Newer Gene Editing Technologies toward HIV Gene Therapy

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
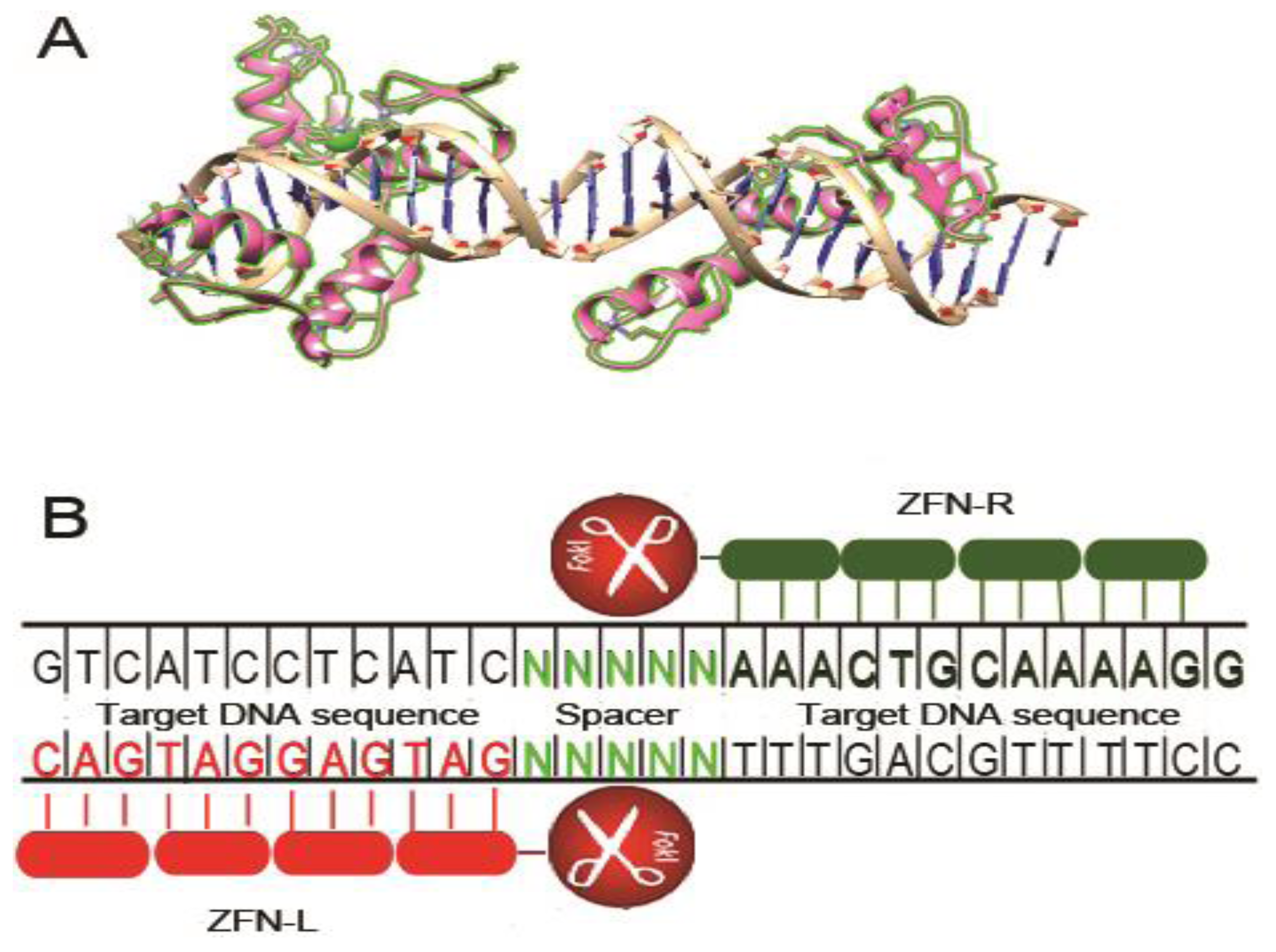
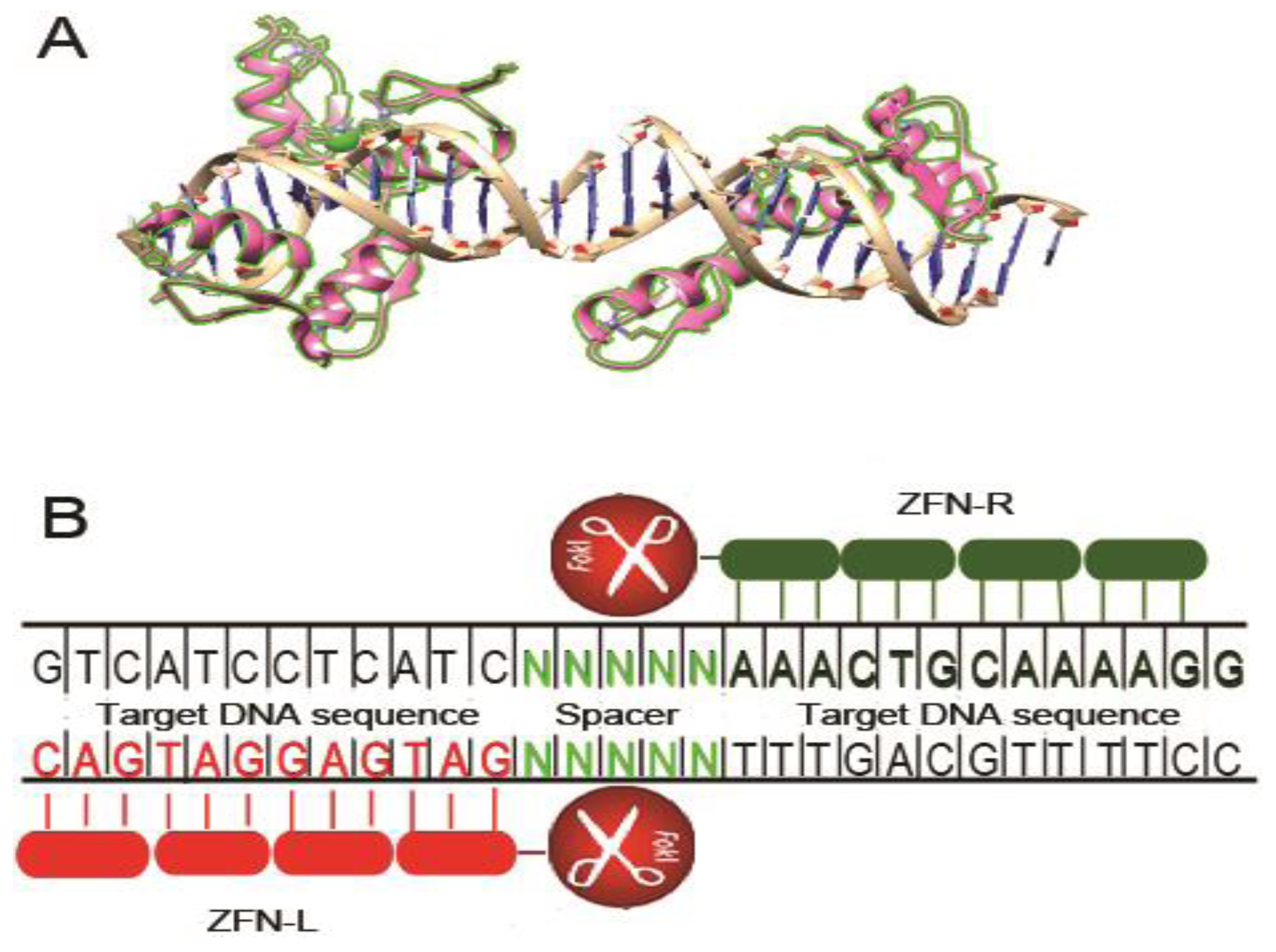
2. Zinc Finger Nucleases
2.1. CCR5 Disruption
2.2. CXCR4 Disruption
2.4. ZFN Delivery Strategies for HIV Gene Therapy
2.5. Specificity of ZFN Targeted Gene Disruption in HIV-1 Therapy
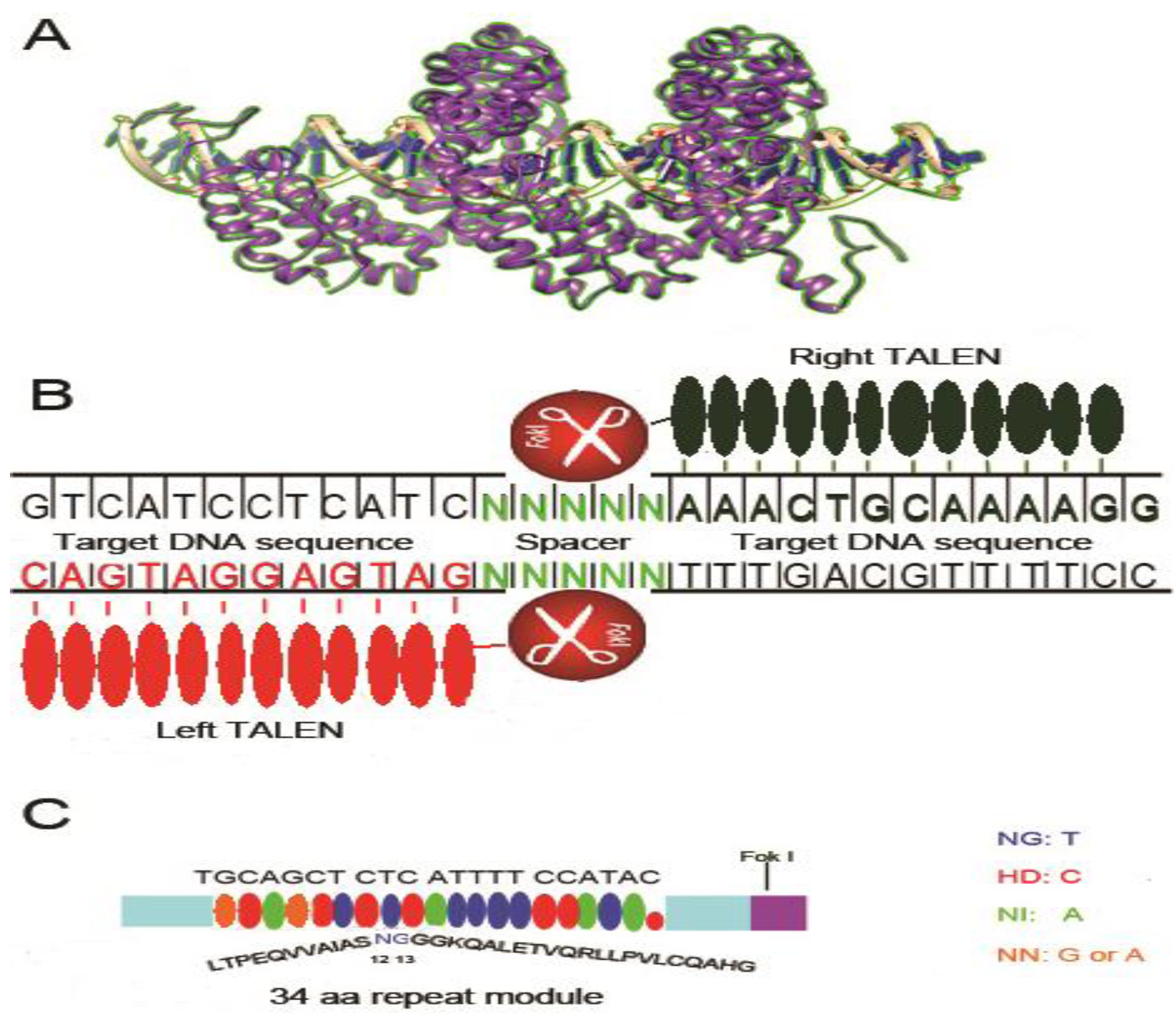
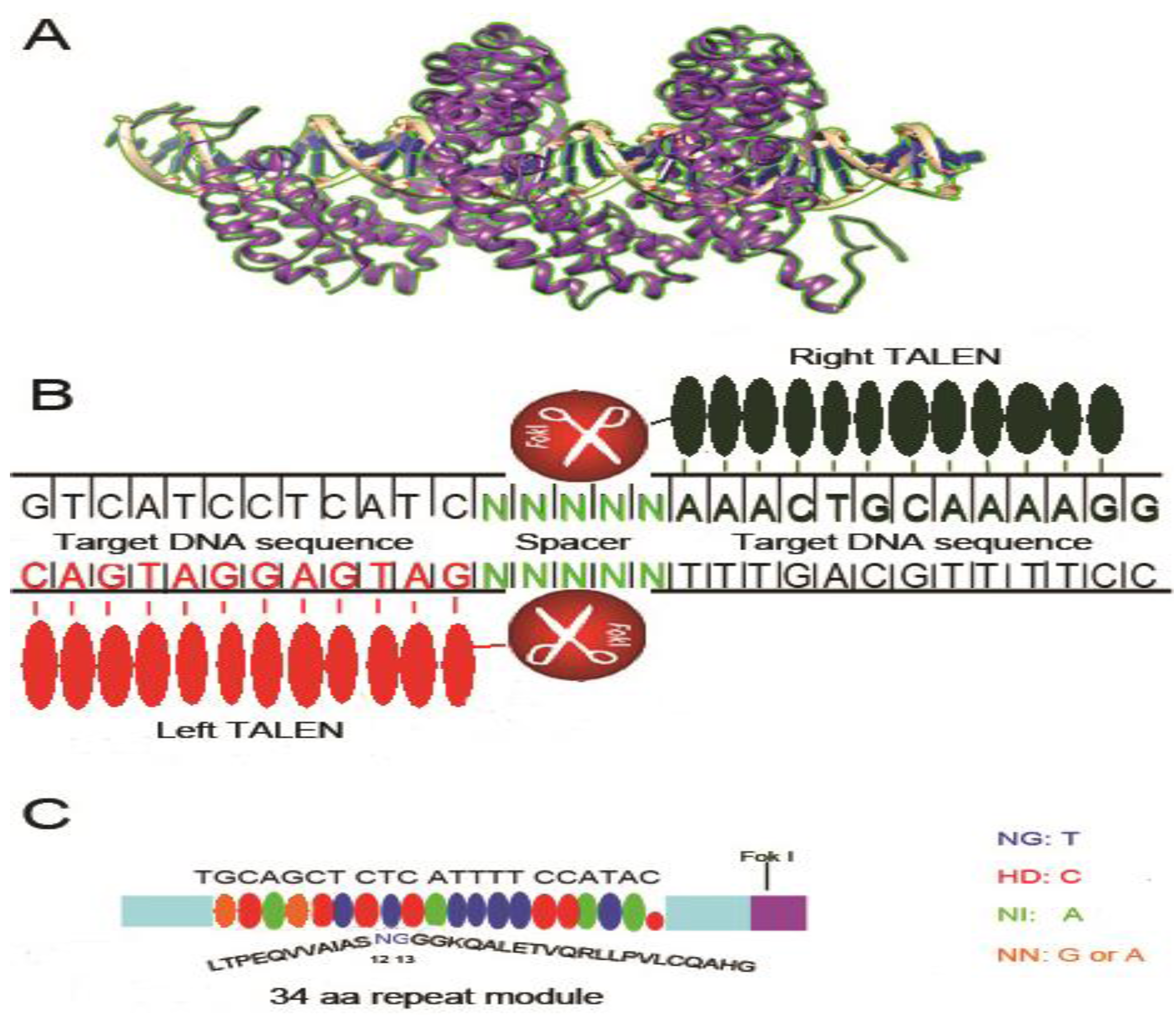
3. Transcription Activator-like Effectors Nucleases (TALENs)
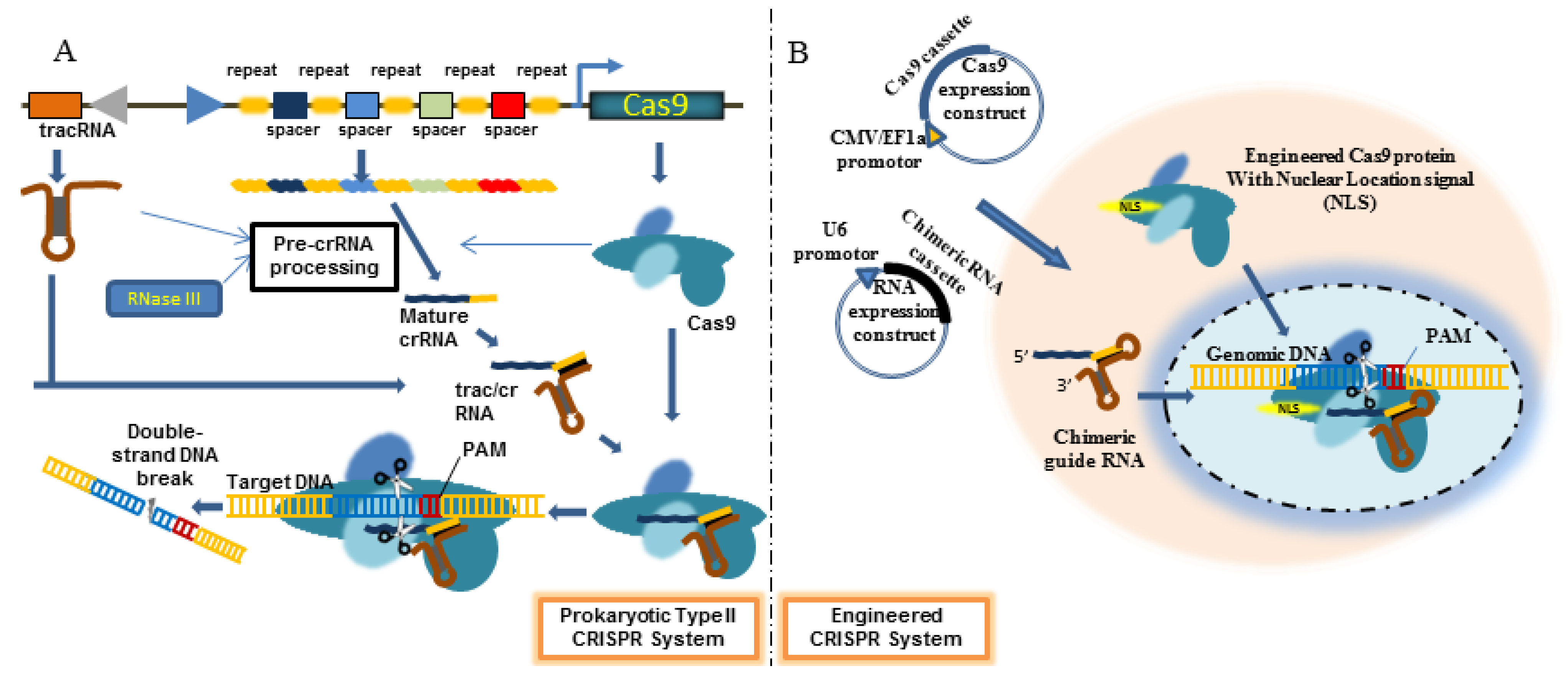
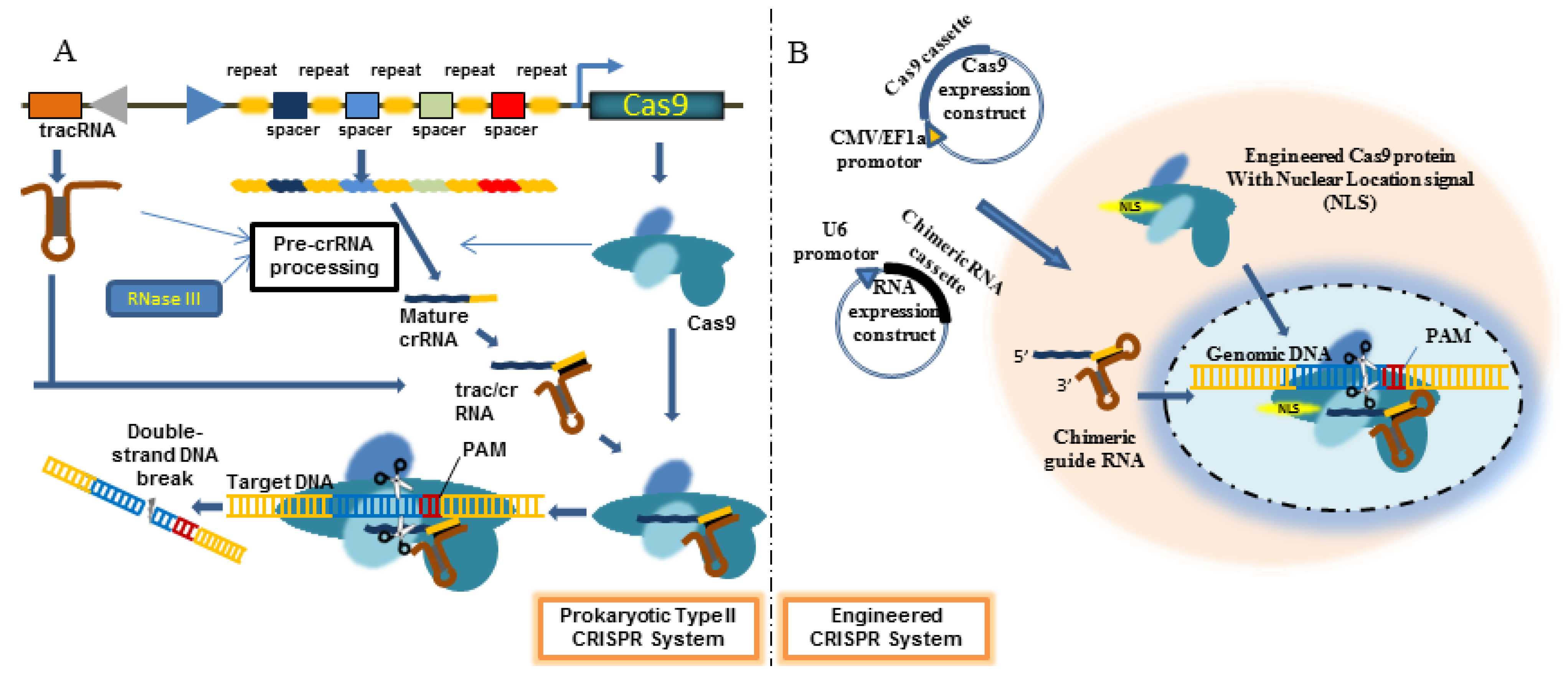
4. Engineered CRISPR/Cas9 System
5. Conclusion and Future Directions
Acknowledgments
Conflicts of Interest
References
- Carr, A. Toxicity of antiretroviral therapy and implications for drug development. Nat. Rev. Drug Discov. 2003, 2, 624–634. [Google Scholar] [CrossRef]
- Zaccarelli, M.; Tozzi, V.; Lorenzini, P.; Trotta, M.P.; Forbici, F.; Visco-Comandini, U.; Gori, C.; Narciso, P.; Perno, C.F.; Antinori, A.; et al. Multiple drug class-wide resistance associated with poorer survival after treatment failure in a cohort of HIV-infected patients. AIDS 2005, 19, 1081–1089. [Google Scholar] [CrossRef]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; Ousterout, D.G.; Gersbach, C.A. Advances in targeted genome editing. Curr. Opin. Chem. Biol. 2012, 16, 268–277. [Google Scholar] [CrossRef]
- Stone, D.; Kiem, H.P.; Jerome, K.R. Targeted gene disruption to cure HIV. Curr. Opin. HIV AIDS 2013, 8, 217–223. [Google Scholar] [CrossRef]
- Dean, M.; Carrington, M.; Winkler, C.; Huttley, G.A.; Smith, M.W.; Allikmets, R.; Goedert, J.J.; Buchbinder, S.P.; Vittinghoff, E.; Gomperts, E.; et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 1996, 273, 1856–1862. [Google Scholar] [CrossRef]
- Liu, R.; Paxton, W.A.; Choe, S.; Ceradini, D.; Martin, S.R.; Horuk, R.; MacDonald, M.E.; Stuhlmann, H.; Koup, R.A.; Landau, N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar] [CrossRef]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef]
- Allers, K.; Hutter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood 2011, 117, 2791–2799. [Google Scholar] [CrossRef]
- Hutter, G.; Thiel, E. Allogeneic transplantation of CCR5-deficient progenitor cells in a patient with HIV infection: An update after 3 years and the search for patient No. 2. AIDS 2011, 25, 273–274. [Google Scholar] [CrossRef]
- Subramanya, S.; Kim, S.S.; Manjunath, N.; Shankar, P. RNA interference-based therapeutics for human immunodeficiency virus HIV-1 treatment: Synthetic siRNA or vector-based shRNA? Expert Opin. Biol. Ther. 2010, 10, 201–213. [Google Scholar] [CrossRef]
- Thomas, K.R.; Capecchi, M.R. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 1987, 51, 503–512. [Google Scholar] [CrossRef]
- Thomas, K.R.; Capecchi, M.R. Targeted disruption of the murine int-1 proto-oncogene resulting in severe abnormalities in midbrain and cerebellar development. Nature 1990, 346, 847–850. [Google Scholar] [CrossRef]
- Mansour, S.L.; Thomas, K.R.; Capecchi, M.R. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: A general strategy for targeting mutations to non-selectable genes. Nature 1988, 336, 348–352. [Google Scholar] [CrossRef]
- Austin, C.P.; Battey, J.F.; Bradley, A.; Bucan, M.; Capecchi, M.; Collins, F.S.; Dove, W.F.; Duyk, G.; Dymecki, S.; Eppig, J.T.; et al. The knockout mouse project. Nat. Genet. 2004, 36, 921–924. [Google Scholar] [CrossRef]
- Isalan, M. Zinc-finger nucleases: How to play two good hands. Nat. Methods 2012, 9, 32–34. [Google Scholar] [CrossRef]
- Pavletich, N.P.; Pabo, C.O. Zinc finger-DNA recognition: Crystal structure of a Zif268-DNA complex at 2.1 A. Science 1991, 252, 809–817. [Google Scholar] [CrossRef]
- Bitinaite, J.; Wah, D.A.; Aggarwal, A.K.; Schildkraut, I. FokI dimerization is required for DNA cleavage. Proc. Natl. Acad. Sci. USA 1998, 95, 10570–10575. [Google Scholar]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008, 283, 1–5. [Google Scholar] [CrossRef]
- Meng, X.; Noyes, M.B.; Zhu, L.J.; Lawson, N.D.; Wolfe, S.A. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 695–701. [Google Scholar] [CrossRef]
- Carroll, D. Progress and prospects: Zinc-finger nucleases as gene therapy agents. Gene Ther. 2008, 15, 1463–1468. [Google Scholar] [CrossRef]
- Doyon, Y.; McCammon, J.M.; Miller, J.C.; Faraji, F.; Ngo, C.; Katibah, G.E.; Amora, R.; Hocking, T.D.; Zhang, L.; Rebar, E.J.; et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 702–708. [Google Scholar] [CrossRef]
- Shukla, V.K.; Doyon, Y.; Miller, J.C.; DeKelver, R.C.; Moehle, E.A.; Worden, S.E.; Mitchell, J.C.; Arnold, N.L.; Gopalan, S.; Meng, X.; et al. Precise genome modification in the crop species Zea mays using zinc-finger nucleases. Nature 2009, 459, 437–441. [Google Scholar] [CrossRef]
- Townsend, J.A.; Wright, D.A.; Winfrey, R.J.; Fu, F.; Maeder, M.L.; Joung, J.K.; Voytas, D.F. High-frequency modification of plant genes using engineered zinc-finger nucleases. Nature 2009, 459, 442–445. [Google Scholar] [CrossRef]
- Jacob, H.J.; Lazar, J.; Dwinell, M.R.; Moreno, C.; Geurts, A.M. Gene targeting in the rat: Advances and opportunities. Trends Genet. 2010, 26, 510–518. [Google Scholar] [CrossRef]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Liu, O.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.L.; et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 808–816. [Google Scholar] [CrossRef]
- Wilen, C.B.; Wang, J.; Tilton, J.C.; Miller, J.C.; Kim, K.A.; Rebar, E.J.; Sherrill-Mix, S.A.; Patro, S.C.; Secreto, A.J.; Jordan, A.P.; et al. Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathog. 2011, 7, e1002020. [Google Scholar] [CrossRef]
- Yuan, J.; Wang, J.; Crain, K.; Fearns, C.; Kim, K.A.; Hua, K.L.; Gregory, P.D.; Holmes, M.C.; Torbett, B.E. Zinc-finger nuclease editing of human cxcr4 promotes HIV-1 CD4(+) T cell resistance and enrichment. Mol. Ther. 2012, 20, 849–859. [Google Scholar] [CrossRef]
- Qu, X.; Wang, P.; Ding, D.; Li, L.; Wang, H.; Ma, L.; Zhou, X.; Liu, S.; Lin, S.; Wang, X.; et al. Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res. 2013, 41, 7771–7782. [Google Scholar] [CrossRef]
- Melum, E.; Karlsen, T.H.; Broome, U.; Thorsby, E.; Schrumpf, E.; Boberg, K.M.; Lie, B.A. The 32-base pair deletion of the chemokine receptor 5 gene (CCR5-Delta32) is not associated with primary sclerosing cholangitis in 363 Scandinavian patients. Tissue Antigens 2006, 68, 78–81. [Google Scholar] [CrossRef]
- Muntinghe, F.L.; Carrero, J.J.; Navis, G.; Stenvinkel, P. TNF-alpha levels are not increased in inflamed patients carrying the CCR5 deletion 32. Cytokine 2011, 53, 16–18. [Google Scholar] [CrossRef]
- Telenti, A. Safety concerns about CCR5 as an antiviral target. Curr. Opin. HIV AIDS 2009, 4, 131–135. [Google Scholar] [CrossRef]
- Mani, M.; Kandavelou, K.; Dy, F.J.; Durai, S.; Chandrasegaran, S. Design, engineering, and characterization of zinc finger nucleases. Biochem. Biophys. Res. Commun. 2005, 335, 447–457. [Google Scholar] [CrossRef]
- Doyon, Y.; Vo, T.D.; Mendel, M.C.; Greenberg, S.G.; Wang, J.; Xia, D.F.; Miller, J.C.; Urnov, F.D.; Gregory, P.D.; Holmes, M.C. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat. Methods 2011, 8, 74–79. [Google Scholar] [CrossRef]
- Voit, R.A.; McMahon, M.A.; Sawyer, S.L.; Porteus, M.H. Generation of an HIV resistant T-cell line by targeted “stacking” of restriction factors. Mol. Ther. 2013, 21, 786–795. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, H.J.; Kim, H.; Cho, S.W.; Kim, J.S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 2009, 19, 1279–1288. [Google Scholar] [CrossRef]
- Maier, D.A.; Brennan, A.L.; Jiang, S.; Binder-Scholl, G.K.; Lee, G.; Plesa, G.; Zheng, Z.; Cotte, J.; Carpenito, C.; Wood, T.; et al. Efficient clinical scale gene modification via zinc finger nuclease-targeted disruption of the HIV co-receptor CCR5. Hum. Gene Ther. 2013, 24, 245–258. [Google Scholar]
- Yao, Y.; Nashun, B.; Zhou, T.; Qin, L.; Qin, L.; Zhao, S.; Xu, J.; Esteban, M.A.; Chen, X. Generation of CD34+ cells from CCR5-disrupted human embryonic and induced pluripotent stem cells. Hum. Gene Ther. 2012, 23, 238–242. [Google Scholar]
- Holt, N.; Wang, J.; Kim, K.; Friedman, G.; Wang, X.; Taupin, V.; Crooks, G.M.; Kohn, D.B.; Gregory, P.D.; Holmes, M.C.; et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010, 28, 839–847. [Google Scholar]
- Lombardo, A.; Genovese, P.; Beausejour, C.M.; Colleoni, S.; Lee, Y.L.; Kim, K.A.; Ando, D.; Urnov, F.D.; Galli, C.; Gregory, P.D.; et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat. Biotechnol. 2007, 25, 1298–1306. [Google Scholar]
- Lombardo, A.; Cesana, D.; Genovese, P.; di Stefano, B.; Provasi, E.; Colombo, D.F.; Neri, M.; Magnani, Z.; Cantore, A.; Lo Riso, P.; et al. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat. Methods 2011, 8, 861–869. [Google Scholar] [CrossRef]
- Li, L.; Krymskaya, L.; Wang, J.; Henley, J.; Rao, A.; Cao, L.F.; Tran, C.A.; Torres-Coronado, M.; Gardner, A.; Gonzalez, N.; et al. Genomic editing of the HIV-1 coreceptor CCR5 in adult hematopoietic stem and progenitor cells using zinc finger nucleases. Mol. Ther. 2013, 21, 1259–1269. [Google Scholar] [CrossRef]
- Melby, T.; Despirito, M.; Demasi, R.; Heilek-Snyder, G.; Greenberg, M.L.; Graham, N. HIV-1 coreceptor use in triple-class treatment-experienced patients: Baseline prevalence, correlates, and relationship to enfuvirtide response. J. Infect. Dis. 2006, 194, 238–246. [Google Scholar] [CrossRef]
- Tilton, J.C.; Wilen, C.B.; Didigu, C.A.; Sinha, R.; Harrison, J.E.; Agrawal-Gamse, C.; Henning, E.A.; Bushman, F.D.; Martin, J.N.; Deeks, S.G.; et al. A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5. J. Virol. 2010, 84, 10863–10876. [Google Scholar] [CrossRef]
- Cradick, T.J.; Keck, K.; Bradshaw, S.; Jamieson, A.C.; McCaffrey, A.P. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol. Ther. 2010, 18, 947–954. [Google Scholar] [CrossRef]
- Wayengera, M. Identity of zinc finger nucleases with specificity to herpes simplex virus type II genomic DNA: Novel HSV-2 vaccine/therapy precursors. Theor. Biol. Med. Model 2011, 8, e23. [Google Scholar] [CrossRef]
- Tanaka, A.; Takeda, S.; Kariya, R.; Matsuda, K.; Urano, E.; Okada, S.; Komano, J. A novel therapeutic molecule against HTLV-1 infection targeting provirus. Leukemia 2013, 27, 1621–1627. [Google Scholar] [CrossRef]
- Wayengera, M. Proviral HIV-genome-wide and pol-gene specific zinc finger nucleases: Usability for targeted HIV gene therapy. Theor. Biol. Med. Model 2011, 8, e26. [Google Scholar] [CrossRef]
- Das, K.T.; Segal, D. Engineered Zinc Finger Nucleases to Inactivate the HIV Genome. In Proceedings of the 19th HIV International Conference, Washington DC, USA, 22–27 July 2012. Abstract TUPEO21.
- He, T.C.; Zhou, S.; da Costa, L.T.; Yu, J.; Kinzler, K.W.; Vogelstein, B. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 1998, 95, 2509–2514. [Google Scholar]
- Cortin, V.; Thibault, J.; Jacob, D.; Garnier, A. High-titer adenovirus vector production in 293S cell perfusion culture. Biotechnol. Prog. 2004, 20, 858–863. [Google Scholar] [CrossRef]
- Duerr, A.; Huang, Y.; Buchbinder, S.; Coombs, R.W.; Sanchez, J.; del Rio, C.; Casapia, M.; Santiago, S.; Gilbert, P.; Corey, L.; et al. Extended follow-up confirms early vaccine-enhanced risk of HIV acquisition and demonstrates waning effect over time among participants in a randomized trial of recombinant adenovirus HIV vaccine (Step Study). J. Infect. Dis. 2012, 206, 258–266. [Google Scholar] [CrossRef]
- Qureshi, H.; Ma, Z.M.; Huang, Y.; Hodge, G.; Thomas, M.A.; DiPasquale, J.; DeSilva, V.; Fritts, L.; Bett, A.J.; Casimiro, D.R.; et al. Low-dose penile SIVmac251 exposure of rhesus macaques infected with adenovirus type 5 (Ad5) and then immunized with a replication-defective Ad5-based SIV gag/pol/nef vaccine recapitulates the results of the phase IIb step trial of a similar HIV-1 vaccine. J. Virol. 2012, 86, 2239–2250. [Google Scholar] [CrossRef]
- Lei, Y.; Lee, C.L.; Joo, K.I.; Zarzar, J.; Liu, Y.; Dai, B.; Fox, V.; Wang, P. Gene editing of human embryonic stem cells via an engineered baculoviral vector carrying zinc-finger nucleases. Mol. Ther. 2011, 19, 942–950. [Google Scholar] [CrossRef]
- Cannon, P.M.; Henley, J.E.; Wood, T.; Trong, L.; Wang, J.; Kim, K.; Yan, J.; Gregory, P.D.; Lee, G.; Holmes, M.C. Electroporation of ZFN mRNA enables efficient CCR5 gene disruption in mobilized blood hematopoietic stem cells at clinical scale. In Proceedings of the ASGCT 16th Annual Meeting, Salt Lake City, UT, USA, 15–18 May 2013. Abstract 183.
- Gaj, T.; Guo, J.; Kato, Y.; Sirk, S.J.; Barbas, C.F., 3rd. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat. Methods 2012, 9, 805–807. [Google Scholar] [CrossRef]
- Bibikova, M.; Golic, M.; Golic, K.G.; Carroll, D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 2002, 161, 1169–1175. [Google Scholar]
- Porteus, M.H.; Baltimore, D. Chimeric nucleases stimulate gene targeting in human cells. Science 2003, 300, e763. [Google Scholar] [CrossRef]
- Alwin, S.; Gere, M.B.; Guhl, E.; Effertz, K.; Barbas, C.F., 3rd; Segal, D.J.; Weitzman, M.D.; Cathomen, T. Custom zinc-finger nucleases for use in human cells. Mol. Ther. 2005, 12, 610–617. [Google Scholar] [CrossRef]
- Pattanayak, V.; Ramirez, C.L.; Joung, J.K.; Liu, D.R. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat. Methods 2011, 8, 765–770. [Google Scholar] [CrossRef]
- Gabriel, R.; Lombardo, A.; Arens, A.; Miller, J.C.; Genovese, P.; Kaeppel, C.; Nowrouzi, A.; Bartholomae, C.C.; Wang, J.; Friedman, G.; et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 2011, 29, 816–823. [Google Scholar] [CrossRef]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef]
- Boch, J.; Bonas, U. Xanthomonas AvrBs3 family-type III effectors: Discovery and function. Annu. Rev. Phytopathol. 2010, 48, 419–436. [Google Scholar] [CrossRef]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar]
- Sander, J.D.; Cade, L.; Khayter, C.; Reyon, D.; Peterson, R.T.; Joung, J.K.; Yeh, J.R. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat. Biotechnol. 2011, 29, 697–698. [Google Scholar] [CrossRef]
- Huang, P.; Xiao, A.; Zhou, M.; Zhu, Z.; Lin, S.; Zhang, B. Heritable gene targeting in zebrafish using customized TALENs. Nat. Biotechnol. 2011, 29, 699–700. [Google Scholar] [CrossRef]
- Reyon, D.; Khayter, C.; Regan, M.R.; Joung, J.K.; Sander, J.D. Engineering designer transcription activator-like effector nucleases (TALENs) by REAL or REAL-Fast assembly. Curr. Protoc. Mol. Biol. 2012. [Google Scholar] [CrossRef]
- Geissler, R.; Scholze, H.; Hahn, S.; Streubel, J.; Bonas, U.; Behrens, S.E.; Boch, J. Transcriptional activators of human genes with programmable DNA-specificity. PLoS One 2011, 6, e19509. [Google Scholar]
- Li, T.; Huang, S.; Zhao, X.; Wright, D.A.; Carpenter, S.; Spalding, M.H.; Weeks, D.P.; Yang, B. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res. 2011, 39, 6315–6325. [Google Scholar]
- Sanjana, N.E.; Cong, L.; Zhou, Y.; Cunniff, M.M.; Feng, G.; Zhang, F. A transcription activator-like effector toolbox for genome engineering. Nat. Protoc. 2012, 7, 171–192. [Google Scholar] [CrossRef]
- Weber, E.; Gruetzner, R.; Werner, S.; Engler, C.; Marillonnet, S. Assembly of designer TAL effectors by Golden Gate cloning. PLoS One 2011, 6, e19722. [Google Scholar]
- Zhang, F.; Cong, L.; Lodato, S.; Kosuri, S.; Church, G.M.; Arlotta, P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 2011, 29, 149–153. [Google Scholar] [Green Version]
- Briggs, A.W.; Rios, X.; Chari, R.; Yang, L.; Zhang, F.; Mali, P.; Church, G.M. Iterative capped assembly: Rapid and scalable synthesis of repeat-module DNA such as TAL effectors from individual monomers. Nucleic Acids Res. 2012, 40, e117. [Google Scholar] [CrossRef]
- Reyon, D.; Tsai, S.Q.; Khayter, C.; Foden, J.A.; Sander, J.D.; Joung, J.K. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 2012, 30, 460–465. [Google Scholar] [CrossRef]
- Schmid-Burgk, J.L.; Schmidt, T.; Kaiser, V.; Honing, K.; Hornung, V. A ligation-independent cloning technique for high-throughput assembly of transcription activator-like effector genes. Nat. Biotechnol. 2013, 31, 76–81. [Google Scholar]
- Mussolino, C.; Morbitzer, R.; Lutge, F.; Dannemann, N.; Lahaye, T.; Cathomen, T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011, 39, 9283–9293. [Google Scholar] [CrossRef]
- Holkers, M.; Maggio, I.; Liu, J.; Janssen, J.M.; Miselli, F.; Mussolino, C.; Recchia, A.; Cathomen, T.; Goncalves, M.A. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013, 41, e63. [Google Scholar] [CrossRef]
- Wiedenheft, B.; Lander, G.C.; Zhou, K.; Jore, M.M.; Brouns, S.J.; van der Oost, J.; Doudna, J.A.; Nogales, E. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature 2011, 477, 486–489. [Google Scholar] [CrossRef]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef]
- Sampson, T.R.; Saroj, S.D.; Llewellyn, A.C.; Tzeng, Y.L.; Weiss, D.S. A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 2013, 497, 254–257. [Google Scholar]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar]
- CRISPR Design. Available online: http://crispr.mit.edu/ (accessed on 12 November 2013).
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3, e2510. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Manjunath, N.; Yi, G.; Dang, Y.; Shankar, P. Newer Gene Editing Technologies toward HIV Gene Therapy. Viruses 2013, 5, 2748-2766. https://doi.org/10.3390/v5112748
Manjunath N, Yi G, Dang Y, Shankar P. Newer Gene Editing Technologies toward HIV Gene Therapy. Viruses. 2013; 5(11):2748-2766. https://doi.org/10.3390/v5112748
Chicago/Turabian StyleManjunath, N., Guohua Yi, Ying Dang, and Premlata Shankar. 2013. "Newer Gene Editing Technologies toward HIV Gene Therapy" Viruses 5, no. 11: 2748-2766. https://doi.org/10.3390/v5112748