Phage Lambda P Protein: Trans-Activation, Inhibition Phenotypes and their Suppression


Abstract
:1. Introduction
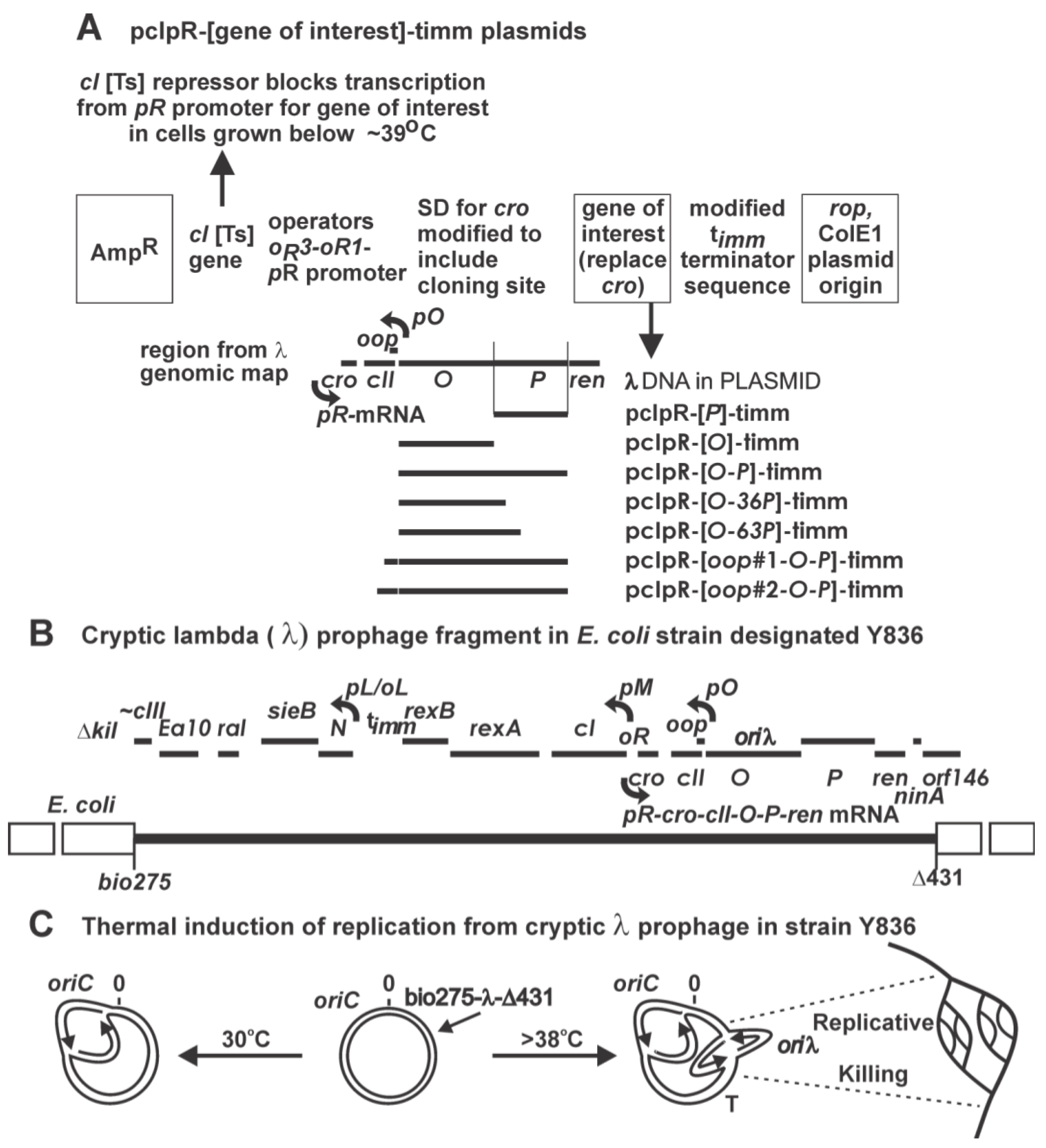
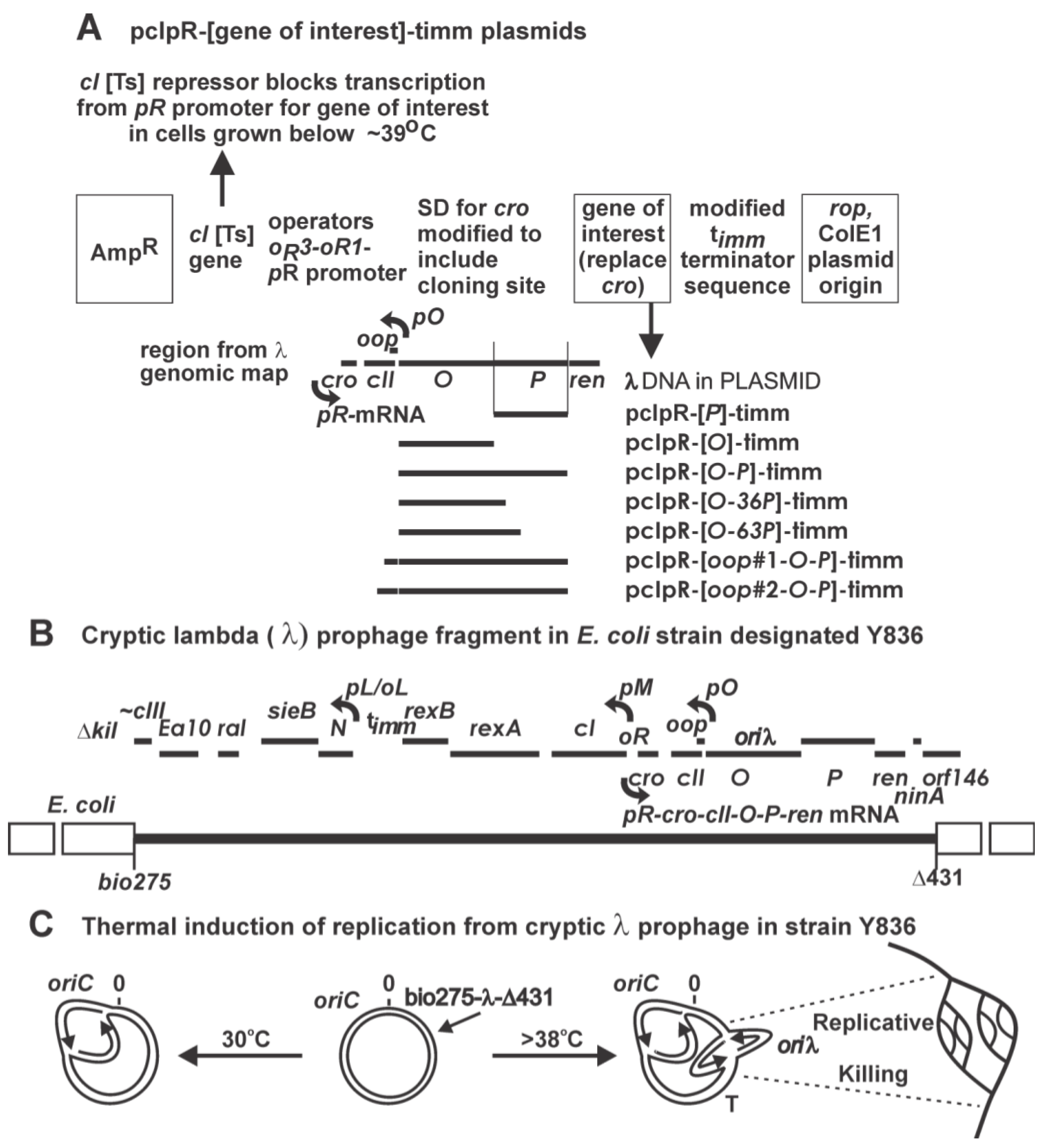
2. Result and Discussions
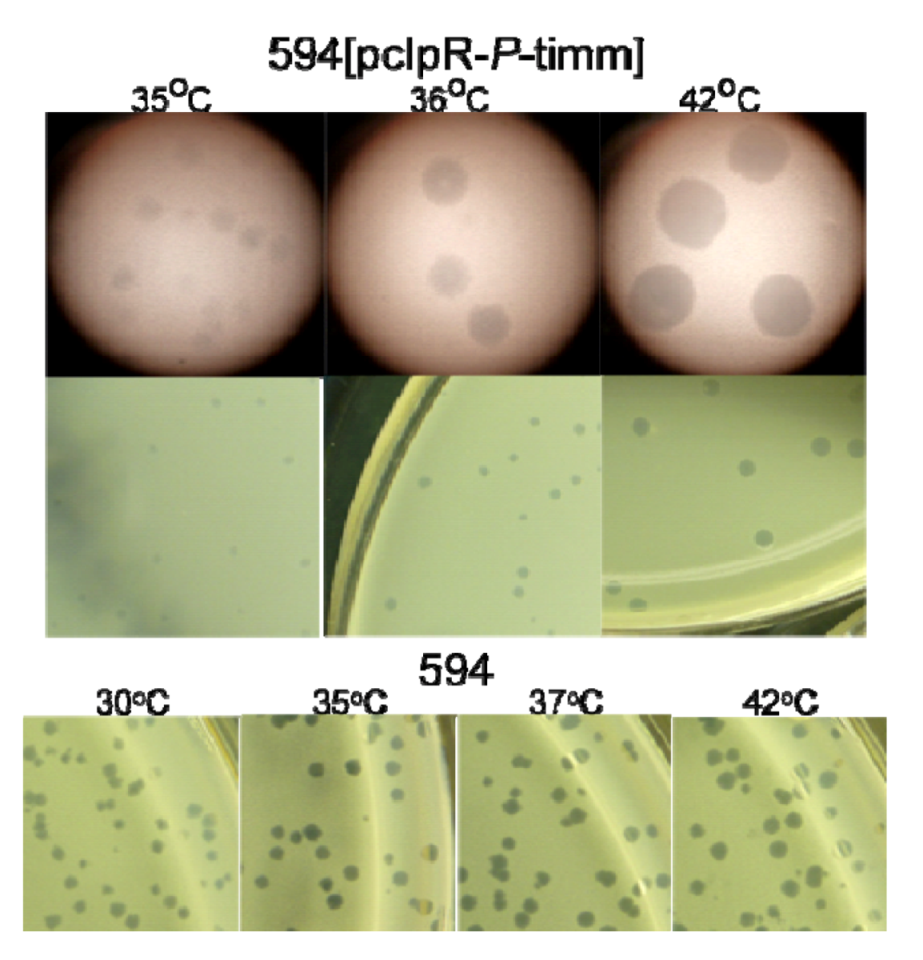
2.1. Transformation of Cells by Plasmids Expressing P, Complementation and Immunity Assays

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| dnaB allele | bp E. coli mutation | AA changed |
|---|---|---|
| grpD55 | 4,263,102 G to A | V256I |
| 4,263,612 G to A | E426K | |
| grpA80 | 4,263,349 G to A | G338E |
| 4,263,612 G to A | E426K |
| Host strains | EOP on host cells incubated at temperature a | |||
| 30 | 37 | 39 | 42 ˚C | |
| 594 | 1.0 | ndb | nd | 0.94 |
| 594 grpD55 | 0.0147 | 4.4 × 10-7 | 4.4 × 10-7 | 4.6 × 10-7 |
| Cells Transformed | Plating Temp. ˚C | Transformation Frequency a | Transformants per µg DNA |
|---|---|---|---|
| 594 | 25 | 5.2 × 10-5 | 1.5 × 105 |
| 37 | <1.7 × 10-8 | 0 | |
| 594(λ wt nin5) | 25 | 7.2 × 10-5 | 4.7 × 104 |
| 37 | 8.5 × 10-6 | 5.5 × 103 |
| Plating host | Incubation temperature | EOP a of λimm434Pam3 |
|---|---|---|
| 594[pcIpR- P-timm] | 25 ˚C | 0.0008 |
| 30 | 0.82 | |
| 37 | 0.93 | |
| 594[pcIpR- PΔ76-timm] | 30 | <0.00003 |
| 37 | 2 × 10-7 |
| Host strains | Plating temperature (˚C) | |||||
|---|---|---|---|---|---|---|
| 25 | 30 | 35 | 37 | 39 | 42 | |
| 594 | 0.5 | 0.9 | 0.9 | 1.0 | 1.0 | 0.4 |
| 594[pcIpR-P-timm] | <4 × 10-10 | <4 × 10-10 | 0.2 | 0.5 | 0.7 | 1.0 |
| 594[pcIpR-O-timm] | <4 × 10-10 | <4 × 10-10 | <4 × 10-10 | 9 × 10-7c | 2 × 10-6c | 0.8 |
| 594[p434’pR-O-timm] | 0.8 | 0.9 | 1.0 | 1.0 | 1.0 | 1.0 |
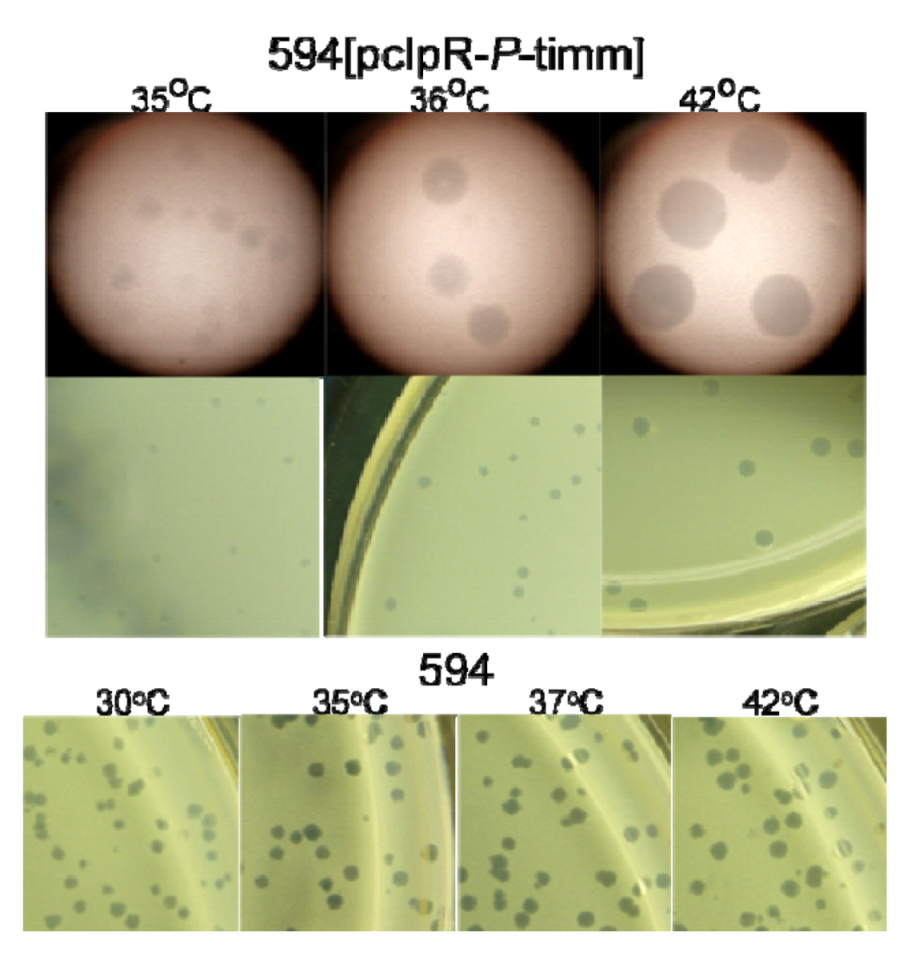
2.1.1. “Trans-activation” of P
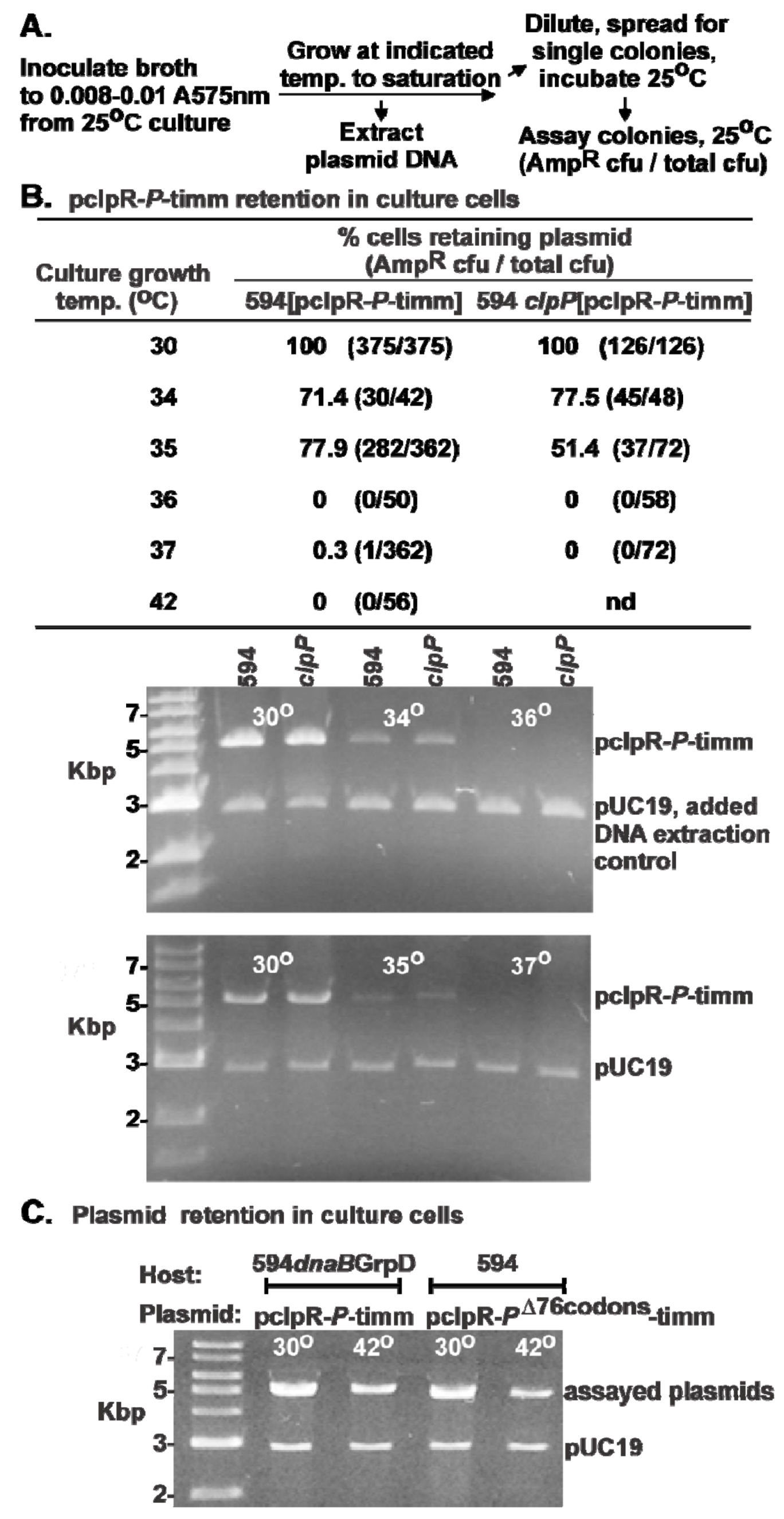
2.2. Influence of P-Expression on Plasmid Retention.

2.3. P-Lethality Suppression
| Host cells and plasmids | Cell viability a and (plasmid retention/cfu; %) b at growth temperature (˚C) | ||
|---|---|---|---|
| 25 | Ave 37 & 39 | 42 | |
| 594[pcIpR- P-timm] | 1.0 | 0.0049 | 0.001 |
| (195/196; ~100) | (0/57; 0) | (0/28; 0) | |
| 594[pcIpR- PSPA-timm] | 1.0 | 0.006 | 0.004 |
| (14/14; 100) | (0/150; 0) | (0/39; 0) | |
| 594[pcIpR- Pπ-timm] | 1.0 | 0.635 | 0.003 |
| (103/103; 100) | (789/793; ~100) | (0/294; 0) | |
| 594[pcIpR- PΔ76-timm] | 1.0 | 0.96 | 0.12 |
| (166/168; 99) | (334/336; 99) | (166/168; 99) | |
| 594 grpD55 [pcIpR-P-timm] | 1.0 | 0.99 | 1.0 |
| (35/35; 100) | (87/88; 99) | (14/14; 100) | |
| 594 grpD55 [pcIpR-PSPA-timm] | 1.0 | 0.75 | 0.57 |
| (42/42; 100) | (88/94; 94) | (18/19; 95) | |
| 594 grpD55 [pcIpR-Pπ-timm] | 1.0 | 0.08 | 0.01 |
| (54/54; 100) | (434/435; ~100) | (5/108; 5) | |
| 594 grpD55 [pcIpR-PΔ76-timm] | 1.0 (nd) | 0.45 (nd) | 0.001 (nd) |
| Re-claim pcIpR-P-timm from 594 grpD55 cultures and transform into 594 dnaB+ cells | |||
| Re-claim from 25˚C cultures c | 1.0 | 0.08 | 0.01 |
| (54/56; 96) | (0/119; 0) | (0/91; 0) | |
| Re-claim from 42˚C cultures d | 1.0 | 0.059 | 0.026 |
| (130/130; 96) | (1/230; 0.4) | (0/201; 0) | |
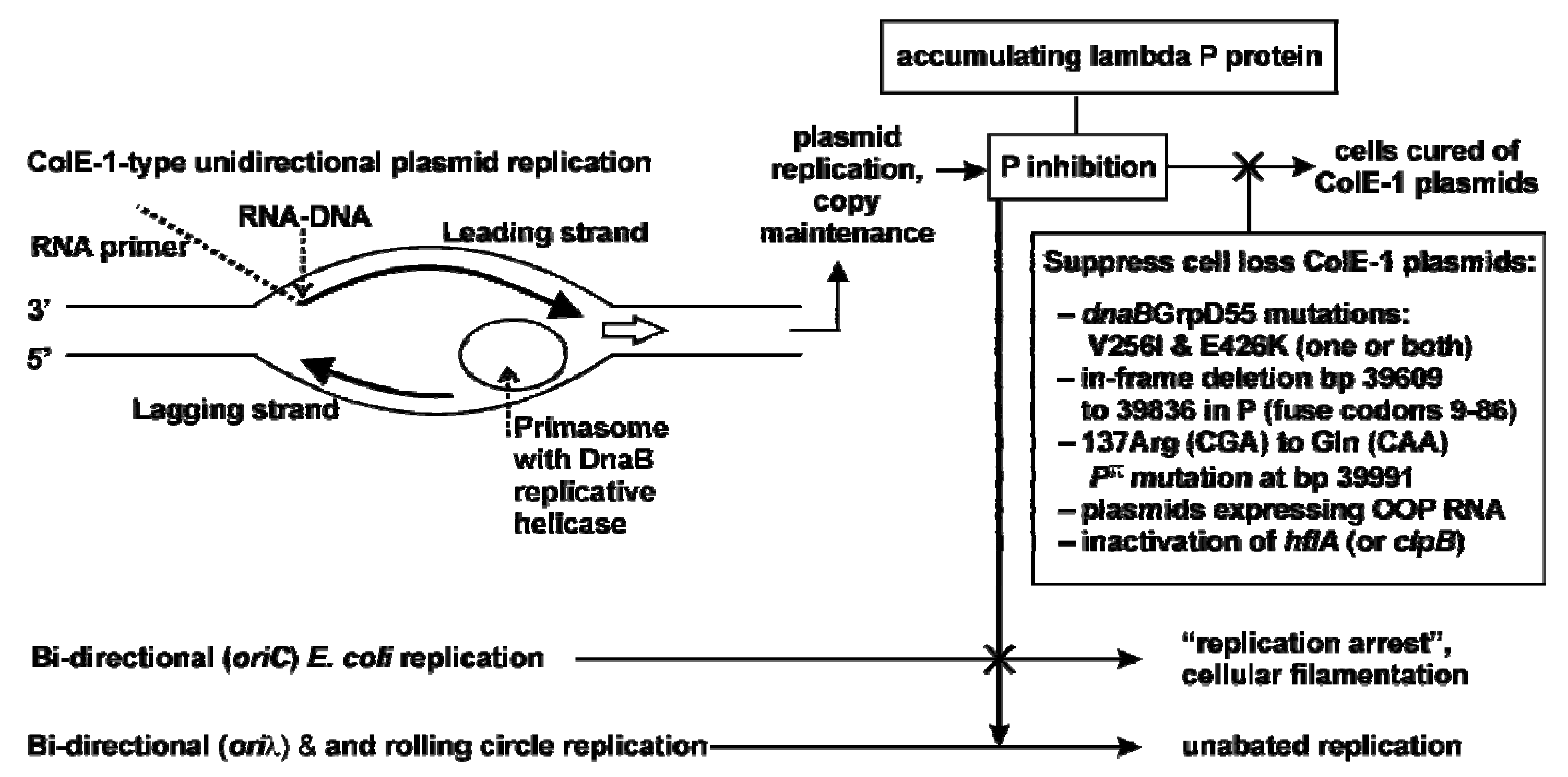
2.3.1. Model for Plasmid Loss
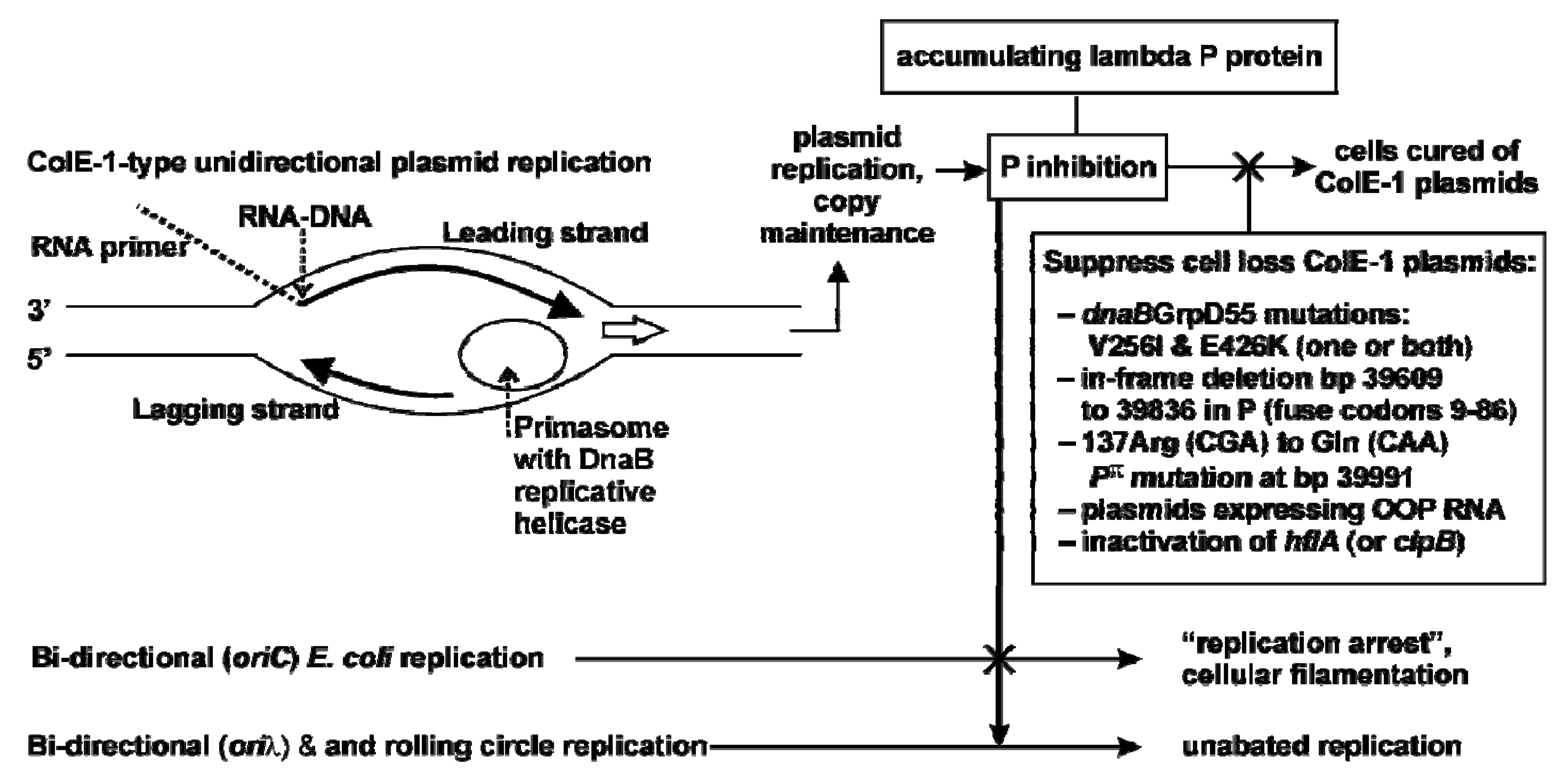
2.4. Influence of P-alteration, or O and OOP RNA Co-Expression on P-lethality and Plasmid Retention
| Plasmids in 594 host cells | Cell viability and (plasmid retention/cfu; %)at growth temperature (˚C) | ||||
|---|---|---|---|---|---|
| 30 | 35 | 37 | 39 | 42 | |
| pcIpR- P-timm | 1.0 | 0.32 | 0.01 | 0.008 | 0.07 |
| (35/35; 100) | (33/33; 100) | (0/35; 0) | (0/35; 0) | (0/35; 0) | |
| pcIpR- O-P-timm | 1.0 | 0.345 | 0.12 | 0.12 | Nd |
| (62/70; 89) | (28/70; 40) | (0/70; 0) | (0/70; 0) | ||
| pcIpR- O-36P-timm | 1.0 | 0.825 | 0.793 | 0.012 | Nd |
| (30/30; 100) | (30/30; 100) | (30/30; 100) | (1/30; 3) | ||
| pcIpR- O-63P-timm | 1.0 | 0.895 | 0.895 | 0.055 | Nd |
| 30/30; 100) | (30/30; 100) | (30/30; 100) | (14/40; 35) | ||
| pcIpR- O-timm | 1.0 | 1.0 | 1.0 | 1.0 | 0.608 |
| (30/30; 100) | (30/30; 100) | (30/30;100) | (30/30; 100) | (29/30; 97) | |
| p434’pR- O-timm b | 1.0 | 1.0 | 1.0 | 1.0 | 0.615 |
| (26/30; 87) | (29/30; 97) | (28/30; 93) | (26/30; 87) | (26/30; 87) | |
| pcIpR- oop#1-O-P-timm c | 1.0 | 0.938 | 0.20 | 0.005 | Nd |
| (120/120; 100) | (120/120; 100) | (98/101; 97) | (115/120; 96) | ||
| pcIpR- oop#2 O-P-timm c | 1.0 | 0.988 | 0.055 | 0.048 | Nd |
| (117/120; 98) | (60/60; 100) | (76/154; 49) | (62/120; 52) | ||
2.5. Replicative (cis) killing; P (trans) Lethality / Inhibition
| A. Cells tolerate short-term exposure to P (short-term interference, not lethality) a | |||
| Strain with plasmid | Incubation at 37 ˚C | Cell viability (AmpR cfu/ total cfu) | Outcome of P expression from plasmid |
| 594[pcIpR-P-timm] | 1 hr | 0.87 (232/242) | most cells recover |
| 2 hr | 0.49 (258/281) | many cells recover | |
| 6 hr | 0.13 (2/173) | high plasmid loss | |
| B. Inducing replication from a trapped cryptic prophage causes Replicative Killing a | |||
| Strains with cryptic prophages | Prophage Induction time | Cell viability | Outcome of prophage Induction |
| Y836[~cIII-cI857-O-P-ren] | 5 min | 0.33 | rapid cell killing |
| 20 min | 0.13 | rapid cell killing | |
| 3 hr | 0.00018 | extensive cell killing | |
| 5 hr | 0.00008 | extensive cell killing | |
| 594[~cIII-cI857-O-P-ren] | 5 min | 0.55 | rapid cell killing |
| 20 min | 0.25 | rapid cell killing | |
| 3 hr | 0.0022 | extensive cell killing | |
| 5 hr | 0.0022 | extensive cell killing | |
| Y836[~cIII-cI857-O-P::kan-ren] | 3 hr | 5.1 b | cell growth |
| 5 hr | 6.1 b | cell growth | |
2.5.1. Contrasting trans P-lethality / inhibition, and cis Replicative Killing
2.6. Does P Expression from pcIpR-P-timm perturb λ Vegetative Growth?
2.6.1. λ Replication and Phage Maturation
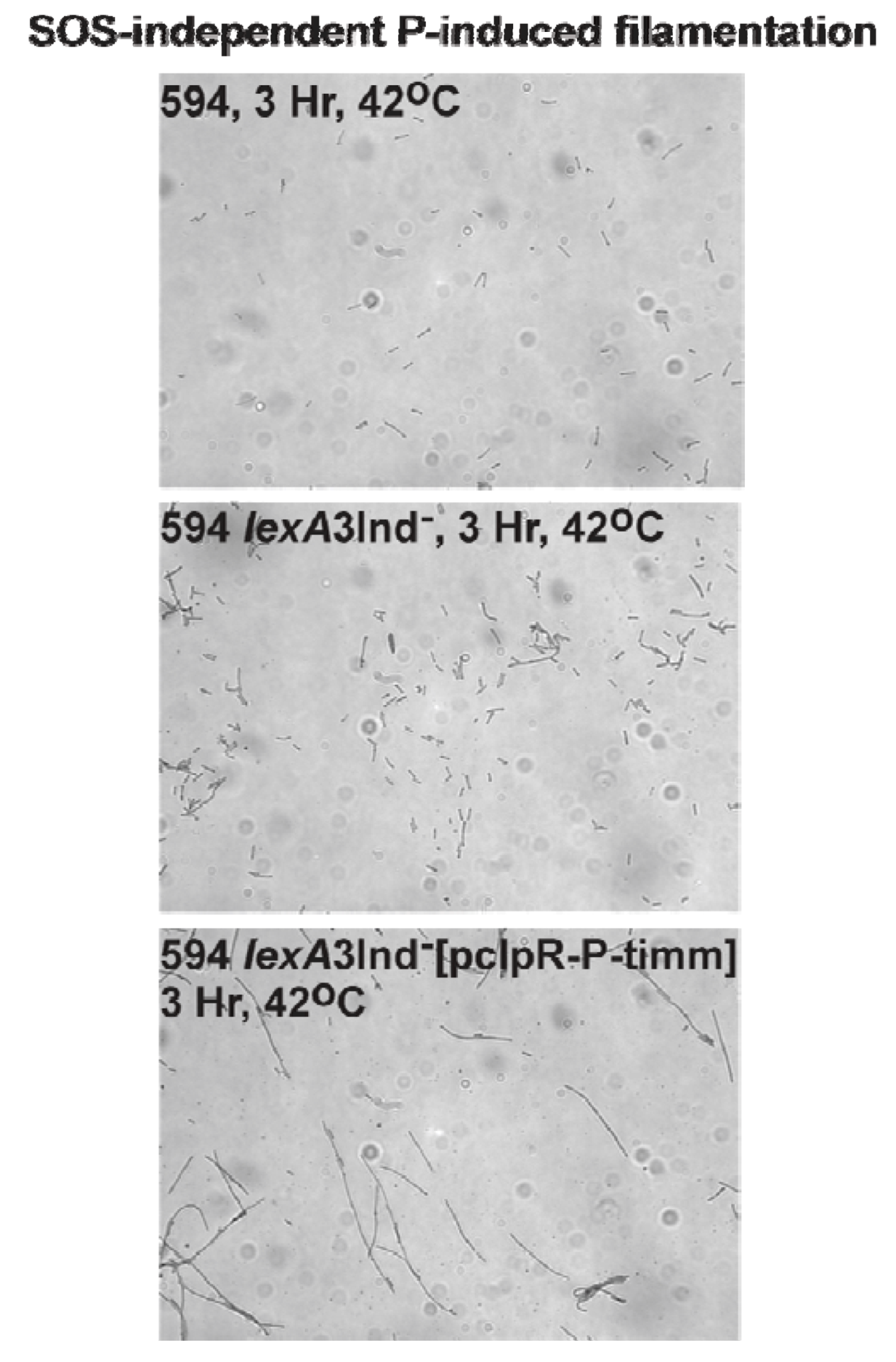
2.7. P-Induced Cellular Filamentation
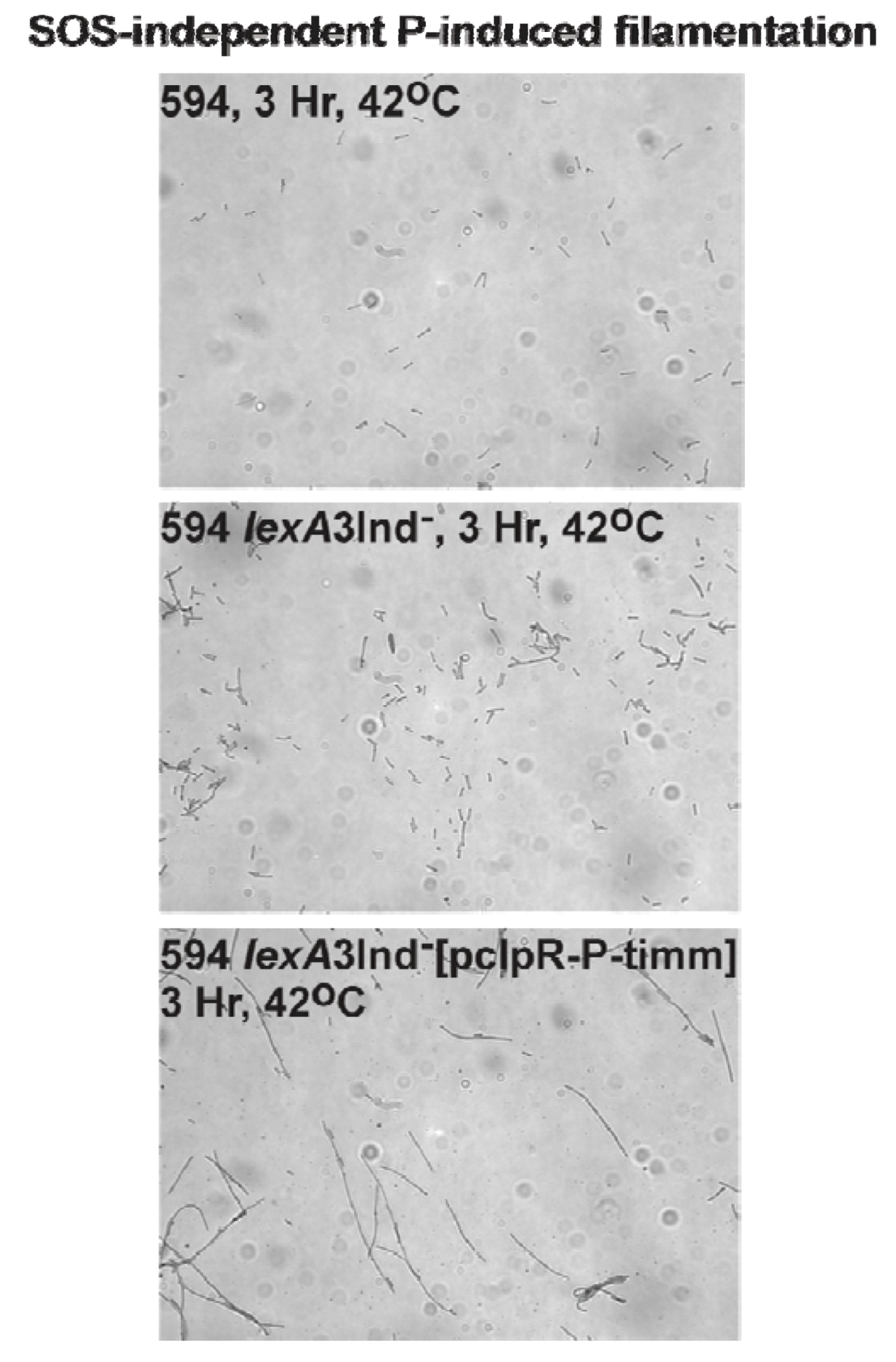
| Strain [plasmid] | Time(Temp) | Relative cell length | Sumcells | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1X | 2X | 3X | 4X | 5X | 6X | 7X | 8X | 9X | ≥10X | |||
| 594 | 0 (25) | 26 | 19 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 45 |
| “ | 1 (42) | 15 | 11 | 11 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 45 |
| “ | 3 (42) | 28 | 15 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 45 |
| “ | 5 (42) | 29 | 14 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 45 |
| 594 lexA3 | 0 (25) | 40 | 32 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 74 |
| “ | 1 (42) | 23 | 16 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 45 |
| “ | 3 (42) | 31 | 13 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 45 |
| “ | 5 (42) | 42 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 45 |
| 594 lexA3[pcIpR-P-timm] | 0 (25) | 71 | 19 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 90 |
| “ | 1 (37) | 21 | 23 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 45 |
| “ | 3 (37) | 7 | 9 | 9 | 6 | 4 | 3 | 2 | 1 | 2 | 2 | 45 |
| “ | 5 (37) | 6 | 20 | 0 | 0 | 2 | 0 | 3 | 3 | 3 | 8 | 45 |
| 594 lexA3[pcIpR-P-timm] | 1 (42) | 9 | 19 | 13 | 3 | 1 | 0 | 0 | 0 | 0 | 0 | 45 |
| “ | 3 (42) | 11 | 1 | 3 | 4 | 4 | 9 | 8 | 2 | 2 | 1 | 45 |
| “ | 5 (42) | 8 | 6 | 7 | 1 | 1 | 0 | 2 | 1 | 2 | 14 | 42 |
| 594 [pHB30] | 0 (30) | 6 | 20 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 30 |
| “ | 1 (42) | 0 | 2 | 10 | 9 | 3 | 4 | 1 | 1 | 0 | 0 | 30 |
| “ | 3 (42) | 2 | 4 | 8 | 3 | 2 | 5 | 2 | 1 | 2 | 1 | 30 |
| “ | 5 (42) | 2 | 8 | 10 | 2 | 2 | 2 | 3 | 0 | 0 | 1 | 30 |
| 594 lexA [pHB30] | 0 (30) | 5 | 17 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 30 |
| “ | 1 (42) | 0 | 6 | 6 | 8 | 4 | 1 | 4 | 0 | 1 | 0 | 30 |
| “ | 3 (42) | 3 | 5 | 2 | 6 | 4 | 0 | 5 | 3 | 1 | 1 | 30 |
| “ | 5 (42) | 1 | 7 | 5 | 6 | 5 | 2 | 1 | 1 | 0 | 2 | 30 |
| 594 ΔrecA | 0 (30) | 16 | 12 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 30 |
| “ | 1 (42) | 4 | 12 | 7 | 4 | 1 | 2 | 0 | 0 | 0 | 0 | 30 |
| “ | 3 (42) | 9 | 16 | 2 | 4 | 2 | 0 | 0 | 0 | 0 | 0 | 30 |
| “ | 5 (42) | 12 | 11 | 4 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 30 |
| 594 ΔrecA [pHB30] | 0 (30) | 8 | 17 | 4 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 30 |
| “ | 1 (42) | 2 | 5 | 8 | 10 | 3 | 1 | 1 | 0 | 0 | 0 | 30 |
| “ | 3 (42) | 5 | 4 | 5 | 3 | 6 | 4 | 2 | 0 | 0 | 1 | 30 |
| “ | 5 (42) | 3 | 5 | 4 | 8 | 0 | 4 | 2 | 0 | 1 | 3 | 30 |
2.7.1. P-Dependent Cellular Filamentation
3. Experimental Section
3.1. Strains Employed
| Bacterial strains | Characteristics or genotype | Source/Ref.’; Hayes lab #a |
|---|---|---|
| 594 | F-lac-3350 galK2 galT22 rpsL179 IN(rrnD-rrnE)1; see [97]; called R594 | [97], SH lab; B10 |
| TC600 | supE, Pm+ | SH lab, B8 |
| Ymel | supF, Pm+ | SH lab, B71 |
| DE407 | lexA3[Ind-] malB::Tn9 TetRsulA211 sfiA11, UVS | D. Ennis; B142 |
| FC40 (=SMR624) | Δ(srlR-recA)306::Tn10 TetR UVS | SM Rosenberg [98]; Y921 |
| AB2834 aroE | grpD55, thi tsxR λR at 42 ˚C from K552 | H. Uchida [32,33]; NB83 |
| W3874 malB5 | dnaB grpA80 lac- StrR λR at 42 ˚C | [32], NB81 |
| W3350 dnaB-grpD55 | grpD55 malF3089::Tn10 TetRλR at 42°C, λrepP22S | [33], NB15 |
| 594 dnaB-grpD55 | grpD55 allele malF3089::Tn10; TetR, λR at 42°C, λrepP22S | [34], NB295 |
| 594 lexA3[Ind-] malB::Tn9 | LexA repressor induction defective | CE, NB293 |
| 594 Δ(srlR-recA)306::Tn10 | deletion of recA TetR UVS | CE, B318 |
| W3350 | F- lac-3350 galK2 galT22 rpsL179 IN(rrnD-rrnE)1 | SH lab, B12 |
| Y836 | SA500(λbio275cI[Ts]857 Δ431) his- | [78,82], NY1049 |
| 594::nadA::Tn10 [~cIII-ren]λ | Tn10 [zbh29 at 16.8 min] bio+ transductant = 594 bio275 (λcIII-cI[Ts]857-O-P-ren) Δ431 | A. Chu, SH lab, NY1057 |
| Y836 P::kan(Bib11t) | SA500 (λbio275 cI[Ts]857 O+P::kan Δ431) his- KanR | SH, NY1153 |
| 594(λcI857Sam7) | λ lysogen defective for cell lysis | C. Marek, SH lab, Y1163 |
| 594(λcI857Sam7)[pcIpR-P-timm] | as above with transformed plasmid | SH lab, P509 |
| 594 clpP::kan | clpP-, KanR from SG22159 | S. Gottesman; [99], NB276 |
| Plasmids | Transformed into strain 594 | Source/Ref.’; Hayes lab #a |
| pUC19 | Wild type AmpR (New England Biolabs) | NP188 |
| pcIpR-P-timm | BamHI-ClaI PCR fragment from λcI857, replacing D-CAP in P459 with λ bp’s 39582-40280 | CH, P466 |
| pcIpR-P::kan-timm | PCR BamHI-ClaI fragment from Y836 P::kan(Bib11t) strain NY1153 | KM, P510 |
| PcIpR-P-SPA-timm b | Replace D in P462 between BamHI and AscI sites with BamHI-P(λ bp’s 39582-40280)-AscI PCR fragment | KM, P467 |
| pcIpR-PΔ76-timm | In-frame deletion76 codons: λbp 39609-39836 in pcIpR-P-timm with HpaI, ligate | KM, P515 |
| pcIpR-Pπ-timm | BamHI-ClaI PCR fragment from λcI72π Lysate #3a, replacing D-CAP in P459 with λ bp’s 39582-40280 | KM, P505 |
| pcIpR-O-timm | BamHI-ClaI PCR fragment from λcI857, replacing D-CAP in P459 with λ bp’s 38686-39582 | [36], CH, P465 |
| P434’pR-O-timm | Constitutive O expression; BamHI-ClaI PCR fragment from λcI857, replacing D-CAP in P459 with λ bp’s 38686-39582 | [36], CH, P494 |
| pcIpR-O-P-timm | BamHI-ClaI PCR fragment from λcI857, replacing D-CAP in P459 with λ bp’s 38686-40280 | CH, P569 |
| pcIpR-O-36P-timm | BamHI-ClaI PCR fragment from λcI857, replacing D-CAP in P459 with λ bp’s 38686-39687 | CH, P565 |
| pcIpR-O-63P-timm | BamHI-ClaI PCR fragment from λcI857, replacing D-CAP in P459 with λ bp’s 38686-39768 | CH, P566 |
| pcIpR-oop#1-O-P-timm | BamHI-ClaI PCR fragment from λcI857, replacing D-CAP in P459 with λ bp’s 38559-40280 | CH, P567 |
| pcIpR-oop#2-O-P-timm | BamHI-ClaI PCR fragment from λcI857, replacing D-CAP in P459 with λ bp’s 38546-40280 | CH, P568 |
| pHB30 | λ bases 34499-34696, 36965-38103, 38814-40806 (see Section 3.2.) | [31,34], SH lab, P8 |
| Bacteriophage | Genotype | Hayes lab lysate # |
| λ wild type (wt) | λpapa | [78], 944,1001 |
| λcI72 | cI- | [78], 951, 999 |
| λnin5 | made from λ wt | [78], CH, 698 |
| Λvir | λv2v1v3 | [78], 260 |
| λcI857 | cI[Ts]857 | [100], 1002 |
| λcI857Sam7 | defective for cell lysis | [101], 963 |
| λimm434Pam3 | imm434, sequenced Pam3 mutation C to T, λ base 39786 (CAG to TAG) | [83], SH lab, 518, 664 |
| λimm434nin5 | imm434, Δnin5 region, forms very turbid plaques at 37˚C | [22], CH, 963 |
3.2. Construction of Expression Vectors for λ Genes, Gel Analysis of Plasmids, Insertion Localization
3.3. Plasmid Transformation; Phage and Culture Assays
3.4. Sequence Analysis of Alleles of dnaB
3.5. Assessing Influence of Modifying P or dnaB on P Lethality and Plasmid Loss
3.6. Contrasting trans P-Lethality / Inhibition and cis Replicative Killing
3.7. SOS-Independent P-Induced Cellular Filamentation
3.8. P-Induced Plasmid Loss
4. Conclusions
Conflict of Interest
Acknowledgements
References
- Tsurimoto, T.; Matsubara, K. Purified bacteriophage lambda O protein binds to four repeating sequences at the lambda replication origin. Nucleic Acids Res. 1981, 9, 1789–1799. [Google Scholar]
- Wickner, S.H. DNA replication proteins of Escherichia coli and phage lambda. Cold Spring Harbor Symp. Quant. Biol. 1979, 43 Pt 1, 303–310. [Google Scholar] [CrossRef]
- LeBowitz, J.H.; McMacken, R. The Escherichia coli DnaB replication protein is a DNA helicase. J. Biol. Chem. 1986, 261, 4738–4748. [Google Scholar]
- McMacken, R.; Ueda, K.; Kornberg, A. Migration of Escherichia coli DnaB protein on the template DNA strand as a mechanism in initiating DNA replication. Proc. Natl. Acad. Sci. U.S.A. 1977, 74, 4190–4194. [Google Scholar] [CrossRef]
- Zyskind, J.W.; Smith, D.W. Novel Escherichia coli dnaB mutant: Direct involvement of the dnaB252 gene product in the synthesis of an origin-ribonucleic acid species during initiaion of a round of deoxyribonucleic acid replication. J. Bacteriol. 1977, 129, 1476–1486. [Google Scholar]
- Bujalowski, W. Expanding the physiological role of the hexameric DnaB helicase. Trends Biochem. Sci. 2003, 28, 116–118. [Google Scholar]
- Kaplan, D.L.; O'Donnell, M. DnaB drives DNA branch migration and dislodges proteins while encircling two DNA strands. Mol. Cell 2002, 10, 647–657. [Google Scholar]
- Arai, K.; Kornberg, A. Mechanism of dnab protein action. Ii. ATP hydrolysis by DnaB protein dependent on single- or double-stranded DNA. J. Biol. Chem. 1981, 256, 5253–5259. [Google Scholar]
- Reha-Krantz, L.J.; Hurwitz, J. The dnaB gene product of Escherichia coli. I. Purification, homogeneity, and physical properties. J. Biol. Chem. 1978, 253, 4043–4050. [Google Scholar]
- Biswas, S.B.; Biswas, E.E. Regulation of dnaB function in DNA replication in Escherichia coli by dnaC and lambda P gene products. J. Biol. Chem. 1987, 262, 7831–7838. [Google Scholar]
- Mallory, J.B.; Alfano, C.; McMacken, R. Host virus interactions in the initiation of bacteriophage lambda DNA replication. Recruitment of Escherichia coli DnaB helicase by lambda P replication protein. J. Biol. Chem. 1990, 265, 13297–13307. [Google Scholar]
- Alfano, C.; McMacken, R. Ordered assembly of nucleoprotein structures at the bacteriophage lambda replication origin during the initiation of DNA replication. J. Biol. Chem. 1989, 264, 10699–10708. [Google Scholar]
- Zylicz, M.; Ang, D.; Liberek, K.; Georgopoulos, C. Initiation of lambda DNA replication with purified host- and bacteriophage-encoded proteins: The role of the DnaK, DnaJ and GrpE heat shock proteins. EMBO J. 1989, 8, 1601–1608. [Google Scholar]
- Nakayama, N.; Arai, N.; Bond, M.W.; Kaziro, Y.; Arai, K. Nucleotide sequence of dnaB and the primary structure of the DnaB protein from Escherichia coli. J. Biol. Chem. 1984, 259, 97–101. [Google Scholar]
- Ueda, K.; McMacken, R.; Kornberg, A. DnaB protein of Escherichia coli. Purification and role in the replication of phix174 DNA. J. Biol. Chem. 1978, 253, 261–269. [Google Scholar]
- Hayes, S.; Bull, H.J.; Tulloch, J. The Rex phenotype of altruistic cell death following infection of a lambda lysogen by T4rII mutants is suppressed by plasmids expressing OOP RNA. Gene 1997, 189, 35–42. [Google Scholar]
- Hayes, S.; Slavcev, R.A. Polarity within pM and pE promoted phage lambda cI-rexA-rexB transcription and its suppression. Can. J. Microbiol. 2005, 51, 37–49. [Google Scholar] [CrossRef]
- Hayes, S.; Szybalski, W. Control of short leftward transcripts from the immunity and ori regions in induced coliphage lambda. Mol. Gen. Gen. 1973, 126, 275–290. [Google Scholar] [CrossRef]
- Landsmann, J.; Kroger, M.; Hobom, G. The rex region of bacteriophage lambda: Two genes under three-way control. Gene 1982, 20, 11–24. [Google Scholar] [CrossRef]
- Hayes, S.; Gamage, L.N.; Hayes, C. Dual expression system for assembling phage lambda display particle (LDP) vaccine to porcine circovirus 2 (PCV2). Vaccine 2010, 28, 6789–6799. [Google Scholar]
- Revet, B.; von Wilcken-Bergmann, B.; Bessert, H.; Barker, A.; Muller-Hill, B. Four dimers of lambda repressor bound to two suitably spaced pairs of lambda operators form octamers and DNA loops over large distances. Curr. Biol. 1999, 9, 151–154. [Google Scholar]
- Hayes, S.; Asai, K.; Chu, A.M.; Hayes, C. NinR- and Red-mediated phage-prophage marker rescue recombination in Escherichia coli: Recovery of a nonhomologous immlambda DNA segment by infecting lambdaimm434 phages. Genetics 2005, 170, 1485–1499. [Google Scholar] [CrossRef]
- Klinkert, J.; Klein, A. Cloning of the replication gene P of bacteriophage lambda: Effects of increased P-protein synthesis on cellular and phage DNA replication. Mol. Gen. Gen. 1979, 171, 219–227. [Google Scholar]
- Maiti, S.; Mukhopadhyay, M.; Mandal, N.C. Bacteriophage lambda P gene shows host killing which is not dependent on lambda DNA replication. Virology 1991, 182, 324–335. [Google Scholar]
- Georgopoulos, C.P.; Herskowitz, I. Escherichia coli mutants blocked in lambda DNA synthesis. In The bacteriophage lambda; Hershey, A.D., Ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, N.Y., 1971; pp. 553–564. [Google Scholar]
- Georgopoulos, C.P. A new bacterial gene (groPC) which affects lambda DNA replication. Mol. Gen. Gen. 1977, 151, 35–39. [Google Scholar] [CrossRef]
- Sunshine, M.; Feiss, M.; Stuart, J.; Yochem, J. A new host gene (groPC) necessary for lambda DNA replication. Mol. Gen. Gen. 1977, 151, 27–34. [Google Scholar] [CrossRef]
- Datta, I.; Sau, S.; Sil, A.K.; Mandal, N.C. The bacteriophage lambda DNA replication protein P inhibits the oriC DNA- and ATP-binding functions of the DNA replication initiator protein DnaA of Escherichia coli. J. Biochem. Mol. Biol. 2005, 38, 97–103. [Google Scholar] [CrossRef]
- Datta, I.; Banik-Maiti, S.; Adhikari, L.; San, S.; Das, N.; Mandal, N.C. The mutation that makes Escherichia coli resistant to lambda P gene-mediated host lethality is located within the DNA initiator gene dnaA of the bacterium. J. Biochem. Mol. Biol. 2005, 38, 89–96. [Google Scholar] [CrossRef]
- Bolivar, F.; Betlach, M.C.; Heyneker, H.L.; Shine, J.; Rodriguez, R.L.; Boyer, H.W. Origin of replication of pBR345 plasmid DNA. Proc. Natl. Acad. Sci. U.S.A. 1977, 74, 5265–5269. [Google Scholar]
- Bull, H.J. Bacteriophage lambda replication-coupled processes: Genetic elements and regulatory choices. University of Saskatchewan: Saskatoon, SK, Canada, 1995. [Google Scholar]
- Saito, H.; Uchida, H. Initiation of the DNA replication of bacteriophage lambda in Escherichia coli K12. J. Mol. Biol. 1977, 113, 1–25. [Google Scholar] [CrossRef]
- Bull, H.J.; Hayes, S. The grpD55 locus of Escherichia coli appears to be an allele of dnaB. Mol. Gen. Gen. 1996, 252, 755–760. [Google Scholar]
- Horbay, M.A. Inhibition phenotype specific for ori-lambda replication dependent phage growth, and a reappraisal of the influence of lambda P expression on Escherichia coli cell metabolism: P-interference phenotype. University of Saskatchewan: Saskatoon, SK, Canada, 2005. [Google Scholar]
- Mandal, N.C.; Lieb, M. Heat-sensitive DNA-binding activity of the cI product of bacteriophage lambda. Mol. Gen. Gen. 1976, 146, 299–302. [Google Scholar]
- Hayes, S.; Horbay, M.A.; Hayes, C. A cI-independent form of replicative inhibition: Turn off of early replication of bacteriophage lambda. PLoS One 2012, 7, e36498. [Google Scholar]
- Herskowitz, I.; Signer, E. Control of transcription from the r strand of bacteriophage lambda. Cold Spring Harbor Symp. Quant. Biol. 1970, 35. [Google Scholar]
- Thomas, R. Control of development in temperate bacteriophages. 3. Which prophage genes are and which are not trans-activable in the presence of immunity? J. Mol. Biol. 1970, 49, 393–404. [Google Scholar] [CrossRef]
- Bejarano, I.; Klemes, Y.; Schoulaker-Schwarz, R.; Engelberg-Kulka, H. Energy-dependent degradation of lambda O protein in Escherichia coli. J. Bacteriol. 1993, 175, 7720–7723. [Google Scholar]
- Wegrzyn, A.; Czyz, A.; Gabig, M.; Wegrzyn, G. ClpP/clpX-mediated degradation of the bacteriophage lambda O protein and regulation of lambda phage and lambda plasmid replication. Arch. Microbiol. 2000, 174, 89–96. [Google Scholar]
- Konieczny, I.; Marszalek, J. The requirement for molecular chaperones in lambda DNA replication is reduced by the mutation pi in lambda P gene, which weakens the interaction between lambda P protein and DnaB helicase. J. Biol. Chem. 1995, 270, 9792–9799. [Google Scholar] [CrossRef]
- Klein, A.; Lanka, E.; Schuster, H. Isolation of a complex between the P protein of phage lambda and the dnaB protein of Escherichia coli. Eur. J. Biochem. 1980, 105, 1–6. [Google Scholar] [CrossRef]
- Sutton, M.D.; Carr, K.M.; Vicente, N.; Kaguni, J.M. Escherichia coli DnaA protein - the N-terminal domain and loading of DnaB helicase at the E. coli chromosomal origin. J. Biol. Chem. 1998, 273, 34255–34262. [Google Scholar]
- Wickner, S.; Hurwitz, J. Interaction of Escherichia coli dnaB and dnaC(D) gene products in vitro. Proc. Natl. Acad. Sci. U.S.A. 1975, 72, 921–925. [Google Scholar] [CrossRef]
- Lu, Y.B.; Ratnakar, P.; Mohanty, B.K.; Bastia, D. Direct physical interaction between DnaG primase and DnaB helicase of Escherichia coli is necessary for optimal synthesis of primer RNA. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 12902–12907. [Google Scholar] [CrossRef]
- Biswas, E.E.; Chen, P.H.; Biswas, S.B. Modulation of enzymatic activities of Escherichia coli dnaB helicase by single-stranded DNA-binding proteins. Nucleic Acids Res. 2002, 30, 2809–2816. [Google Scholar] [CrossRef]
- Gao, D.X.; McHenry, C.S. Tau binds and organizes Escherichia coli replication proteins through distinct domains - domain IV, located within the unique C terminus of Tau, binds the replication fork helicase, DnaB. J. Biol. Chem. 2001, 276, 4441–4446. [Google Scholar] [CrossRef]
- McKinney, M.D.; Wechsler, J.A. RNA polymerase interaction with DnaB protein and lambda P protein during lambda replication. J. Virol. 1983, 48, 551–554. [Google Scholar]
- Maiti, S.; Das, B.; Mandal, N.C. Isolation and preliminary characterization of Escherichia coli mutants resistant to lethal action of the bacteriophage lambda P gene. Virology 1991, 182, 351–352. [Google Scholar] [CrossRef]
- del Solar, G.; Giraldo, R.; Ruiz-Echevarria, M.J.; Espinosa, M.; Diaz-Orejas, R. Replication and control of circular bacterial plasmids. Microbio. Mol. Biol. Rev. 1998, 62, 434–464. [Google Scholar]
- Tomizawa, J.; Sakakibara, Y.; Kakefuda, T. Replication of colicin E1 plasmid DNA in cell extracts. Origin and direction of replication. Proc. Natl. Acad. Sci. U.S.A. 1974, 71, 2260–2264. [Google Scholar] [CrossRef]
- Tomizawa, J.I.; Sakakibara, Y.; Kakefuda, T. Replication of colicin E1 plasmid DNA added to cell extracts. Proc. Natl. Acad. Sci. U.S.A. 1975, 72, 1050–1054. [Google Scholar]
- Dasgupta, S.; Masukata, H.; Tomizawa, J. Multiple mechanisms for initiation of ColE1 DNA replication: DNA synthesis in the presence and absence of ribonuclease H. Cell 1987, 51, 1113–1122. [Google Scholar]
- Kogoma, T. Absence of Rnase H allows replication of pBR322 in Escherichia coli mutants lacking DNA polymerase I. Proc. Natl. Acad. Sci. U.S.A. 1984, 81, 7845–7849. [Google Scholar] [CrossRef]
- Naito, S.; Uchida, H. Rnase H and replication of ColE1 DNA in Escherichia coli. J. Bacteriol. 1986, 166, 143–147. [Google Scholar]
- Tomizawa, J. Two distinct mechanisms of synthesis of DNA fragments on colicin E1 plasmid DNA. Nature 1975, 257, 253–254. [Google Scholar]
- Itoh, T.; Tomizawa, J. Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 2450–2454. [Google Scholar]
- Masukata, H.; Tomizawa, J. A mechanism of formation of a persistent hybrid between elongating RNA and template DNA. Cell 1990, 62, 331–338. [Google Scholar]
- Hillenbrand, G.; Staudenbauer, W.L. Discriminatory function of ribonuclease H in the selective initiation of plasmid DNA replication. Nucleic Acids Res. 1982, 10, 833–853. [Google Scholar]
- Bird, R.E.; Tomizawa, J. Ribonucleotide-deoxyribonucleotide linkages at the origin of DNA replication of colicin E1 plasmid. J. Mol. Biol. 1978, 120, 137–143. [Google Scholar]
- Tomizawa, J.I.; Ohmori, H.; Bird, R.E. Origin of replication of colicin E1 plasmid DNA. Proc. Natl. Acad. Sci. U.S.A. 1977, 74, 1865–1869. [Google Scholar]
- Masai, H.; Nomura, N.; Kubota, Y.; Arai, K. Roles of phi x174 type primosome- and G4 type primase-dependent primings in initiation of lagging and leading strand syntheses of DNA replication. J. Biol. Chem. 1990, 265, 15124–15133. [Google Scholar]
- Allen, J.M.; Simcha, D.M.; Ericson, N.G.; Alexander, D.L.; Marquette, J.T.; Van Biber, B.P.; Troll, C.J.; Karchin, R.; Bielas, J.H.; Loeb, L.A.; et al. Roles of DNA polymerase I in leading and lagging-strand replication defined by a high-resolution mutation footprint of ColE1 plasmid replication. Nucleic Acids Res. 2011, 39, 7020–7033. [Google Scholar] [CrossRef]
- Masai, H.; Arai, K. Dnaa- and pria-dependent primosomes: Two distinct replication complexes for replication of Escherichia coli chromosome. Front. Bios. 1996, 1, d48–d58. [Google Scholar]
- Nomura, N.; Ray, D.S. Expression of a DNA strand initiation sequence of ColE1 plasmid in a single-stranded DNA phage. Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 6566–6570. [Google Scholar]
- Minden, J.S.; Marians, K.J. Replication of pBR322 DNA in vitro with purified proteins. Requirement for topoisomerase I in the maintenance of template specificity. J. Biol. Chem. 1985, 260, 9316–9325. [Google Scholar]
- Seufert, W.; Dobrinski, B.; Lurz, R.; Messer, W. Functionality of the dnaA protein binding site in DNA replication is orientation-dependent. J. Biol. Chem. 1988, 263, 2719–2723. [Google Scholar]
- Seufert, W.; Messer, W. DnaA protein binding to the plasmid origin region can substitute for primosome assembly during replication of pBR322 in vitro. Cell 1987, 48, 73–78. [Google Scholar] [CrossRef]
- McCool, J.D.; Sandler, S.J. Effects of mutations involving cell division, recombination, and chromosome dimer resolution on a priA2::Kan mutant. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 8203–8210. [Google Scholar] [CrossRef]
- Bramhill, D.; Kornberg, A. Duplex opening by DnaA protein at novel sequences in initiation of replication at the origin of the E. coli chromosome. Cell 1988, 52, 743–755. [Google Scholar] [CrossRef]
- Marians, K.J. Pria-directed replication fork restart in Escherichia coli. Trends Biochem. Sci. 2000, 25, 185–189. [Google Scholar] [CrossRef]
- Zeghouf, M.; Li, J.; Butland, G.; Borkowska, A.; Canadien, V.; Richards, D.; Beattie, B.; Emili, A.; Greenblatt, J.F. Sequential peptide affinity (SPA) system for the identification of mammalian and bacterial protein complexes. J. Proteome. Res. 2004, 3, 463–468. [Google Scholar]
- Reiser, W.; Leibrecht, I.; Klein, A. Structure and function of mutants in the P gene of bacteriophage lambda leading to the pi phenotype. Mol. Gen. Gen. 1983, 192, 430–435. [Google Scholar] [CrossRef]
- Yates, J.L.; Gette, W.R.; Furth, M.E.; Nomura, M. Effects of ribosomal mutations on the read-through of a chain termination signal: Studies on the synthesis of bacteriophage lambda O gene protein in vitro. Proc. Natl. Acad. Sci. U.S.A. 1977, 74, 689–693. [Google Scholar] [CrossRef]
- Tsurimoto, T.; Hase, T.; Matsubara, H.; Matsubara, K. Bacteriophage lambda initiators: Preparation from a strain that overproduces the O and P proteins. Mol. Gen. Gen. 1982, 187, 79–86. [Google Scholar]
- Krinke, L.; Wulff, D.L. Rnase iii-dependent hydrolysis of lambda cII-O gene mRNA mediated by lambda OOP antisense RNA. Genes Dev. 1990, 4, 2223–2233. [Google Scholar] [CrossRef]
- Krinke, L.; Wulff, D.L. The cleavage specificity of Rnase III. Nucleic Acids Res. 1990, 18, 4809–4815. [Google Scholar]
- Hayes, S.; Hayes, C. Spontaneous lambda oR mutations suppress inhibition of bacteriophage growth by nonimmune exclusion phenotype of defective lambda prophage. J. Virol. 1986, 58, 835–842. [Google Scholar]
- Miwa, T.; Akaboshi, E.; Matsubara, K. Instability of bacteriophage lambda initiator O and P proteins in DNA replication. J. Biochem. 1983, 94, 331–338. [Google Scholar]
- Wyatt, W.M.; Inokuchi, H. Stability of lambda O and P replication functions. Virology 1974, 58, 313–315. [Google Scholar]
- Hayes, S. Mutations suppressing loss of replication control. Genetic analysis of bacteriophage lambda-dependent replicative killing, replication initiation, and mechanisms of mutagenesis. In DNA replication and mutagenesis; Moses, R.E., Summers, W.C., Eds.; American Society for Microbiology: Washington, D.C., 1988; pp. 367–377. [Google Scholar]
- Hayes, S.; Duncan, D.; Hayes, C. Alcohol treatment of defective lambda lysogens is deletionogenic. Mol. Gen. Gen. 1990, 222, 17–24. [Google Scholar]
- Hayes, S.; Hayes, C.; Bull, H.J.; Pelcher, L.A.; Slavcev, R.A. Acquired mutations in phage lambda genes O or P that enable constitutive expression of a cryptic lambda-N+cI[Ts]cro- prophage in E. coli cells shifted from 30 degreesC to 42 degreesC, accompanied by loss of immlambda and Rex+ phenotypes and emergence of a non-immune exclusion-state. Gene 1998, 223, 115–128. [Google Scholar]
- Kaplan, D.L.; O'Donnell, M. DnaB drives DNA branch migration and dislodges proteins while encircling two DNA strands. Mol. Cell 2002, 10, 647–657. [Google Scholar]
- Horbay, M.A.; McCrea, R.P.E.; Hayes, S. OOP RNA: A regulatory pivot in temperate lambdoid phage development. In Modern bacteriophage biology and biotechnology; Wegrzyn, G., Ed.; Research Signpost: Kerala, India, 2006; pp. 37–57. [Google Scholar]
- Walker, G.C. Mutagenesis and inducible responses to deoxyribonucleic acid damage in Escherichia coli. Microbiol. Rev. 1984, 48, 60–93. [Google Scholar]
- Horii, T.; Ogawa, T.; Nakatani, T.; Hase, T.; Matsubara, H.; Ogawa, H. Regulation of SOS functions: Purification of E. coli LexA protein and determination of its specific site cleaved by the RecA protein. Cell 1981, 27, 515–522. [Google Scholar] [CrossRef]
- Markham, B.E.; Little, J.W.; Mount, D.W. Nucleotide sequence of the lexA gene of Escherichia coli K-12. Nucleic Acids Res. 1981, 9, 4149–4161. [Google Scholar] [CrossRef]
- Gottesman, S.; Halpern, E.; Trisler, P. Role of SulA and SulB in filamentation by lon mutants of Escherichia coli K-12. J. Bacteriol. 1981, 148, 265–273. [Google Scholar]
- Schoemaker, J.M.; Gayda, R.C.; Markovitz, A. Regulation of cell division in Escherichia coli: SOS induction and cellular location of the SulA protein, a key to Lon-associated filamentation and death. J. Bacteriol. 1984, 158, 551–561. [Google Scholar]
- Hill, T.M.; Sharma, B.; Valjavec-Gratian, M.; Smith, J. Sfi-independent filamentation in Escherichia coli is LexA dependent and requires DNA damage for induction. J. Bacteriol. 1997, 179, 1931–1939. [Google Scholar]
- Hirota, Y.; Jacob, F.; Ryter, A.; Buttin, G.; Nakai, T. On the process of cellular division in Escherichia coli. I. Asymmetrical cell division and production of deoxyribonucleic acid-less bacteria. J. Mol. Biol. 1968, 35, 175–192. [Google Scholar] [CrossRef]
- Mulder, E.; Woldringh, C.L. Actively replicating nucleoids influence positioning of division sites in Escherichia coli filaments forming cells lacking DNA. J. Bacteriol. 1989, 171, 4303–4314. [Google Scholar]
- Bukau, B.; Walker, G.C. Delta dnaK52 mutants of Escherichia coli have defects in chromosome segregation and plasmid maintenance at normal growth temperatures. J. Bacteriol. 1989, 171, 6030–6038. [Google Scholar]
- Baharoglu, Z.; Lestini, R.; Duigou, S.; Michel, B. RNA polymerase mutations that facilitate replication progression in the rep uvrD recF mutant lacking two accessory replicative helicases. Mol. Microbiol. 2010, 77, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Trautinger, B.W.; Lloyd, R.G. Modulation of DNA repair by mutations flanking the DNA channel through RNA polymerase. EMBO J. 2002, 21, 6944–6953. [Google Scholar]
- Bachmann, B.J. Derivations and genotypes of some mutant derivatives of Escherichia coli K-12. In Escherichia coli and Salmonella typhimurium: Cellular and molecular biology; Neidhardt, F.C., Ingraham, J.I., Low, K.B., Magasanik, B., Schaechter, M., Umbargr, H.E., Eds.; American Society for Microbiology: Washington, D.C., 1987; Vol. 2, pp. 1192–1219. [Google Scholar]
- Harris, R.S.; Longerich, S.; Rosenberg, S.M. Recombination in adaptive mutation. Science 1994, 264, 258–260. [Google Scholar]
- Slavcev, R.A.; Hayes, S. Blocking the T4 lysis inhibition phenotype. Gene 2003, 321, 163–171. [Google Scholar]
- Hayes, S.; Hayes, C. Control of lambda repressor prophage and establishment transcription by the product of gene tof. Mol. Gen. Gen. 1978, 164, 63–76. [Google Scholar] [CrossRef]
- Hayes, S. Control of the initiation of lambda replication, oop, lit and repressor establishment RNA synthesis. In DNA synthesis, present and future; Molineux, I., Kohiyama, M., Eds.; Plenum Press: New York, 1978; pp. 127–142. [Google Scholar]
- Grosschedl, R.; Schwarz, E. Nucleotide sequence of the cro-cII-oop region of bacteriophage 434 DNA. Nucleic Acids Res. 1979, 6, 867–881. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hayes, S.; Erker, C.; Horbay, M.A.; Marciniuk, K.; Wang, W.; Hayes, C. Phage Lambda P Protein: Trans-Activation, Inhibition Phenotypes and their Suppression. Viruses 2013, 5, 619-653. https://doi.org/10.3390/v5020619
Hayes S, Erker C, Horbay MA, Marciniuk K, Wang W, Hayes C. Phage Lambda P Protein: Trans-Activation, Inhibition Phenotypes and their Suppression. Viruses. 2013; 5(2):619-653. https://doi.org/10.3390/v5020619
Chicago/Turabian StyleHayes, Sidney, Craig Erker, Monique A. Horbay, Kristen Marciniuk, Wen Wang, and Connie Hayes. 2013. "Phage Lambda P Protein: Trans-Activation, Inhibition Phenotypes and their Suppression" Viruses 5, no. 2: 619-653. https://doi.org/10.3390/v5020619
APA StyleHayes, S., Erker, C., Horbay, M. A., Marciniuk, K., Wang, W., & Hayes, C. (2013). Phage Lambda P Protein: Trans-Activation, Inhibition Phenotypes and their Suppression. Viruses, 5(2), 619-653. https://doi.org/10.3390/v5020619