||
| BM-derived MSC | bone marrow derived mesenchymal stem cell | mouse | Fukuoka-1 | [110] |
| BM-derived MSC-like | bone marrow derived mesenchymal stem cell like | mouse | Fukuoka-1 | [114] |
5. PrPSc Uptake During the Infection Process

6. Early Steps of Prion Infection
7. PrPSc Formation in Persistently Infected Cells
8. GAGs As Co-Factors for PrPSc Formation
9. Cell-To-Cell Transmission of Prions
| Prion-infected donor cell line | Prion strain | Intercellular prion spreading | PrPSc secreted | References |
|---|---|---|---|---|
| N2a | 22L | Yes, via conditioned medium | Yes, associated with exosomes | [36] |
| N2a | RML | No or inefficient | Not determined | [97,196] |
| SMB | Chandler | Yes, via direct cell contact | Not determined | [149] |
| HpL3-4* | 22L | Yes, via conditioned medium | Not determined | [121] |
| NIH/3T3 | 22L | Yes, via conditioned medium | Yes, associated with exosomes | [198] |
| CAD | 139A | Yes, via TNTs | Not determined | [200] |
| GT1 | RML | Yes, via conditioned medium | Not determined | [97] |
| GT1 | FU CJD | Yes, via conditioned medium | Not determined | [201] |
| GT1 | M1000 | Yes | Yes, associated with exosomes | [99] |
| ovRK13/ RoV9 | PG127 | Yes (inefficiently) | Yes, associated with exosomes | [37,197] |
| moRK13 | M1000 | Yes | Yes, associated with exosomes | [99] |
| Mov | PG127 | Yes, via close proximity of cells | Yes, associated with exosomes | [37,111,197] |
| SN56 | Chandler | Yes, via conditioned medium | Yes | [202] |
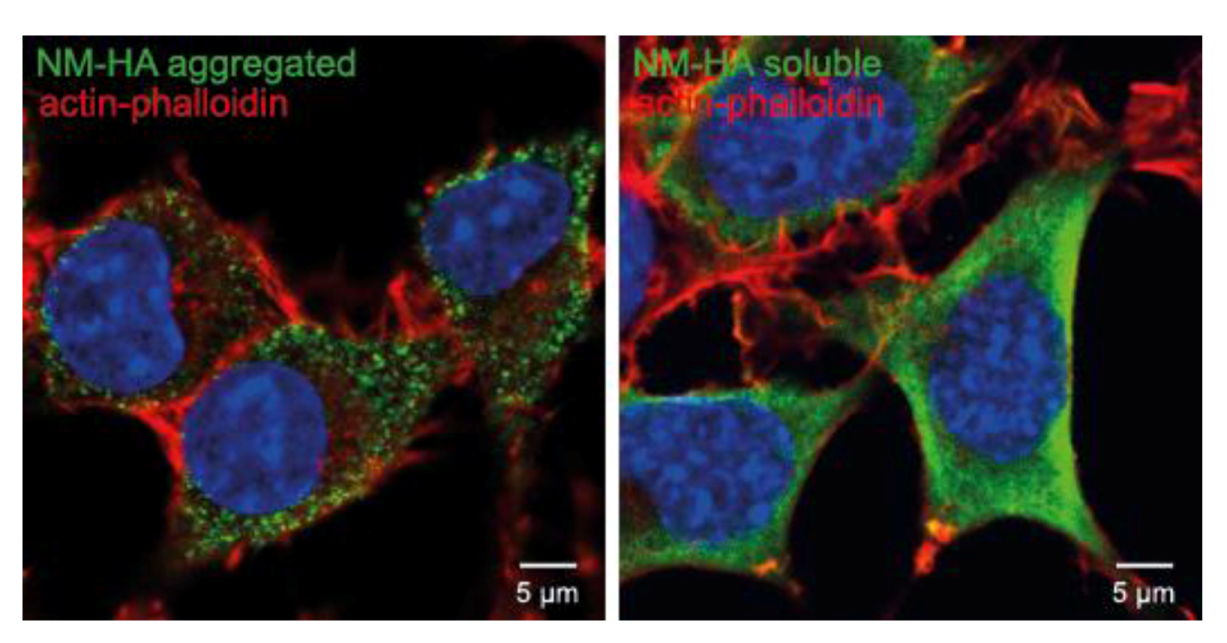
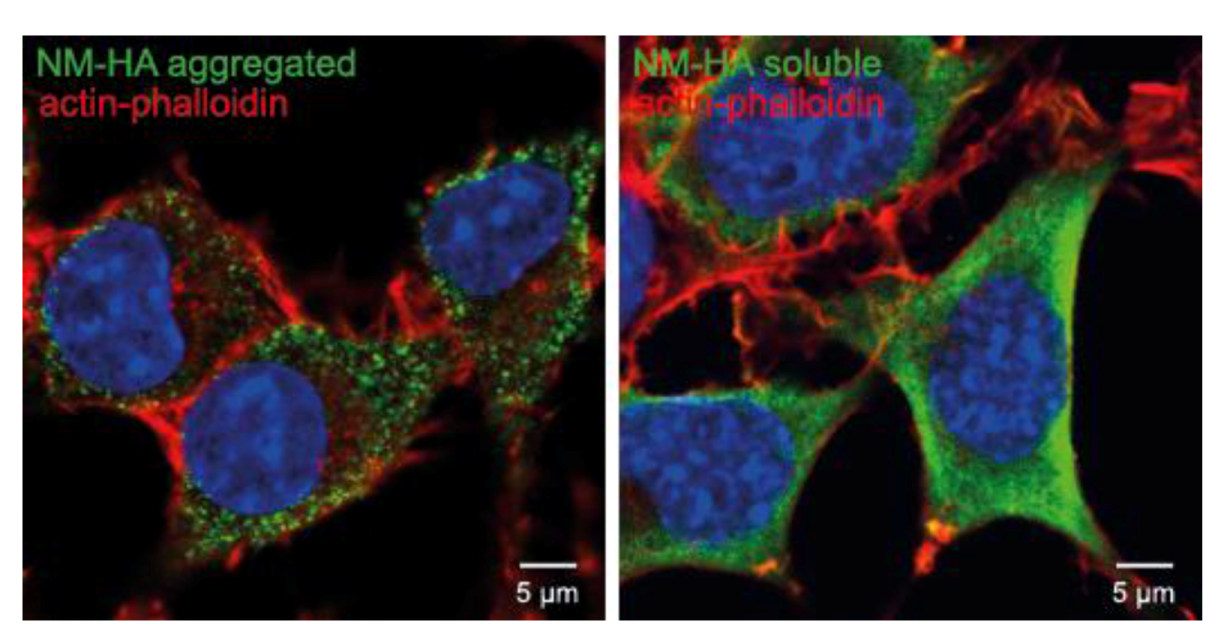
10. Other Protein Aggregates Can Spread and Propagate in Cell Culture
11. Concluding Remarks
Acknowledgments
Conflict of Interest
References
- Beekes, M.; McBride, P.A. The spread of prions through the body in naturally acquired transmissible spongiform encephalopathies. FEBS J. 2007, 274, 588–605. [Google Scholar] [CrossRef]
- Bolton, D.C.; McKinley, M.P.; Prusiner, S.B. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Pattison, I.H.; Millson, G.C. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J. Comp. Pathol. 1961, 71, 101–109. [Google Scholar] [CrossRef]
- Bessen, R.A.; Marsh, R.F. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J. Virol. 1992, 66, 2096–2101. [Google Scholar]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have prp(sc) molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar]
- Asante, E.A.; Linehan, J.M.; Desbruslais, M.; Joiner, S.; Gowland, I.; Wood, A.L.; Welch, J.; Hill, A.F.; Lloyd, S.E.; Wadsworth, J.D.; et al. Bse prions propagate as either variant cjd-like or sporadic cjd-like prion strains in transgenic mice expressing human prion protein. EMBO J. 2002, 21, 6358–6366. [Google Scholar] [CrossRef]
- Cali, I.; Castellani, R.; Alshekhlee, A.; Cohen, Y.; Blevins, J.; Yuan, J.; Langeveld, J.P.; Parchi, P.; Safar, J.G.; Zou, W.Q.; et al. Co-existence of scrapie prion protein types 1 and 2 in sporadic creutzfeldt-jakob disease: Its effect on the phenotype and prion-type characteristics. Brain 2009, 132, 2643–2658. [Google Scholar] [CrossRef]
- Weissmann, C. Birth of a prion: Spontaneous generation revisited. Cell. 2005, 122, 165–168. [Google Scholar] [CrossRef]
- Chesebro, B.; Race, R.; Wehrly, K.; Nishio, J.; Bloom, M.; Lechner, D.; Bergstrom, S.; Robbins, K.; Mayer, L.; Keith, J.M.; et al. Identification of scrapie prion protein-specific mrna in scrapie-infected and uninfected brain. Nature 1985, 315, 331–333. [Google Scholar] [CrossRef]
- Basler, K.; Oesch, B.; Scott, M.; Westaway, D.; Walchli, M.; Groth, D.F.; McKinley, M.P.; Prusiner, S.B.; Weissmann, C. Scrapie and cellular prp isoforms are encoded by the same chromosomal gene. Cell. 1986, 46, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Robakis, N.K.; Sawh, P.R.; Wolfe, G.C.; Rubenstein, R.; Carp, R.I.; Innis, M.A. Isolation of a cdna clone encoding the leader peptide of prion protein and expression of the homologous gene in various tissues. Proc. Natl. Acad. Sci. USA 1986, 83, 6377–6381. [Google Scholar] [CrossRef]
- Sparkes, R.S.; Simon, M.; Cohn, V.H.; Fournier, R.E.; Lem, J.; Klisak, I.; Heinzmann, C.; Blatt, C.; Lucero, M.; Mohandas, T.; et al. Assignment of the human and mouse prion protein genes to homologous chromosomes. Proc. Natl. Acad. Sci. USA 1986, 83, 7358–7362. [Google Scholar]
- Oesch, B.; Westaway, D.; Prusiner, S.B. Prion protein genes: Evolutionary and functional aspects. Curr. Top. Microbiol. Immunol. 1991, 172, 109–124. [Google Scholar]
- Schatzl, H.M.; Da Costa, M.; Taylor, L.; Cohen, F.E.; Prusiner, S.B. Prion protein gene variation among primates. J. Mol. Biol 1995, 245, 362–374. [Google Scholar] [CrossRef]
- Wopfner, F.; Weidenhofer, G.; Schneider, R.; von Brunn, A.; Gilch, S.; Schwarz, T.F.; Werner, T.; Schatzl, H.M. Analysis of 27 mammalian and 9 avian prps reveals high conservation of flexible regions of the prion protein. J. Mol. Biol. 1999, 289, 1163–1178. [Google Scholar] [CrossRef]
- Linden, R.; Martins, V.R.; Prado, M.A.; Cammarota, M.; Izquierdo, I.; Brentani, R.R. Physiology of the prion protein. Physiol. Rev. 2008, 88, 673–728. [Google Scholar] [CrossRef]
- Brown, D.R.; Schmidt, B.; Kretzschmar, H.A. Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature 1996, 380, 345–347. [Google Scholar] [CrossRef]
- Moser, M.; Colello, R.J.; Pott, U.; Oesch, B. Developmental expression of the prion protein gene in glial cells. Neuron 1995, 14, 509–517. [Google Scholar] [CrossRef]
- Haraguchi, T.; Fisher, S.; Olofsson, S.; Endo, T.; Groth, D.; Tarentino, A.; Borchelt, D.R.; Teplow, D.; Hood, L.; Burlingame, A.; et al. Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch. Biochem. Biophys. 1989, 274, 1–13. [Google Scholar] [CrossRef]
- Turk, E.; Teplow, D.B.; Hood, L.E.; Prusiner, S.B. Purification and properties of the cellular and scrapie hamster prion proteins. Eur. J. Biochem. 1988, 176, 21–30. [Google Scholar] [CrossRef]
- Stahl, N.; Baldwin, M.A.; Hecker, R.; Pan, K.M.; Burlingame, A.L.; Prusiner, S.B. Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemistry 1992, 31, 5043–5053. [Google Scholar]
- Taraboulos, A.; Scott, M.; Semenov, A.; Avrahami, D.; Laszlo, L.; Prusiner, S.B. Cholesterol depletion and modification of cooh-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell. Biol 1995, 129, 121–132. [Google Scholar] [CrossRef]
- Vey, M.; Pilkuhn, S.; Wille, H.; Nixon, R.; DeArmond, S.J.; Smart, E.J.; Anderson, R.G.; Taraboulos, A.; Prusiner, S.B. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc. Natl. Acad. Sci. USA 1996, 93, 14945–14949. [Google Scholar]
- Baron, G.S.; Wehrly, K.; Dorward, D.W.; Chesebro, B.; Caughey, B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of prp-res (prp(sc)) into contiguous membranes. EMBO J. 2002, 21, 1031–1040. [Google Scholar] [CrossRef]
- Walmsley, A.R.; Watt, N.T.; Taylor, D.R.; Perera, W.S.; Hooper, N.M. Alpha-cleavage of the prion protein occurs in a late compartment of the secretory pathway and is independent of lipid rafts. Mol. Cell. Neurosci. 2009, 40, 242–248. [Google Scholar] [CrossRef]
- Sarnataro, D.; Campana, V.; Paladino, S.; Stornaiuolo, M.; Nitsch, L.; Zurzolo, C. Prp(c) association with lipid rafts in the early secretory pathway stabilizes its cellular conformation. Mol. Biol. Cell. 2004, 15, 4031–4042. [Google Scholar] [CrossRef]
- Mironov, A., Jr.; Latawiec, D.; Wille, H.; Bouzamondo-Bernstein, E.; Legname, G.; Williamson, R.A.; Burton, D.; DeArmond, S.J.; Prusiner, S.B.; Peters, P.J. Cytosolic prion protein in neurons. J. Neurosci. 2003, 23, 7183–7193. [Google Scholar]
- Vincent, B.; Paitel, E.; Frobert, Y.; Lehmann, S.; Grassi, J.; Checler, F. Phorbol ester-regulated cleavage of normal prion protein in hek293 human cells and murine neurons. J. Biol. Chem. 2000, 275, 35612–35616. [Google Scholar]
- Vincent, B.; Paitel, E.; Saftig, P.; Frobert, Y.; Hartmann, D.; De Strooper, B.; Grassi, J.; Lopez-Perez, E.; Checler, F. The disintegrins adam10 and tace contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J. Biol. Chem. 2001, 276, 37743–37746. [Google Scholar]
- Parkin, E.T.; Watt, N.T.; Turner, A.J.; Hooper, N.M. Dual mechanisms for shedding of the cellular prion protein. J. Biol. Chem. 2004, 279, 11170–11178. [Google Scholar]
- Alfa Cisse, M.; Sunyach, C.; Slack, B.E.; Fisher, A.; Vincent, B.; Checler, F. M1 and m3 muscarinic receptors control physiological processing of cellular prion by modulating adam17 phosphorylation and activity. J. Neurosci. 2007, 27, 4083–4092. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Rogers, M.; Stahl, N.; Telling, G.; Prusiner, S.B. Release of the cellular prion protein from cultured cells after loss of its glycoinositol phospholipid anchor. Glycobiology 1993, 3, 319–329. [Google Scholar] [CrossRef]
- Starke, R.; Harrison, P.; Drummond, O.; Macgregor, I.; Mackie, I.; Machin, S. The majority of cellular prion protein released from endothelial cells is soluble. Transfusion 2003, 43, 677–678, author reply 678.. [Google Scholar] [CrossRef]
- Alais, S.; Simoes, S.; Baas, D.; Lehmann, S.; Raposo, G.; Darlix, J.L.; Leblanc, P. Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biol. Cell 2008, 100, 603–615. [Google Scholar] [CrossRef]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar]
- Rane, N.S.; Yonkovich, J.L.; Hegde, R.S. Protection from cytosolic prion protein toxicity by modulation of protein translocation. EMBO J. 2004, 23, 4550–4559. [Google Scholar] [CrossRef]
- Crozet, C.; Vezilier, J.; Delfieu, V.; Nishimura, T.; Onodera, T.; Casanova, D.; Lehmann, S.; Beranger, F. The truncated 23–230 form of the prion protein localizes to the nuclei of inducible cell lines independently of its nuclear localization signals and is not cytotoxic. Mol. Cell. Neurosci. 2006, 32, 315–323. [Google Scholar] [CrossRef]
- Linden, R.; Cordeiro, Y.; Lima, L.M. Allosteric function and dysfunction of the prion protein. Cell. Mol. Life Sci. 2012, 69, 1105–1124. [Google Scholar] [CrossRef]
- Gibbings, D.; Leblanc, P.; Jay, F.; Pontier, D.; Michel, F.; Schwab, Y.; Alais, S.; Lagrange, T.; Voinnet, O. Human prion protein binds argonaute and promotes accumulation of microrna effector complexes. Nat. Struct. Mol. Biol. 2012, 19, 517–524, S511. [Google Scholar] [CrossRef]
- Cheng, F.; Lindqvist, J.; Haigh, C.L.; Brown, D.R.; Mani, K. Copper-dependent co-internalization of the prion protein and glypican-1. J. Neurochem. 2006, 98, 1445–1457. [Google Scholar] [CrossRef]
- Watt, N.T.; Taylor, D.R.; Kerrigan, T.L.; Griffiths, H.H.; Rushworth, J.V.; Whitehouse, I.J.; Hooper, N.M. Prion protein facilitates uptake of zinc into neuronal cells. Nat. Commun. 2012, 3, 1134. [Google Scholar] [CrossRef]
- Graner, E.; Mercadante, A.F.; Zanata, S.M.; Forlenza, O.V.; Cabral, A.L.; Veiga, S.S.; Juliano, M.A.; Roesler, R.; Walz, R.; Minetti, A.; et al. Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res. Mol. 2000, 76, 85–92. [Google Scholar] [CrossRef]
- Zanata, S.M.; Lopes, M.H.; Mercadante, A.F.; Hajj, G.N.; Chiarini, L.B.; Nomizo, R.; Freitas, A.R.; Cabral, A.L.; Lee, K.S.; Juliano, M.A.; et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002, 21, 3307–3316. [Google Scholar]
- Santuccione, A.; Sytnyk, V.; Leshchyns'ka, I.; Schachner, M. Prion protein recruits its neuronal receptor ncam to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell. Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.C.; Steele, A.D.; Lindquist, S.; Lodish, H.F. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc. Natl. Acad. Sci. USA 2006, 103, 2184–2189. [Google Scholar] [CrossRef]
- Steele, A.D.; Emsley, J.G.; Ozdinler, P.H.; Lindquist, S.; Macklis, J.D. Prion protein (prpc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 3416–3421. [Google Scholar]
- Gabus, C.; Auxilien, S.; Pechoux, C.; Dormont, D.; Swietnicki, W.; Morillas, M.; Surewicz, W.; Nandi, P.; Darlix, J.L. The prion protein has DNA strand transfer properties similar to retroviral nucleocapsid protein. J. Mol. Biol. 2001, 307, 1011–1021. [Google Scholar] [CrossRef]
- Gabus, C.; Derrington, E.; Leblanc, P.; Chnaiderman, J.; Dormont, D.; Swietnicki, W.; Morillas, M.; Surewicz, W.K.; Marc, D.; Nandi, P.; et al. The prion protein has rna binding and chaperoning properties characteristic of nucleocapsid protein ncp7 of hiv-1. J. Biol. Chem. 2001, 276, 19301–19309. [Google Scholar]
- Cordeiro, Y.; Machado, F.; Juliano, L.; Juliano, M.A.; Brentani, R.R.; Foguel, D.; Silva, J.L. DNA converts cellular prion protein into the beta-sheet conformation and inhibits prion peptide aggregation. J. Biol. Chem. 2001, 276, 49400–49409. [Google Scholar]
- Lima, L.M.; Cordeiro, Y.; Tinoco, L.W.; Marques, A.F.; Oliveira, C.L.; Sampath, S.; Kodali, R.; Choi, G.; Foguel, D.; Torriani, I.; et al. Structural insights into the interaction between prion protein and nucleic acid. Biochemistry 2006, 45, 9180–9187. [Google Scholar]
- Morris, R.J.; Parkyn, C.J.; Jen, A. Traffic of prion protein between different compartments on the neuronal surface, and the propagation of prion disease. FEBS Lett. 2006, 580, 5565–5571. [Google Scholar] [CrossRef]
- Sunyach, C.; Jen, A.; Deng, J.; Fitzgerald, K.T.; Frobert, Y.; Grassi, J.; McCaffrey, M.W.; Morris, R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 2003, 22, 3591–3601. [Google Scholar] [CrossRef]
- Caetano, F.A.; Lopes, M.H.; Hajj, G.N.; Machado, C.F.; Pinto Arantes, C.; Magalhaes, A.C.; Vieira Mde, P.; Americo, T.A.; Massensini, A.R.; Priola, S.A.; et al. Endocytosis of prion protein is required for erk1/2 signaling induced by stress-inducible protein 1. J. Neurosci 2008, 28, 6691–6702. [Google Scholar]
- Magalhaes, A.C.; Silva, J.A.; Lee, K.S.; Martins, V.R.; Prado, V.F.; Ferguson, S.S.; Gomez, M.V.; Brentani, R.R.; Prado, M.A. Endocytic intermediates involved with the intracellular trafficking of a fluorescent cellular prion protein. J. Biol. Chem. 2002, 277, 33311–33318. [Google Scholar]
- Prado, M.A.; Alves-Silva, J.; Magalhaes, A.C.; Prado, V.F.; Linden, R.; Martins, V.R.; Brentani, R.R. Prpc on the road: Trafficking of the cellular prion protein. J. Neurochem. 2004, 88, 769–781. [Google Scholar] [CrossRef]
- Campana, V.; Sarnataro, D.; Zurzolo, C. The highways and byways of prion protein trafficking. Trends Cell. Biol. 2005, 15, 102–111. [Google Scholar] [CrossRef]
- Stuermer, C.A.; Langhorst, M.F.; Wiechers, M.F.; Legler, D.F.; Von Hanwehr, S.H.; Guse, A.H.; Plattner, H. Prpc capping in t cells promotes its association with the lipid raft proteins reggie-1 and reggie-2 and leads to signal transduction. FASEB J. 2004, 18, 1731–1733. [Google Scholar]
- Shyng, S.L.; Huber, M.T.; Harris, D.A. A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J. Biol. Chem. 1993, 268, 15922–15928. [Google Scholar]
- Godsave, S.F.; Wille, H.; Kujala, P.; Latawiec, D.; DeArmond, S.J.; Serban, A.; Prusiner, S.B.; Peters, P.J. Cryo-immunogold electron microscopy for prions: Toward identification of a conversion site. J. Neurosci. 2008, 28, 12489–12499. [Google Scholar]
- Peters, P.J.; Mironov, A.., Jr.; Peretz, D.; van Donselaar, E.; Leclerc, E.; Erpel, S.; DeArmond, S.J.; Burton, D.R.; Williamson, R.A.; Vey, M.; et al. Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J. Cell. Biol. 2003, 162, 703–717. [Google Scholar] [CrossRef]
- Baumann, M.H.; Kallijarvi, J.; Lankinen, H.; Soto, C.; Haltia, M. Apolipoprotein e includes a binding site which is recognized by several amyloidogenic polypeptides. Biochem. J. 2000, 349, 77–84. [Google Scholar] [CrossRef]
- Kang, Y.S.; Zhao, X.; Lovaas, J.; Eisenberg, E.; Greene, L.E. Clathrin-independent internalization of normal cellular prion protein in neuroblastoma cells is associated with the arf6 pathway. J. Cell. Sci. 2009, 122, 4062–4069. [Google Scholar] [CrossRef]
- Marella, M.; Lehmann, S.; Grassi, J.; Chabry, J. Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular prp release. J. Biol. Chem. 2002, 277, 25457–25464. [Google Scholar]
- Taylor, D.R.; Watt, N.T.; Perera, W.S.; Hooper, N.M. Assigning functions to distinct regions of the n-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J. Cell Sci. 2005, 118, 5141–5153. [Google Scholar] [CrossRef]
- Watt, N.T.; Hooper, N.M. Reactive oxygen species (ros)-mediated beta-cleavage of the prion protein in the mechanism of the cellular response to oxidative stress. Biochem. Soc. Trans. 2005, 33, 1123–1125. [Google Scholar] [CrossRef]
- Martins, V.R.; Graner, E.; Garcia-Abreu, J.; de Souza, S.J.; Mercadante, A.F.; Veiga, S.S.; Zanata, S.M.; Neto, V.M.; Brentani, R.R. Complementary hydropathy identifies a cellular prion protein receptor. Nat. Med. 1997, 3, 1376–1382. [Google Scholar] [CrossRef]
- Pauly, P.C.; Harris, D.A. Copper stimulates endocytosis of the prion protein. J. Biol. Chem. 1998, 273, 33107–33110. [Google Scholar] [CrossRef]
- Madore, N.; Smith, K.L.; Graham, C.H.; Jen, A.; Brady, K.; Hall, S.; Morris, R. Functionally different gpi proteins are organized in different domains on the neuronal surface. EMBO J. 1999, 18, 6917–6926. [Google Scholar] [CrossRef]
- Galvan, C.; Camoletto, P.G.; Dotti, C.G.; Aguzzi, A.; Ledesma, M.D. Proper axonal distribution of prp(c) depends on cholesterol-sphingomyelin-enriched membrane domains and is developmentally regulated in hippocampal neurons. Mol. Cell. Neurosci. 2005, 30, 304–315. [Google Scholar] [CrossRef]
- Sarnataro, D.; Caputo, A.; Casanova, P.; Puri, C.; Paladino, S.; Tivodar, S.S.; Campana, V.; Tacchetti, C.; Zurzolo, C. Lipid rafts and clathrin cooperate in the internalization of prp in epithelial frt cells. PLoS One 2009, 4, e5829. [Google Scholar]
- Kirchhausen, T. Clathrin. Annu Rev. Biochem. 2000, 69, 699–727. [Google Scholar] [CrossRef]
- Gauczynski, S.; Peyrin, J.M.; Haik, S.; Leucht, C.; Hundt, C.; Rieger, R.; Krasemann, S.; Deslys, J.P.; Dormont, D.; Lasmezas, C.I.; et al. The 37-kda/67-kda laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 2001, 20, 5863–5875. [Google Scholar] [CrossRef]
- Gauczynski, S.; Hundt, C.; Leucht, C.; Weiss, S. Interaction of prion proteins with cell surface receptors, molecular chaperones, and other molecules. Adv. Protein. Chem. 2001, 57, 229–272. [Google Scholar] [CrossRef]
- Rieger, R.; Edenhofer, F.; Lasmezas, C.I.; Weiss, S. The human 37-kda laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat. Med. 1997, 3, 1383–1388. [Google Scholar] [CrossRef]
- Hundt, C.; Peyrin, J.M.; Haik, S.; Gauczynski, S.; Leucht, C.; Rieger, R.; Riley, M.L.; Deslys, J.P.; Dormont, D.; Lasmezas, C.I.; et al. Identification of interaction domains of the prion protein with its 37-kda/67-kda laminin receptor. EMBO J. 2001, 20, 5876–5886. [Google Scholar] [CrossRef]
- Douville, P.J.; Harvey, W.J.; Carbonetto, S. Isolation and partial characterization of high affinity laminin receptors in neural cells. J. Biol. Chem. 1988, 263, 14964–14969. [Google Scholar]
- Watts, J.C.; Huo, H.; Bai, Y.; Ehsani, S.; Jeon, A.H.; Shi, T.; Daude, N.; Lau, A.; Young, R.; Xu, L.; et al. Interactome analyses identify ties of prp and its mammalian paralogs to oligomannosidic n-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog. 2009, 5, e1000608. [Google Scholar] [CrossRef]
- Nykjaer, A.; Willnow, T.E. The low-density lipoprotein receptor gene family: A cellular swiss army knife? Trends Cell. Biol. 2002, 12, 273–280. [Google Scholar] [CrossRef]
- Wu, L.; Gonias, S.L. The low-density lipoprotein receptor-related protein-1 associates transiently with lipid rafts. J. Cell. Biochem. 2005, 96, 1021–1033. [Google Scholar] [CrossRef]
- Taylor, D.R.; Hooper, N.M. The low-density lipoprotein receptor-related protein 1 (lrp1) mediates the endocytosis of the cellular prion protein. Biochem. J. 2007, 402, 17–23. [Google Scholar] [CrossRef]
- Parkyn, C.J.; Vermeulen, E.G.; Mootoosamy, R.C.; Sunyach, C.; Jacobsen, C.; Oxvig, C.; Moestrup, S.; Liu, Q.; Bu, G.; Jen, A.; et al. Lrp1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J. Cell. Sci. 2008, 121, 773–783. [Google Scholar] [CrossRef]
- Wang, S.; Herndon, M.E.; Ranganathan, S.; Godyna, S.; Lawler, J.; Argraves, W.S.; Liau, G. Internalization but not binding of thrombospondin-1 to low density lipoprotein receptor-related protein-1 requires heparan sulfate proteoglycans. J. Cell. Biochem. 2004, 91, 766–776. [Google Scholar] [CrossRef]
- Yin, S.; Yu, S.; Li, C.; Wong, P.; Chang, B.; Xiao, F.; Kang, S.C.; Yan, H.; Xiao, G.; Grassi, J.; et al. Prion proteins with insertion mutations have altered n-terminal conformation and increased ligand binding activity and are more susceptible to oxidative attack. J. Biol. Chem. 2006, 281, 10698–10705. [Google Scholar]
- Pan, T.; Wong, B.S.; Liu, T.; Li, R.; Petersen, R.B.; Sy, M.S. Cell-surface prion protein interacts with glycosaminoglycans. Biochem. J. 2002, 368, 81–90. [Google Scholar] [CrossRef]
- Warner, R.G.; Hundt, C.; Weiss, S.; Turnbull, J.E. Identification of the heparan sulfate binding sites in the cellular prion protein. J. Biol. Chem. 2002, 277, 18421–18430. [Google Scholar]
- Gabizon, R.; Meiner, Z.; Halimi, M.; Ben-Sasson, S.A. Heparin-like molecules bind differentially to prion-proteins and change their intracellular metabolic fate. J. Cell. Physiol. 1993, 157, 319–325. [Google Scholar] [CrossRef]
- Caughey, B.; Raymond, G.J. Sulfated polyanion inhibition of scrapie-associated prp accumulation in cultured cells. J. Virol. 1993, 67, 643–650. [Google Scholar]
- Shyng, S.L.; Lehmann, S.; Moulder, K.L.; Harris, D.A. Sulfated glycans stimulate endocytosis of the cellular isoform of the prion protein, prpc, in cultured cells. J. Biol. Chem. 1995, 270, 30221–30229. [Google Scholar]
- Ben-Zaken, O.; Tzaban, S.; Tal, Y.; Horonchik, L.; Esko, J.D.; Vlodavsky, I.; Taraboulos, A. Cellular heparan sulfate participates in the metabolism of prions. J. Biol. Chem. 2003, 278, 40041–40049. [Google Scholar]
- Gilch, S.; Winklhofer, K.F.; Groschup, M.H.; Nunziante, M.; Lucassen, R.; Spielhaupter, C.; Muranyi, W.; Riesner, D.; Tatzelt, J.; Schatzl, H.M. Intracellular re-routing of prion protein prevents propagation of prp(sc) and delays onset of prion disease. EMBO J. 2001, 20, 3957–3966. [Google Scholar] [CrossRef]
- Clarke, M.C.; Haig, D.A. Multiplication of scrapie agent in cell culture. Res. Vet. Sci 1970, 11, 500–501. [Google Scholar]
- Caughey, B.; Race, R.E.; Ernst, D.; Buchmeier, M.J.; Chesebro, B. Prion protein biosynthesis in scrapie-infected and uninfected neuroblastoma cells. J. Virol. 1989, 63, 175–181. [Google Scholar]
- Butler, D.A.; Scott, M.R.; Bockman, J.M.; Borchelt, D.R.; Taraboulos, A.; Hsiao, K.K.; Kingsbury, D.T.; Prusiner, S.B. Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. J. Virol 1988, 62, 1558–1564. [Google Scholar]
- Nishida, N.; Harris, D.A.; Vilette, D.; Laude, H.; Frobert, Y.; Grassi, J.; Casanova, D.; Milhavet, O.; Lehmann, S. Successful transmission of three mouse-adapted scrapie strains to murine neuroblastoma cell lines overexpressing wild-type mouse prion protein. J. Virol. 2000, 74, 320–325. [Google Scholar] [CrossRef]
- Schatzl, H.M.; Laszlo, L.; Holtzman, D.M.; Tatzelt, J.; DeArmond, S.J.; Weiner, R.I.; Mobley, W.C.; Prusiner, S.B. A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. J. Virol. 1997, 71, 8821–8831. [Google Scholar]
- Iwamaru, Y.; Takenouchi, T.; Ogihara, K.; Hoshino, M.; Takata, M.; Imamura, M.; Tagawa, Y.; Hayashi-Kato, H.; Ushiki-Kaku, Y.; Shimizu, Y.; et al. Microglial cell line established from prion protein-overexpressing mice is susceptible to various murine prion strains. J. Virol. 2007, 81, 1524–1527. [Google Scholar]
- Vella, L.J.; Sharples, R.A.; Lawson, V.A.; Masters, C.L.; Cappai, R.; Hill, A.F. Packaging of prions into exosomes is associated with a novel pathway of prp processing. J. Pathol. 2007, 211, 582–590. [Google Scholar] [CrossRef]
- Courageot, M.P.; Daude, N.; Nonno, R.; Paquet, S.; Di Bari, M.A.; Le Dur, A.; Chapuis, J.; Hill, A.F.; Agrimi, U.; Laude, H.; et al. A cell line infectible by prion strains from different species. J. Gen. Virol. 2008, 89, 341–347. [Google Scholar] [CrossRef]
- Lawson, V.A.; Vella, L.J.; Stewart, J.D.; Sharples, R.A.; Klemm, H.; Machalek, D.M.; Masters, C.L.; Cappai, R.; Collins, S.J.; Hill, A.F. Mouse-adapted sporadic human creutzfeldt-jakob disease prions propagate in cell culture. Int. J. Biochem. Cell. Biol. 2008, 40, 2793–2801. [Google Scholar] [CrossRef]
- Vilette, D.; Andreoletti, O.; Archer, F.; Madelaine, M.F.; Vilotte, J.L.; Lehmann, S.; Laude, H. Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc. Natl. Acad. Sci. USA 2001, 98, 4055–4059. [Google Scholar]
- Sabuncu, E.; Petit, S.; Le Dur, A.; Lan Lai, T.; Vilotte, J.L.; Laude, H.; Vilette, D. Prp polymorphisms tightly control sheep prion replication in cultured cells. J. Virol. 2003, 77, 2696–2700. [Google Scholar]
- Neale, M.H.; Mountjoy, S.J.; Edwards, J.C.; Vilette, D.; Laude, H.; Windl, O.; Saunders, G.C. Infection of cell lines with experimental and natural ovine scrapie agents. J. Virol. 2010, 84, 2444–2452. [Google Scholar]
- Bian, J.; Napier, D.; Khaychuck, V.; Angers, R.; Graham, C.; Telling, G. Cell-based quantification of chronic wasting disease prions. J. Virol. 2010, 84, 8322–8326. [Google Scholar] [CrossRef]
- Clarke, M.C.; Millson, G.C. Infection of a cell line of mouse l fibroblasts with scrapie agent. Nature 1976, 261, 144–145. [Google Scholar] [CrossRef]
- Vorberg, I.; Raines, A.; Story, B.; Priola, S.A. Susceptibility of common fibroblast cell lines to transmissible spongiform encephalopathy agents. J. Infect. Dis. 2004, 189, 431–439. [Google Scholar] [CrossRef]
- Dlakic, W.M.; Grigg, E.; Bessen, R.A. Prion infection of muscle cells in vitro. J. Virol. 2007, 81, 4615–4624. [Google Scholar] [CrossRef]
- Kim, H.J.; Tark, D.S.; Lee, Y.H.; Kim, M.J.; Lee, W.Y.; Cho, I.S.; Sohn, H.J.; Yokoyama, T. Establishment of a cell line persistently infected with chronic wasting disease prions. J. Vet. Med. Sci. 2012, 74, 1377–1380. [Google Scholar] [CrossRef]
- Akimov, S.; Vasilyeva, I.; Yakovleva, O.; McKenzie, C.; Cervenakova, L. Murine bone marrow stromal cell culture with features of mesenchymal stem cells susceptible to mouse-adapted human tse agent, fukuoka-1. Folia. Neuropathol. 2009, 47, 205–214. [Google Scholar]
- Cronier, S.; Laude, H.; Peyrin, J.M. Prions can infect primary cultured neurons and astrocytes and promote neuronal cell death. Proc. Natl. Acad. Sci. USA 2004, 101, 12271–12276. [Google Scholar] [CrossRef]
- Giri, R.K.; Young, R.; Pitstick, R.; DeArmond, S.J.; Prusiner, S.B.; Carlson, G.A. Prion infection of mouse neurospheres. Proc. Natl. Acad. Sci. USA 2006, 103, 3875–3880. [Google Scholar]
- Milhavet, O.; Casanova, D.; Chevallier, N.; McKay, R.D.; Lehmann, S. Neural stem cell model for prion propagation. Stem. Cell. 2006, 24, 2284–2291. [Google Scholar] [CrossRef]
- Cervenakova, L.; Akimov, S.; Vasilyeva, I.; Yakovleva, O.; McKenzie, C.; Cervenak, J.; Piccardo, P.; Asher, D.M. Fukuoka-1 strain of transmissible spongiform encephalopathy agent infects murine bone marrow-derived cells with features of mesenchymal stem cells. Transfusion 2011, 51, 1755–1768. [Google Scholar] [CrossRef]
- Herva, M.E.; Relano-Gines, A.; Villa, A.; Torres, J.M. Prion infection of differentiated neurospheres. J. Neurosci. Meth. 2010, 188, 270–275. [Google Scholar] [CrossRef]
- Polymenidou, M.; Trusheim, H.; Stallmach, L.; Moos, R.; Julius, C.; Miele, G.; Lenz-Bauer, C.; Aguzzi, A. Canine mdck cell lines are refractory to infection with human and mouse prions. Vaccine 2008, 26, 2601–2614. [Google Scholar] [CrossRef] [Green Version]
- Gibson, P.E.; Bell, T.M.; Field, E.J. Failure of the scrapie agent to replicate in l5178y mouse leukaemic cells. Res. Vet. Sci. 1972, 13, 95–96. [Google Scholar]
- Klohn, P.C.; Stoltze, L.; Flechsig, E.; Enari, M.; Weissmann, C. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc. Natl. Acad. Sci. USA 2003, 100, 11666–11671. [Google Scholar]
- Ladogana, A.; Liu, Q.; Xi, Y.G.; Pocchiari, M. Proteinase-resistant protein in human neuroblastoma cells infected with brain material from creutzfeldt-jakob patient. Lancet 1995, 345, 594–595. [Google Scholar]
- Lewis, V.; Hill, A.F.; Haigh, C.L.; Klug, G.M.; Masters, C.L.; Lawson, V.A.; Collins, S.J. Increased proportions of c1 truncated prion protein protect against cellular m1000 prion infection. J. Neuropathol. Exp. Neurol. 2009, 68, 1125–1135. [Google Scholar] [CrossRef]
- Maas, E.; Geissen, M.; Groschup, M.H.; Rost, R.; Onodera, T.; Schatzl, H.; Vorberg, I.M. Scrapie infection of prion protein-deficient cell line upon ectopic expression of mutant prion proteins. J. Biol. Chem. 2007, 282, 18702–18710. [Google Scholar]
- McNally, K.L.; Ward, A.E.; Priola, S.A. Cells expressing anchorless prion protein are resistant to scrapie infection. J. Virol. 2009, 83, 4469–4475. [Google Scholar] [CrossRef]
- Bosque, P.J.; Ryou, C.; Telling, G.; Peretz, D.; Legname, G.; DeArmond, S.J.; Prusiner, S.B. Prions in skeletal muscle. Proc. Natl. Acad. Sci. USA 2002, 99, 3812–3817. [Google Scholar]
- Bosque, P.J.; Prusiner, S.B. Cultured cell sublines highly susceptible to prion infection. J. Virol. 2000, 74, 4377–4386. [Google Scholar] [CrossRef]
- Mahal, S.P.; Baker, C.A.; Demczyk, C.A.; Smith, E.W.; Julius, C.; Weissmann, C. Prion strain discrimination in cell culture: The cell panel assay. Proc. Natl. Acad. Sci. USA 2007, 104, 20908–20913. [Google Scholar]
- Bach, C.; Gilch, S.; Rost, R.; Greenwood, A.D.; Horsch, M.; Hajj, G.N.; Brodesser, S.; Facius, A.; Schadler, S.; Sandhoff, K.; et al. Prion-induced activation of cholesterogenic gene expression by srebp2 in neuronal cells. J. Biol. Chem. 2009, 284, 31260–31269. [Google Scholar]
- Race, R.E.; Fadness, L.H.; Chesebro, B. Characterization of scrapie infection in mouse neuroblastoma cells. J. Gen. Virol 1987, 68, 1391–1399. [Google Scholar]
- Kocisko, D.A.; Caughey, B. Searching for anti-prion compounds: Cell-based high-throughput in vitro assays and animal testing strategies. Methods Enzymol. 2006, 412, 223–234. [Google Scholar] [CrossRef]
- Raymond, G.J.; Olsen, E.A.; Lee, K.S.; Raymond, L.D.; Bryant, P.K., 3rd; Baron, G.S.; Caughey, W.S.; Kocisko, D.A.; McHolland, L.E.; Favara, C.; et al. Inhibition of protease-resistant prion protein formation in a transformed deer cell line infected with chronic wasting disease. J. Virol. 2006, 80, 596–604. [Google Scholar] [CrossRef]
- Chasseigneaux, S.; Pastore, M.; Britton-Davidian, J.; Manie, E.; Stern, M.H.; Callebert, J.; Catalan, J.; Casanova, D.; Belondrade, M.; Provansal, M.; et al. Genetic heterogeneity versus molecular analysis of prion susceptibility in neuroblasma n2a sublines. Arch. Virol. 2008, 153, 1693–1702. [Google Scholar] [CrossRef]
- Taraboulos, A.; Serban, D.; Prusiner, S.B. Scrapie prion proteins accumulate in the cytoplasm of persistently infected cultured cells. J. Cell. Biol. 1990, 110, 2117–2132. [Google Scholar]
- Vorberg, I.; Raines, A.; Priola, S.A. Acute formation of protease-resistant prion protein does not always lead to persistent scrapie infection in vitro. J. Biol. Chem. 2004, 279, 29218–29225. [Google Scholar]
- Greil, C.S.; Vorberg, I.M.; Ward, A.E.; Meade-White, K.D.; Harris, D.A.; Priola, S.A. Acute cellular uptake of abnormal prion protein is cell type and scrapie-strain independent. Virology 2008, 379, 284–293. [Google Scholar] [CrossRef]
- Goold, R.; Rabbanian, S.; Sutton, L.; Andre, R.; Arora, P.; Moonga, J.; Clarke, A.R.; Schiavo, G.; Jat, P.; Collinge, J.; et al. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat. Commun. 2011, 2, 281. [Google Scholar] [CrossRef]
- Bian, J.; Nazor, K.E.; Angers, R.; Jernigan, M.; Seward, T.; Centers, A.; Green, M.; Telling, G.C. Gfp-tagged prp supports compromised prion replication in transgenic mice. Biochem. Biophys. Res. Comm. 2006, 340, 894–900. [Google Scholar] [CrossRef]
- Magalhaes, A.C.; Baron, G.S.; Lee, K.S.; Steele-Mortimer, O.; Dorward, D.; Prado, M.A.; Caughey, B. Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J. Neurosci. 2005, 25, 5207–5216. [Google Scholar] [CrossRef]
- Mahal, S.P.; Demczyk, C.A.; Smith, E.W., Jr.; Klohn, P.C.; Weissmann, C. Assaying prions in cell culture: The standard scrapie cell assay (ssca) and the scrapie cell assay in end point format (scepa). Methods Mol. Biol. 2008, 459, 49–68. [Google Scholar] [CrossRef]
- Arellano-Anaya, Z.E.; Savistchenko, J.; Mathey, J.; Huor, A.; Lacroux, C.; Andreoletti, O.; Vilette, D. A simple, versatile and sensitive cell-based assay for prions from various species. PLoS One 2011, 6, e20563. [Google Scholar]
- Arjona, A.; Simarro, L.; Islinger, F.; Nishida, N.; Manuelidis, L. Two creutzfeldt-jakob disease agents reproduce prion protein-independent identities in cell cultures. Proc. Natl. Acad. Sci. USA 2004, 101, 8768–8773. [Google Scholar] [CrossRef]
- Enari, M.; Flechsig, E.; Weissmann, C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc. Natl. Acad. Sci. USA 2001, 98, 9295–9299. [Google Scholar]
- Markovits, P.; Dautheville, C.; Dormont, D.; Dianoux, L.; Latarjet, R. In vitro propagation of the scrapie agent. I. Transformation of mouse glia and neuroblastoma cells after infection with the mouse-adapted scrapie strain c-506. Acta. Neuropathol. 1983, 60, 75–80. [Google Scholar] [CrossRef]
- Ostlund, P.; Lindegren, H.; Pettersson, C.; Bedecs, K. Up-regulation of functionally impaired insulin-like growth factor-1 receptor in scrapie-infected neuroblastoma cells. J. Biol. Chem. 2001, 276, 36110–36115. [Google Scholar]
- Race, R. The scrapie agent in vitro. Curr Top. Microbiol. Immunol. 1991, 172, 181–193. [Google Scholar] [CrossRef]
- Scott, M.R.; Kohler, R.; Foster, D.; Prusiner, S.B. Chimeric prion protein expression in cultured cells and transgenic mice. Protein. Sci. 1992, 1, 986–997. [Google Scholar] [CrossRef]
- Miyazawa, K.; Emmerling, K.; Manuelidis, L. High cjd infectivity remains after prion protein is destroyed. J. Cell. Biochem. 2011, 112, 3630–3637. [Google Scholar] [CrossRef]
- Baron, T.G.; Biacabe, A.G.; Bencsik, A.; Langeveld, J.P. Transmission of new bovine prion to mice. Emerg. Infect. Dis. 2006, 12, 1125–1128. [Google Scholar] [CrossRef]
- Nunziante, M.; Ackermann, K.; Dietrich, K.; Wolf, H.; Gadtke, L.; Gilch, S.; Vorberg, I.; Groschup, M.; Schatzl, H.M. Proteasomal dysfunction and endoplasmic reticulum stress enhance trafficking of prion protein aggregates through the secretory pathway and increase accumulation of pathologic prion protein. J. Biol. Chem. 2011, 286, 33942–33953. [Google Scholar]
- Birkett, C.R.; Hennion, R.M.; Bembridge, D.A.; Clarke, M.C.; Chree, A.; Bruce, M.E.; Bostock, C.J. Scrapie strains maintain biological phenotypes on propagation in a cell line in culture. EMBO J. 2001, 20, 3351–3358. [Google Scholar] [CrossRef]
- Kanu, N.; Imokawa, Y.; Drechsel, D.N.; Williamson, R.A.; Birkett, C.R.; Bostock, C.J.; Brockes, J.P. Transfer of scrapie prion infectivity by cell contact in culture. Curr. Biol. 2002, 12, 523–530. [Google Scholar] [CrossRef]
- Dron, M.; Dandoy-Dron, F.; Farooq Salamat, M.K.; Laude, H. Proteasome inhibitors promote the sequestration of prpsc into aggresomes within the cytosol of prion-infected cad neuronal cells. J. Gen. Virol. 2009, 90, 2050–2060. [Google Scholar] [CrossRef]
- Browning, S.; Baker, C.A.; Smith, E.; Mahal, S.P.; Herva, M.E.; Demczyk, C.A.; Li, J.; Weissmann, C. Abrogation of complex glycosylation by swainsonine results in strain- and cell-specific inhibition of prion replication. J. Biol. Chem. 2011, 286, 40962–40973. [Google Scholar]
- Julius, C.; Hutter, G.; Wagner, U.; Seeger, H.; Kana, V.; Kranich, J.; Klohn, P.C.; Weissmann, C.; Miele, G.; Aguzzi, A. Transcriptional stability of cultured cells upon prion infection. J. Mol. Biol. 2008, 375, 1222–1233. [Google Scholar] [CrossRef]
- Rubenstein, R.; Deng, H.; Race, R.E.; Ju, W.; Scalici, C.L.; Papini, M.C.; Kascsak, R.J.; Carp, R.I. Demonstration of scrapie strain diversity in infected pc12 cells. J. Gen. Virol 1992, 73, 3027–3031. [Google Scholar]
- Rubenstein, R.; Carp, R.I.; Callahan, S.M. In vitro replication of scrapie agent in a neuronal model: Infection of pc12 cells. J. Gen. Virol. 1984, 65, 2191–2198. [Google Scholar] [CrossRef]
- Rubenstein, R.; Deng, H.; Scalici, C.L.; Papini, M.C. Alterations in neurotransmitter-related enzyme activity in scrapie-infected pc12 cells. J. Gen. Virol. 1991, 72, 1279–1285. [Google Scholar] [CrossRef]
- Follet, J.; Lemaire-Vieille, C.; Blanquet-Grossard, F.; Podevin-Dimster, V.; Lehmann, S.; Chauvin, J.P.; Decavel, J.P.; Varea, R.; Grassi, J.; Fontes, M.; et al. Prp expression and replication by schwann cells: Implications in prion spreading. J. Virol. 2002, 76, 2434–2439. [Google Scholar]
- Archer, F.; Bachelin, C.; Andreoletti, O.; Besnard, N.; Perrot, G.; Langevin, C.; Le Dur, A.; Vilette, D.; Baron-Van Evercooren, A.; Vilotte, J.L.; et al. Cultured peripheral neuroglial cells are highly permissive to sheep prion infection. J. Virol. 2004, 78, 482–490. [Google Scholar]
- Morel, E.; Andrieu, T.; Casagrande, F.; Gauczynski, S.; Weiss, S.; Grassi, J.; Rousset, M.; Dormont, D.; Chambaz, J. Bovine prion is endocytosed by human enterocytes via the 37 kda/67 kda laminin receptor. Am. J. Pathol. 2005, 167, 1033–1042. [Google Scholar] [CrossRef]
- Hijazi, N.; Kariv-Inbal, Z.; Gasset, M.; Gabizon, R. Prpsc incorporation to cells requires endogenous glycosaminoglycan expression. J. Biol. Chem. 2005, 280, 17057–17061. [Google Scholar]
- Horonchik, L.; Tzaban, S.; Ben-Zaken, O.; Yedidia, Y.; Rouvinski, A.; Papy-Garcia, D.; Barritault, D.; Vlodavsky, I.; Taraboulos, A. Heparan sulfate is a cellular receptor for purified infectious prions. J. Biol. Chem. 2005, 280, 17062–17067. [Google Scholar]
- Mohan, J.; Hopkins, J.; Mabbott, N.A. Skin-derived dendritic cells acquire and degrade the scrapie agent following in vitro exposure. Immunology 2005, 116, 122–133. [Google Scholar] [CrossRef]
- Paquet, S.; Daude, N.; Courageot, M.P.; Chapuis, J.; Laude, H.; Vilette, D. Prpc does not mediate internalization of prpsc but is required at an early stage for de novo prion infection of rov cells. J. Virol. 2007, 81, 10786–10791. [Google Scholar] [CrossRef]
- Langevin, C.; Gousset, K.; Costanzo, M.; Richard-Le Goff, O.; Zurzolo, C. Characterization of the role of dendritic cells in prion transfer to primary neurons. Biochem. J. 2010, 431, 189–198. [Google Scholar] [CrossRef]
- Jen, A.; Parkyn, C.J.; Mootoosamy, R.C.; Ford, M.J.; Warley, A.; Liu, Q.; Bu, G.; Baskakov, I.V.; Moestrup, S.; McGuinness, L.; et al. Neuronal low-density lipoprotein receptor-related protein 1 binds and endocytoses prion fibrils via receptor cluster 4. J. Cell. Sci. 2010, 123, 246–255. [Google Scholar] [CrossRef]
- Schonberger, O.; Horonchik, L.; Gabizon, R.; Papy-Garcia, D.; Barritault, D.; Taraboulos, A. Novel heparan mimetics potently inhibit the scrapie prion protein and its endocytosis. Biochem. Biophys. Res. Commun. 2003, 312, 473–479. [Google Scholar] [CrossRef]
- Gauczynski, S.; Nikles, D.; El-Gogo, S.; Papy-Garcia, D.; Rey, C.; Alban, S.; Barritault, D.; Lasmezas, C.I.; Weiss, S. The 37-kda/67-kda laminin receptor acts as a receptor for infectious prions and is inhibited by polysulfated glycanes. J. Infect. Dis. 2006, 194, 702–709. [Google Scholar] [CrossRef]
- Shmakov, A.N.; Bode, J.; Kilshaw, P.J.; Ghosh, S. Diverse patterns of expression of the 67-kd laminin receptor in human small intestinal mucosa: Potential binding sites for prion proteins? J. Pathol. 2000, 191, 318–322. [Google Scholar] [CrossRef]
- Prusiner, S.B.; McKinley, M.P.; Bowman, K.A.; Bolton, D.C.; Bendheim, P.E.; Groth, D.F.; Glenner, G.G. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell 1983, 35, 349–358. [Google Scholar]
- Lofgren, K.; Wahlstrom, A.; Lundberg, P.; Langel, U.; Graslund, A.; Bedecs, K. Antiprion properties of prion protein-derived cell-penetrating peptides. FASEB J. 2008, 22, 2177–2184. [Google Scholar] [CrossRef]
- Taylor, D.R.; Whitehouse, I.J.; Hooper, N.M. Glypican-1 mediates both prion protein lipid raft association and disease isoform formation. PLoS Pathog. 2009, 5, e1000666. [Google Scholar] [CrossRef]
- Hooper, N.M. Glypican-1 facilitates prion conversion in lipid rafts. J. Neurochem. 2011, 116, 721–725. [Google Scholar] [CrossRef]
- Wadia, J.S.; Schaller, M.; Williamson, R.A.; Dowdy, S.F. Pathologic prion protein infects cells by lipid-raft dependent macropinocytosis. PLoS One 2008, 3, e3314. [Google Scholar] [CrossRef]
- Taylor, D.R.; Hooper, N.M. The prion protein and lipid rafts. Mol. Membr. Biol. 2006, 23, 89–99. [Google Scholar]
- Deleault, N.R.; Harris, B.T.; Rees, J.R.; Supattapone, S. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA 2007, 104, 9741–9746. [Google Scholar]
- Abid, K.; Morales, R.; Soto, C. Cellular factors implicated in prion replication. FEBS Lett. 2010, 584, 2409–2414. [Google Scholar] [CrossRef]
- Caughey, B.; Raymond, G.J.; Ernst, D.; Race, R.E. N-terminal truncation of the scrapie-associated form of prp by lysosomal protease(s): Implications regarding the site of conversion of prp to the protease-resistant state. J. Virol. 1991, 65, 6597–6603. [Google Scholar]
- Borchelt, D.R.; Taraboulos, A.; Prusiner, S.B. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 1992, 267, 16188–16199. [Google Scholar]
- Taraboulos, A.; Raeber, A.J.; Borchelt, D.R.; Serban, D.; Prusiner, S.B. Synthesis and trafficking of prion proteins in cultured cells. Mol. Biol. Cell. 1992, 3, 851–863. [Google Scholar]
- Naslavsky, N.; Stein, R.; Yanai, A.; Friedlander, G.; Taraboulos, A. Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J. Biol. Chem. 1997, 272, 6324–6331. [Google Scholar]
- Stahl, N.; Baldwin, M.A.; Burlingame, A.L.; Prusiner, S.B. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry 1990, 29, 8879–8884. [Google Scholar] [CrossRef]
- Veith, N.M.; Plattner, H.; Stuermer, C.A.; Schulz-Schaeffer, W.J.; Burkle, A. Immunolocalisation of prpsc in scrapie-infected n2a mouse neuroblastoma cells by light and electron microscopy. Eur. J. Cell Biol. 2009, 88, 45–63. [Google Scholar] [CrossRef]
- Mange, A.; Crozet, C.; Lehmann, S.; Beranger, F. Scrapie-like prion protein is translocated to the nuclei of infected cells independently of proteasome inhibition and interacts with chromatin. J. Cell Sci. 2004, 117, 2411–2416. [Google Scholar] [CrossRef]
- Lehmann, S.; Harris, D.A. Mutant and infectious prion proteins display common biochemical properties in cultured cells. J. Biol. Chem. 1996, 271, 1633–1637. [Google Scholar] [CrossRef]
- Caughey, B.; Raymond, G.J. The scrapie-associated form of prp is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J. Biol. Chem. 1991, 266, 18217–18223. [Google Scholar]
- Beranger, F.; Mange, A.; Goud, B.; Lehmann, S. Stimulation of prp(c) retrograde transport toward the endoplasmic reticulum increases accumulation of prp(sc) in prion-infected cells. J. Biol. Chem. 2002, 277, 38972–38977. [Google Scholar]
- Vorberg, I.; Chan, K.; Priola, S.A. Deletion of beta-strand and alpha-helix secondary structure in normal prion protein inhibits formation of its protease-resistant isoform. J. Virol. 2001, 75, 10024–10032. [Google Scholar] [CrossRef]
- Marijanovic, Z.; Caputo, A.; Campana, V.; Zurzolo, C. Identification of an intracellular site of prion conversion. PLoS Pathog. 2009, 5, e1000426. [Google Scholar] [CrossRef]
- McKinley, M.P.; Taraboulos, A.; Kenaga, L.; Serban, D.; Stieber, A.; DeArmond, S.J.; Prusiner, S.B.; Gonatas, N. Ultrastructural localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells. Lab. Invest. 1991, 65, 622–630. [Google Scholar]
- Pimpinelli, F.; Lehmann, S.; Maridonneau-Parini, I. The scrapie prion protein is present in flotillin-1-positive vesicles in central- but not peripheral-derived neuronal cell lines. Eur. J. Neurosci. 2005, 21, 2063–2072. [Google Scholar] [CrossRef]
- McKinley, M.P.; Meyer, R.K.; Kenaga, L.; Rahbar, F.; Cotter, R.; Serban, A.; Prusiner, S.B. Scrapie prion rod formation in vitro requires both detergent extraction and limited proteolysis. J. Virol. 1991, 65, 1340–1351. [Google Scholar]
- Snow, A.D.; Kisilevsky, R.; Willmer, J.; Prusiner, S.B.; DeArmond, S.J. Sulfated glycosaminoglycans in amyloid plaques of prion diseases. Acta. Neuropathol. 1989, 77, 337–342. [Google Scholar] [CrossRef]
- Adjou, K.T.; Simoneau, S.; Sales, N.; Lamoury, F.; Dormont, D.; Papy-Garcia, D.; Barritault, D.; Deslys, J.P.; Lasmezas, C.I. A novel generation of heparan sulfate mimetics for the treatment of prion diseases. J. Gen. Virol. 2003, 84, 2595–2603. [Google Scholar] [CrossRef]
- Wong, C.; Xiong, L.W.; Horiuchi, M.; Raymond, L.; Wehrly, K.; Chesebro, B.; Caughey, B. Sulfated glycans and elevated temperature stimulate prp(sc)-dependent cell-free formation of protease-resistant prion protein. EMBO J. 2001, 20, 377–386. [Google Scholar] [CrossRef]
- Murayama, Y.; Yoshioka, M.; Masujin, K.; Okada, H.; Iwamaru, Y.; Imamura, M.; Matsuura, Y.; Fukuda, S.; Onoe, S.; Yokoyama, T.; et al. Sulfated dextrans enhance in vitro amplification of bovine spongiform encephalopathy prp(sc) and enable ultrasensitive detection of bovine prp(sc). PLoS One 2010, 5. [Google Scholar]
- Yokoyama, T.; Takeuchi, A.; Yamamoto, M.; Kitamoto, T.; Ironside, J.W.; Morita, M. Heparin enhances the cell-protein misfolding cyclic amplification efficiency of variant creutzfeldt-jakob disease. Neurosci. Lett. 2011, 498, 119–123. [Google Scholar] [CrossRef]
- Ghaemmaghami, S.; Phuan, P.W.; Perkins, B.; Ullman, J.; May, B.C.; Cohen, F.E.; Prusiner, S.B. Cell division modulates prion accumulation in cultured cells. Proc. Natl. Acad. Sci. USA 2007, 104, 17971–17976. [Google Scholar]
- Paquet, S.; Langevin, C.; Chapuis, J.; Jackson, G.S.; Laude, H.; Vilette, D. Efficient dissemination of prions through preferential transmission to nearby cells. J. Gen. Virol. 2007, 88, 706–713. [Google Scholar]
- Leblanc, P.; Alais, S.; Porto-Carreiro, I.; Lehmann, S.; Grassi, J.; Raposo, G.; Darlix, J.L. Retrovirus infection strongly enhances scrapie infectivity release in cell culture. EMBO J. 2006, 25, 2674–2685. [Google Scholar] [CrossRef]
- Gousset, K.; Zurzolo, C. Tunnelling nanotubes: A highway for prion spreading? Prion 2009, 3, 94–98. [Google Scholar] [CrossRef]
- Gousset, K.; Schiff, E.; Langevin, C.; Marijanovic, Z.; Caputo, A.; Browman, D.T.; Chenouard, N.; de Chaumont, F.; Martino, A.; Enninga, J.; et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell. Biol. 2009, 11, 328–336. [Google Scholar] [CrossRef]
- Nishida, N.; Katamine, S.; Manuelidis, L. Reciprocal interference between specific cjd and scrapie agents in neural cell cultures. Science 2005, 310, 493–496. [Google Scholar] [CrossRef]
- Baron, G.S.; Magalhaes, A.C.; Prado, M.A.; Caughey, B. Mouse-adapted scrapie infection of sn56 cells: Greater efficiency with microsome-associated versus purified prp-res. J. Virol. 2006, 80, 2106–2117. [Google Scholar]
- Krammer, C.; Schatzl, H.M.; Vorberg, I. Prion-like propagation of cytosolic protein aggregates: Insights from cell culture models. Prion 2009, 3, 206–212. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O'Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell. Biol 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Munch, C.; O'Brien, J.; Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3548–3553. [Google Scholar] [CrossRef]
- Kfoury, N.; Holmes, B.B.; Jiang, H.; Holtzman, D.M.; Diamond, M.I. Trans-cellular propagation of tau aggregation by fibrillar species. J. Biol. Chem. 2012, 287, 19440–19451. [Google Scholar]
- Hansen, C.; Angot, E.; Bergstrom, A.L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. Alpha-synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 2011, 121, 715–725. [Google Scholar] [CrossRef]
- Ren, P.H.; Lauckner, J.E.; Kachirskaia, I.; Heuser, J.E.; Melki, R.; Kopito, R.R. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell. Biol. 2009, 11, 219–225. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic parkinson's disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O'Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar]
- Krammer, C.; Kryndushkin, D.; Suhre, M.H.; Kremmer, E.; Hofmann, A.; Pfeifer, A.; Scheibel, T.; Wickner, R.B.; Schatzl, H.M.; Vorberg, I. The yeast sup35nm domain propagates as a prion in mammalian cells. Proc. Natl. Acad. Sci. USA 2009, 106, 462–467. [Google Scholar]
- Munch, C.; Bertolotti, A. Self-propagation and transmission of misfolded mutant sod1: Prion or prion-like phenomenon? Cell. Cycle. 2011, 10, 1711. [Google Scholar] [CrossRef]
- Kocisko, D.A.; Engel, A.L.; Harbuck, K.; Arnold, K.M.; Olsen, E.A.; Raymond, L.D.; Vilette, D.; Caughey, B. Comparison of protease-resistant prion protein inhibitors in cell cultures infected with two strains of mouse and sheep scrapie. Neurosci. Lett. 2005, 388, 106–111. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Grassmann, A.; Wolf, H.; Hofmann, J.; Graham, J.; Vorberg, I. Cellular Aspects of Prion Replication In Vitro. Viruses 2013, 5, 374-405. https://doi.org/10.3390/v5010374
Grassmann A, Wolf H, Hofmann J, Graham J, Vorberg I. Cellular Aspects of Prion Replication In Vitro. Viruses. 2013; 5(1):374-405. https://doi.org/10.3390/v5010374
Chicago/Turabian StyleGrassmann, Andrea, Hanna Wolf, Julia Hofmann, James Graham, and Ina Vorberg. 2013. "Cellular Aspects of Prion Replication In Vitro" Viruses 5, no. 1: 374-405. https://doi.org/10.3390/v5010374
APA StyleGrassmann, A., Wolf, H., Hofmann, J., Graham, J., & Vorberg, I. (2013). Cellular Aspects of Prion Replication In Vitro. Viruses, 5(1), 374-405. https://doi.org/10.3390/v5010374