Foamy Virus Budding and Release


Abstract
:1. Introduction
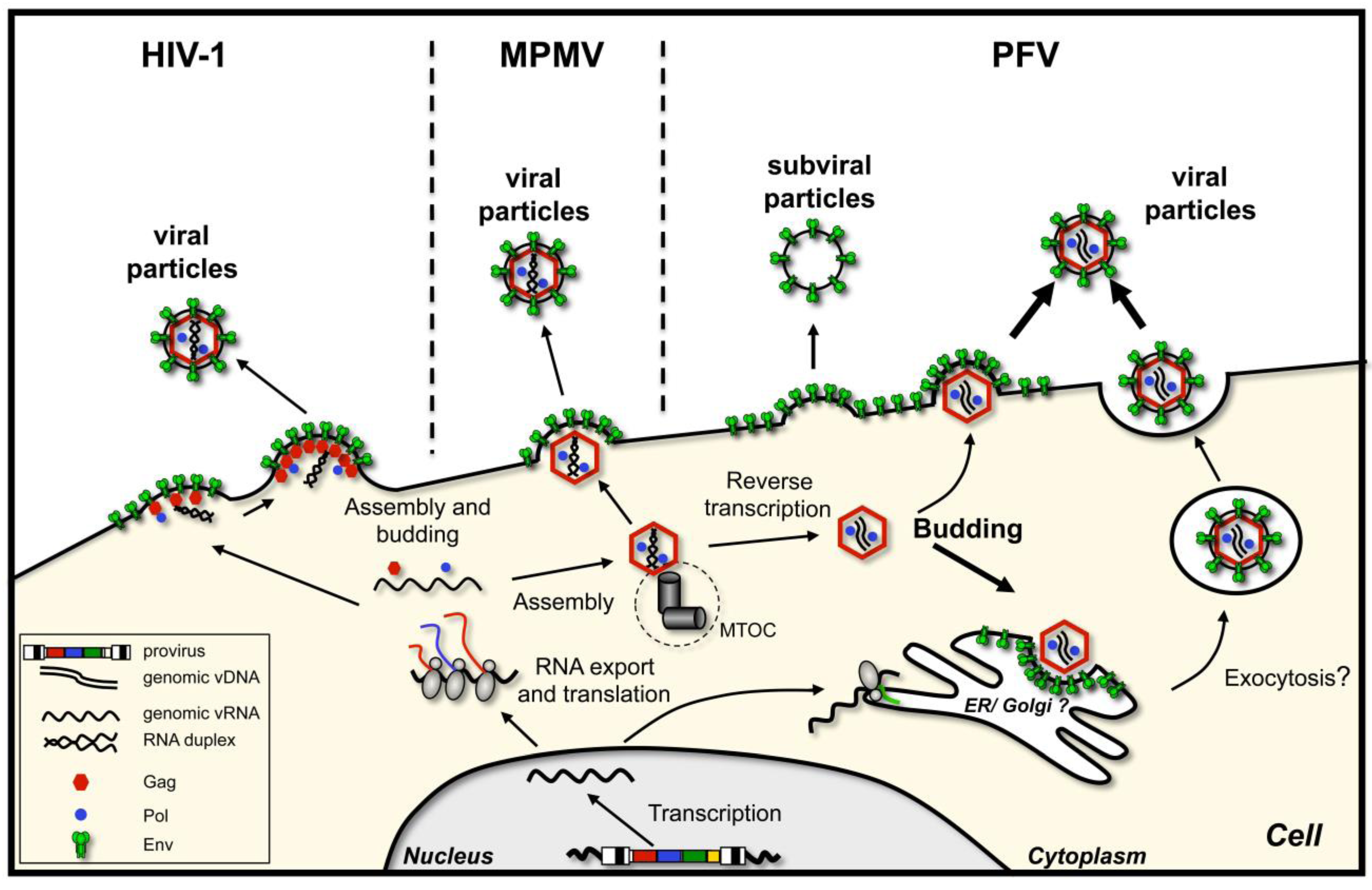
- Particle assembly occurs at the plasma membrane, in regions enriched in envelope glycoprotein. Oligomerization of capsid protein leads to formation of the particle, its growth and bending of the plasma membrane, after which the assembled virion pinches off. This is characteristic of C-type retroviruses, such as human immunodeficiency virus type 1 (HIV-1) [3] or murine leukemia virus (MuLV) [4]. As mentioned, here capsid assembly and virion budding occur simultaneously. However, importantly, neither the presence of the glycoprotein nor its interaction with the capsid is a prerequisite for virus release, although they might enhance particle release.
- Immature capsids with encapsidated RNA genome assemble first in the cytoplasm. Then they travel to the budding site, in some cases assisted by the viral glycoprotein. The budding site can be either plasma membrane or an intracellular compartment (such as endoplasmic reticulum (ER) or Golgi), where the envelope protein is localized. This mechanism characterizes B/D-type retroviruses such as Mason-Pfizer monkey virus (MPMV) and mouse mammary tumor virus (MMTV) [5,6,7].
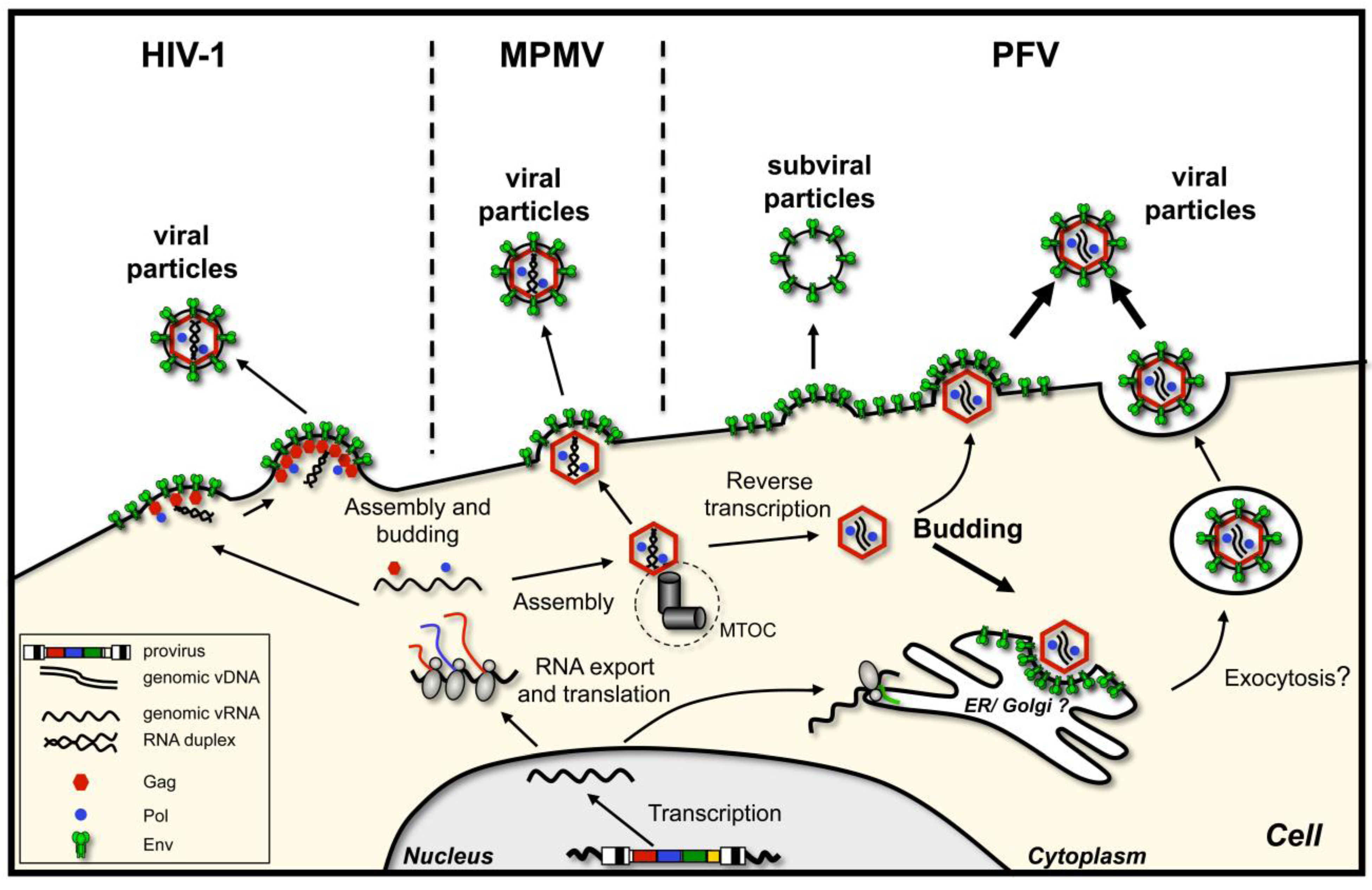
2. Foamy Virus Budding, an Overview

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus species | Retroviroviridae | Hepadnaviridae | ||
|---|---|---|---|---|
| Human Immunodeficiency Virus Type 1 (HIV-1) | Rous Sarcoma Virus (RSV) | Prototype Foamy Virus (PFV) | Hepatitis B virus (HBV) | |
| Glycoprotein organization | Polyprotein precursor (gp160): Surface glycoprotein (gp120SU) Transmembrane glycoprotein (gp41TM) | Polyprotein precursor (gp95): Surface glycoprotein (gp85SU) Transmembrane glycoprotein (gp37TM) | Polyprotein precursor (gp130): Surface glycoprotein (gp80 SU) Transmembrane glycoprotein (gp48 TM) Leader peptide (gp18LP) | Hepatitis B surface antigen (HBsAg): Small (S) protein (226aa) Middle (M) protein (226aa + preS2 domain 55aa) Large (L) protein (226aa + preS2 domain 55aa + preS1 domain 108aa or 119aa) |
| Capsid organization | Gag precursor (Pr55Gag): Matrix (p17MA); Capsid (p24CA); Spacer p2 Nucleocapsid (p7NC); Spacer p1; p6 domain | Gag-PR precursor (Pr76Gag-PR): Matrix (p17MA); p2a, p2b; pp10; Capsid (p27CA); Spacer; Nucleocapsid (p12NC); Protease (p15PR) | Gag precursor (p71Gag): p68Gag; p3Gag | Hepatitis B core protein (p22HBc): 183-185aa |
| Capsid: membrane targeting domain | Present in MA subunit | Present in MA subunit | No | Not known |
| Budding type | Type C morphogenesis: Assembly takes place at the plasma membrane; particle release from plasma membrane or plasma membrane-derived membranes | Type C morphogenesis: Assembly takes placeat the plasma membrane; particle release from plasma membrane or plasma membrane-derived membranes | Type B/D morphogenesis: Capsid preassembly at the MTOC in the cytoplasm; budding from intracellular membranes (ER/Golgi) and plasma membrane | Nucleocapsid-formation in cytosol; Budding from intracellular membranes (ER) |
| ESCRT-dependent budding process | Yes ESCRT I, III AIP1/Alix Vps4A/B | Yes ESCRT II, III AIP1/Alix Vps4A/B | Yes ESCRT I, III Vps4A/B | Yes ESCRT II, III Vps4A/B |
| L domain | Gag (p6): PTAP; YPXL | Gag (p2b): PPPY; LYPSL | Gag (p71, p68): PSAP | Core: PPAY; K96? |
| ESCRT interaction partner | Tsg101; AIP1/Alix; | AIP1/Alix; (Nedd4) | Tsg101 | (Nedd4); (γ2-adaptin) |
| Virus like particles | Yes | Yes | No | No, but release of naked capsids |
| Subviral particles | No | No | Yes, low amounts | Yes, high amounts |
| Budding requires | Capsid (Gag) protein only | Capsid (Gag) protein only | Capsid (Gag) and Envelope protein (Env) necessary | Capsid (Core) and Envelope protein (L and S) necessary vDNA synthesis |
| Place of interaction (Capsid-Envelope) | plasma membrane | plasma membrane | trans-Golgi network | ER |
| Pseudotyping | yes | yes | yes, but only with a artificial heterodimerizer system | no |
3. Glycoprotein-Dependent Particle Release
3.1. FV Capsid- and Glycoprotein Biosynthesis
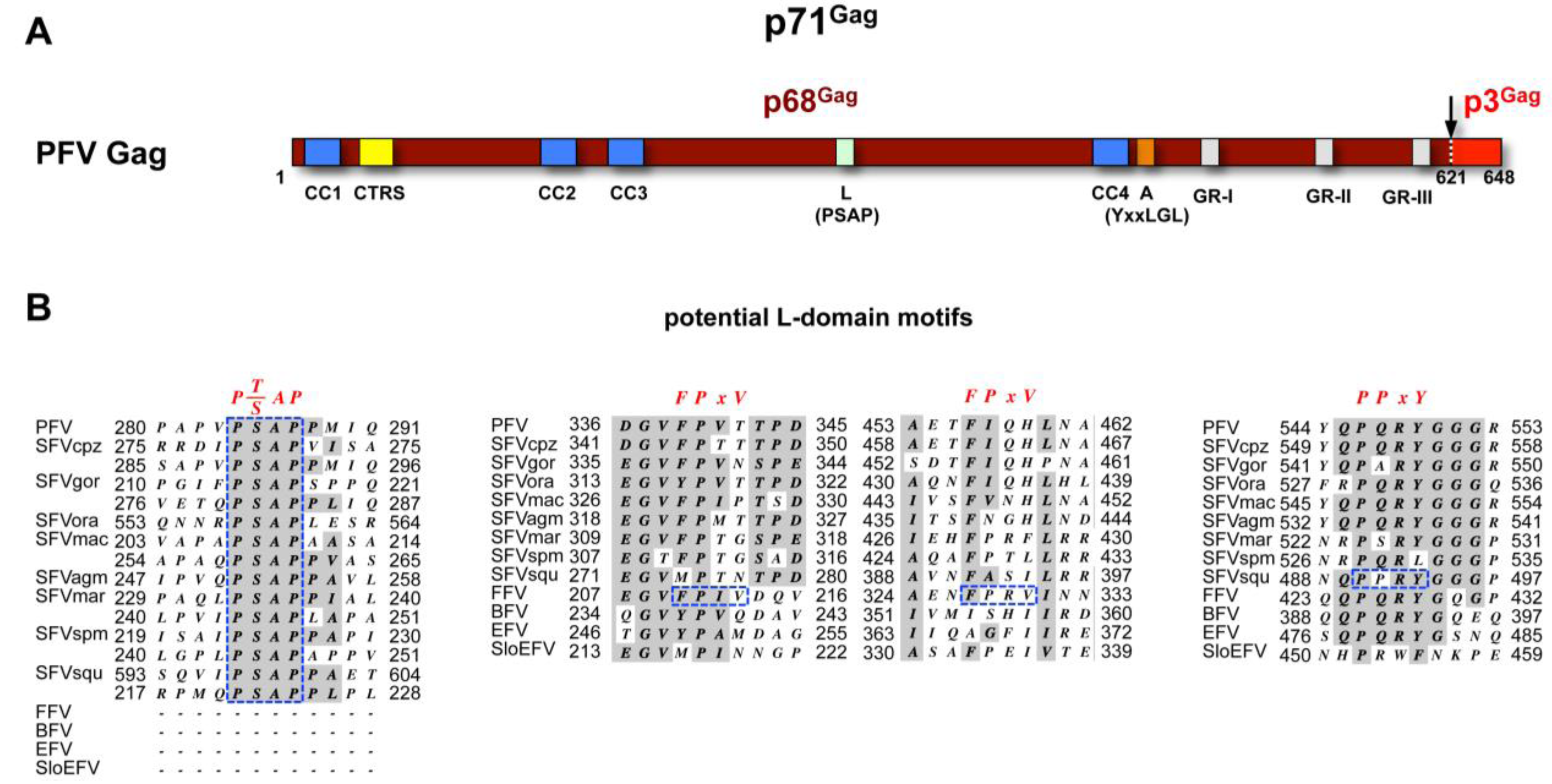
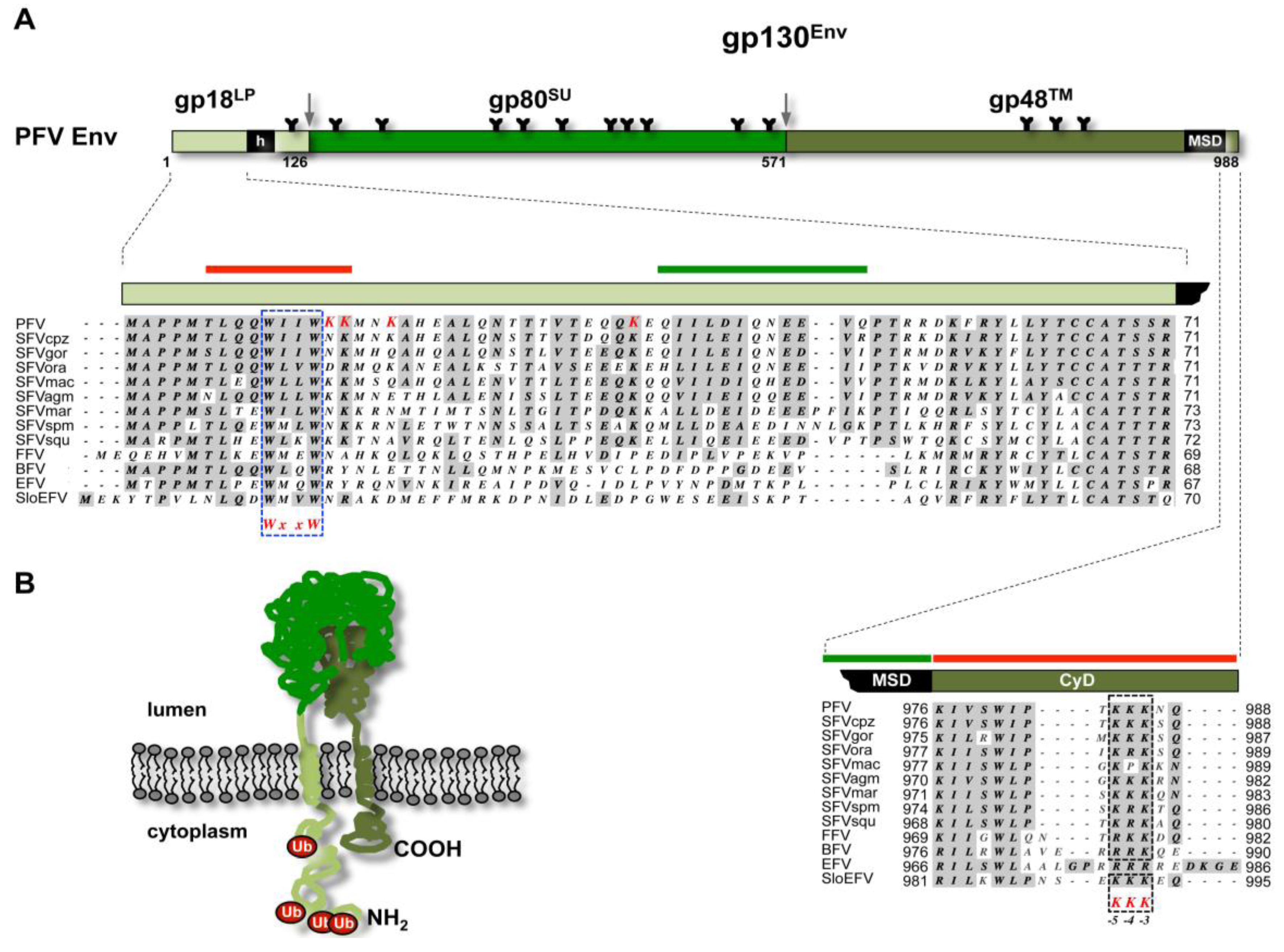
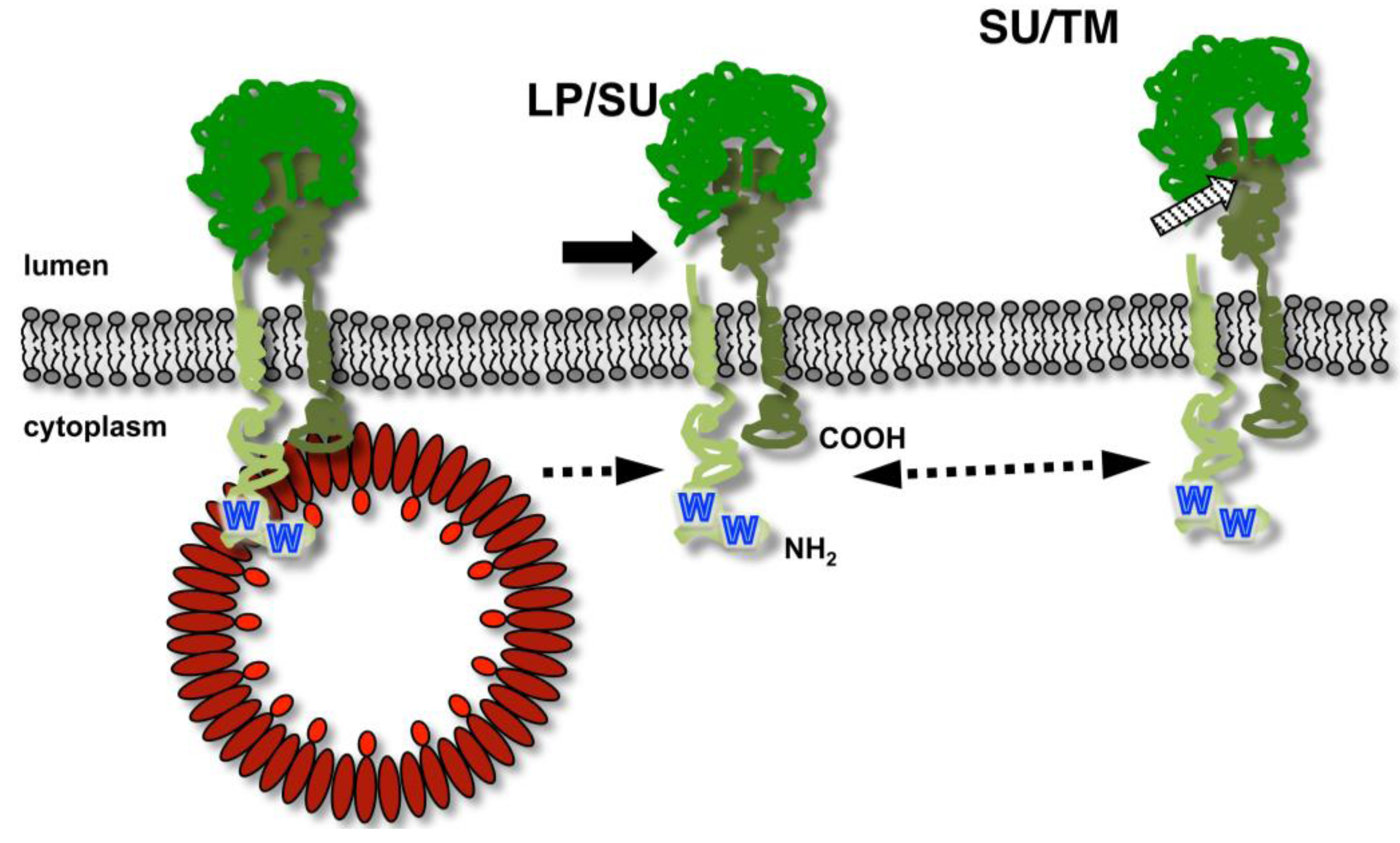
3.2. Subcellular Localization of FV Budding
3.3. Details of FV Gag-Env Interaction

4. Cellular Factors Involved in FV Budding
4.1. The ESCRT Machinery
4.2. The Ubiquitination Machinery
5. Alternative and Artificial Budding of FV Particle Structures
5.2. Glycoprotein-Independent Capsid Membrane Targeting
5.3. Pseudotyping
6. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Martin-Serrano, J.; Neil, S.J. Host factors involved in retroviral budding and release. Nat. Rev. Microbiol. 2011, 9, 519–531. [Google Scholar]
- Welsch, S.; Muller, B.; Krausslich, H.G. More than one door—Budding of enveloped viruses through cellular membranes. FEBS Lett. 2007, 581, 2089–2097. [Google Scholar] [CrossRef]
- Weiss, E.R.; Gottlinger, H. The role of cellular factors in promoting HIV budding. J. Mol. Biol. 2011, 410, 525–533. [Google Scholar]
- Suomalainen, M.; Hultenby, K.; Garoff, H. Targeting of Moloney murine leukemia virus gag precursor to the site of virus budding. J. Cell Biol. 1996, 135, 1841–1852. [Google Scholar] [CrossRef]
- Choi, G.; Park, S.; Choi, B.; Hong, S.; Lee, J.; Hunter, E.; Rhee, S.S. Identification of a cytoplasmic targeting/retention signal in a retroviral Gag polyprotein. J. Virol. 1999, 73, 5431–5437. [Google Scholar]
- Sfakianos, J.N.; LaCasse, R.A.; Hunter, E. The M-PMV cytoplasmic targeting-retention signal directs nascent Gag polypeptides to a pericentriolar region of the cell. Traffic 2003, 4, 660–670. [Google Scholar]
- Stansell, E.; Apkarian, R.; Haubova, S.; Diehl, W.E.; Tytler, E.M.; Hunter, E. Basic residues in the Mason-Pfizer monkey virus gag matrix domain regulate intracellular trafficking and capsid-membrane interactions. J. Virol. 2007, 81, 8977–8988. [Google Scholar]
- Patient, R.; Hourioux, C.; Roingeard, P. Morphogenesis of hepatitis B virus and its subviral envelope particles. Cell. Microbiol. 2009, 11, 1561–1570. [Google Scholar] [CrossRef] [Green Version]
- Bruss, V. Hepatitis B virus morphogenesis. World J. Gastroenterol. 2007, 13, 65–73. [Google Scholar]
- Linial, M.L.; Fan, H.; Hahn, B.; Löwer, R.; Neil, J.; Quackenbush, S.; Rethwilm, A.; Sonigo, P.; Stoye, J.P.; Tristem, M. Retroviridae. In Virus Taxonomy; Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., Eds.; Elsevier Academic Press: London, UK, 2005; pp. 421–440. [Google Scholar]
- Rethwilm, A. The replication strategy of foamy viruses. Curr. Top. Microbiol. Immunol. 2003, 277, 1–26. [Google Scholar] [CrossRef]
- Pietschmann, T.; Heinkelein, M.; Heldmann, M.; Zentgraf, H.; Rethwilm, A.; Lindemann, D. Foamy virus capsids require the cognate envelope protein for particle export. J. Virol. 1999, 73, 2613–2621. [Google Scholar]
- Fischer, N.; Heinkelein, M.; Lindemann, D.; Enssle, J.; Baum, C.; Werder, E.; Zentgraf, H.; Müller, J.G.; Rethwilm, A. Foamy virus particle formation. J. Virol. 1998, 72, 1610–1615. [Google Scholar]
- Baldwin, D.N.; Linial, M.L. The roles of Pol and Env in the assembly pathway of human foamy virus. J. Virol. 1998, 72, 3658–3665. [Google Scholar]
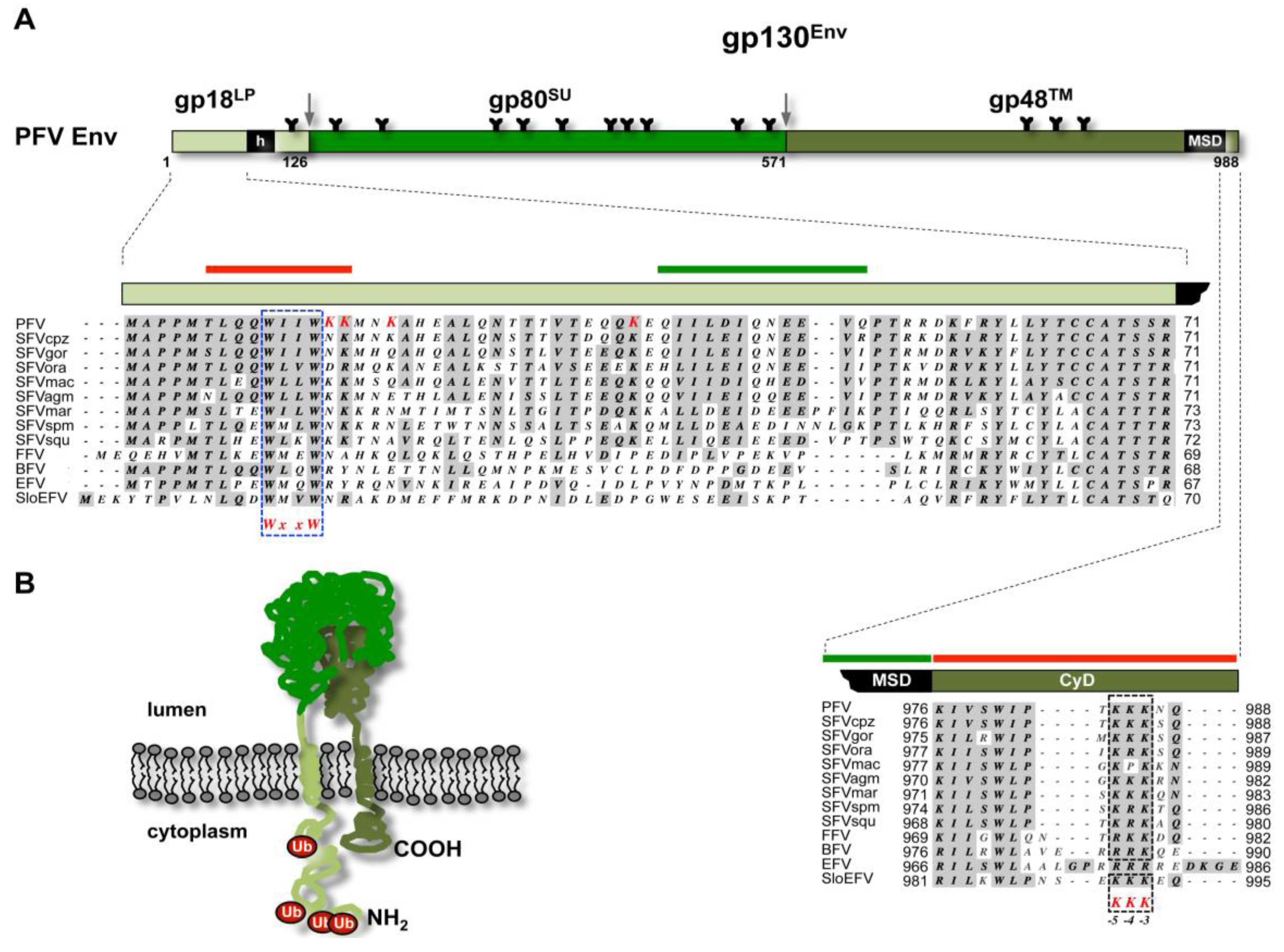
- Lindemann, D.; Pietschmann, T.; Picard-Maureau, M.; Berg, A.; Heinkelein, M.; Thurow, J.; Knaus, P.; Zentgraf, H.; Rethwilm, A. A particle-associated glycoprotein signal peptide essential for virus maturation and infectivity. J. Virol. 2001, 75, 5762–5771. [Google Scholar] [CrossRef]
- Pietschmann, T.; Zentgraf, H.; Rethwilm, A.; Lindemann, D. An evolutionarily conserved positively charged amino acid in the putative membrane-spanning domain of the foamy virus envelope protein controls fusion activity. J. Virol. 2000, 74, 4474–4482. [Google Scholar] [CrossRef]
- Flügel, R.M.; Pfrepper, K.I. Proteolytic processing of foamy virus Gag and Pol proteins. Curr. Top. Microbiol. Immunol. 2003, 277, 63–88. [Google Scholar]
- Cartellieri, M.; Rudolph, W.; Herchenröder, O.; Lindemann, D.; Rethwilm, A. Determination of the relative amounts of Gag and Pol proteins in foamy virus particles. Retrovirology 2005, 2, e44. [Google Scholar]
- Morozov, V.A.; Copeland, T.D.; Nagashima, K.; Gonda, M.A.; Oroszlan, S. Protein composition and morphology of human foamy virus intracellular cores and extracellular particles. Virology 1997, 228, 307–317. [Google Scholar] [CrossRef]
- Hahn, H.; Baunach, G.; Bräutigam, S.; Mergia, A.; Neumann-Haefelin, D.; Daniel, M.D.; McClure, M.O.; Rethwilm, A. Reactivity of primate sera to foamy virus Gag and Bet proteins. J. Gen. Virol. 1994, 75, 2635–2644. [Google Scholar] [CrossRef]
- Netzer, K.O.; Rethwilm, A.; Maurer, B.; ter Meulen, V. Identification of the major immunogenic structural proteins of human foamy virus. J. Gen. Virol. 1990, 71, 1237–1241. [Google Scholar] [CrossRef]
- Enssle, J.; Fischer, N.; Moebes, A.; Mauer, B.; Smola, U.; Rethwilm, A. Carboxy-terminal cleavage of the human foamy virus Gag precursor molecule is an essential step in the viral life cycle. J. Virol. 1997, 71, 7312–7317. [Google Scholar]
- Katzourakis, A.; Gifford, R.J.; Tristem, M.; Gilbert, M.T.; Pybus, O.G. Macroevolution of complex retroviruses. Science 2009, 325, 1512. [Google Scholar] [CrossRef]
- Geiselhart, V.; Schwantes, A.; Bastone, P.; Frech, M.; Löchelt, M. Features of the Env leader protein and the N-terminal Gag domain of feline foamy virus important for virus morphogenesis. Virology 2003, 310, 235–244. [Google Scholar] [CrossRef]
- Wilk, T.; Geiselhart, V.; Frech, M.; Fuller, S.D.; Flügel, R.M.; Löchelt, M. Specific interaction of a novel foamy virus env leader protein with the N-terminal gag domain. J. Virol. 2001, 75, 7995–8007. [Google Scholar] [CrossRef]
- Geiselhart, V.; Bastone, P.; Kempf, T.; Schnolzer, M.; Löchelt, M. Furin-mediated cleavage of the feline foamy virus Env leader protein. J. Virol. 2004, 78, 13573–13581. [Google Scholar]
- Duda, A.; Stange, A.; Luftenegger, D.; Stanke, N.; Westphal, D.; Pietschmann, T.; Eastman, S.W.; Linial, M.L.; Rethwilm, A.; Lindemann, D. Prototype foamy virus envelope glycoprotein leader peptide processing is mediated by a furin-like cellular protease, but cleavage is not essential for viral infectivity. J. Virol. 2004, 78, 13865–13870. [Google Scholar] [CrossRef]
- Sun, Y.; Wen, D.D.; Liu, Q.M.; Yi, X.F.; Wang, T.T.; Wei, L.L.; Li, Z.; Liu, W.H.; He, X.H. Comparative analysis of the envelope glycoproteins of foamy viruses. Acta Virol. 2012, 56, 283–291. [Google Scholar] [CrossRef]
- Bansal, A.; Shaw, K.L.; Edwards, B.H.; Goepfert, P.A.; Mulligan, M.J. Characterization of the R572T point mutant of a putative cleavage site in human foamy virus Env. J. Virol. 2000, 74, 2949–2954. [Google Scholar] [CrossRef]
- Li, Y.; Luo, L.; Thomas, D.Y.; Kang, C.Y. Control of expression, glycosylation, and secretion of HIV-1 gp120 by homologous and heterologous signal sequences. Virology 1994, 204, 266–278. [Google Scholar] [CrossRef]
- Sommerfelt, M.A.; Petteway, S.R., Jr.; Dreyer, G.B.; Hunter, E. Effect of retroviral proteinase inhibitors on Mason-Pfizer monkey virus maturation and transmembrane glycoprotein cleavage. J. Virol. 1992, 66, 4220–4227. [Google Scholar]
- Green, N.; Shinnick, T.M.; Witte, O.; Ponticelli, A.; Sutcliffe, J.G.; Lerner, R.A. Sequence-specific antibodies show that maturation of Moloney leukemia virus envelope polyprotein involves removal of a COOH-terminal peptide. Proc. Natl. Acad. Sci. USA 1981, 78, 6023–6027. [Google Scholar]
- Lüftenegger, D.; Picard-Maureau, M.; Stanke, N.; Rethwilm, A.; Lindemann, D. Analysis and function of prototype foamy virus envelope N glycosylation. J. Virol. 2005, 79, 7664–7672. [Google Scholar] [CrossRef]
- Pinter, A.; Honnen, W.J. O-linked glycosylation of retroviral envelope gene products. J. Virol. 1988, 62, 1016–1021. [Google Scholar]
- Stansell, E.; Canis, K.; Haslam, S.M.; Dell, A.; Desrosiers, R.C. Simian immunodeficiency virus from the sooty mangabey and rhesus macaque is modified with O-linked carbohydrate. J. Virol. 2011, 85, 582–595. [Google Scholar] [CrossRef]
- Voss, M.; Fukumori, A.; Kuhn, P.H.; Kunzel, U.; Klier, B.; Grammer, G.; Haug-Kroper, M.; Kremmer, E.; Lichtenthaler, S.F.; Steiner, H.; et al. Foamy Virus Envelope Protein Is a Substrate for Signal Peptide Peptidase-like 3 (SPPL3). J. Biol. Chem. 2012, 287, 43401–43409. [Google Scholar] [CrossRef]
- Byun, H.; Halani, N.; Mertz, J.A.; Ali, A.F.; Lozano, M.M.; Dudley, J.P. Retroviral Rem protein requires processing by signal peptidase and retrotranslocation for nuclear function. Proc. Natl. Acad. Sci. USA 2010, 107, 12287–12292. [Google Scholar]
- Dultz, E.; Hildenbeutel, M.; Martoglio, B.; Hochman, J.; Dobberstein, B.; Kapp, K. The signal peptide of the mouse mammary tumor virus Rem protein is released from the endoplasmic reticulum membrane and accumulates in nucleoli. J. Biol. Chem. 2008, 283, 9966–7996. [Google Scholar] [CrossRef]
- Yu, S.F.; Eastman, S.W.; Linial, M.L. Foamy virus capsid assembly occurs at a pericentriolar region through a cytoplasmic targeting/retention signal in Gag. Traffic 2006, 7, 966–977. [Google Scholar] [CrossRef]
- Eastman, S.W.; Linial, M.L. Identification of a conserved residue of foamy virus Gag required for intracellular capsid assembly. J. Virol. 2001, 75, 6857–6864. [Google Scholar] [CrossRef]
- Tobaly-Tapiero, J.; Bittoun, P.; Neves, M.; Guillemin, M.C.; Lecellier, C.H.; Puvion-Dutilleul, F.; Gicquel, B.; Zientara, S.; Giron, M.L.; de The, H.; et al. Isolation and characterization of an equine foamy virus. J. Virol. 2000, 74, 4064–4073. [Google Scholar] [CrossRef]
- Goepfert, P.A.; Wang, G.; Mulligan, M.J. Identification of an ER retrieval signal in a retroviral glycoprotein. Cell 1995, 82, 543–544. [Google Scholar] [CrossRef]
- Wang, G.; Mulligan, M.J. Comparative sequence analysis and predictions for the envelope glycoproteins of foamy viruses. J. Gen. Virol. 1999, 80, 245–254. [Google Scholar]
- Goepfert, P.A.; Shaw, K.; Wang, G.; Bansal, A.; Edwards, B.H.; Mulligan, M.J. An endoplasmic reticulum retrieval signal partitions human foamy virus maturation to intracytoplasmic membranes. J. Virol. 1999, 73, 7210–7217. [Google Scholar]
- Welsch, S.; Groot, F.; Krausslich, H.G.; Keppler, O.T.; Sattentau, Q.J. Architecture and regulation of the HIV-1 assembly and holding compartment in macrophages. J. Virol. 2011, 85, 7922–7927. [Google Scholar] [CrossRef]
- Jouvenet, N.; Neil, S.J.; Bess, C.; Johnson, M.C.; Virgen, C.A.; Simon, S.M.; Bieniasz, P.D. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006, 4, e435. [Google Scholar] [CrossRef]
- Reh, J.; Stange, A.; Götz, A.; Rönitz, M.; Große, A.; Lindemann, D. An N-terminal putative coiled-coil domain of Prototype Foamy Virus Gag is essential for the specific interaction with the glycoprotein required for particle egress. Submitted for publication.
- Berg, A.; Pietschmann, T.; Rethwilm, A.; Lindemann, D. Determinants of foamy virus envelope glycoprotein mediated resistance to superinfection. Virology 2003, 314, 243–252. [Google Scholar] [CrossRef]
- Liu, Y.; Kim, Y.B.; Lochelt, M. N-terminally myristoylated feline foamy virus Gag allows Env-independent budding of sub-viral particles. Viruses 2011, 3, 2223–2237. [Google Scholar] [CrossRef]
- Stirnnagel, K.; Lüftenegger, D.; Stange, A.; Swiersy, A.; Müllers, E.; Reh, J.; Stanke, N.; Grosse, A.; Chiantia, S.; Keller, H.; et al. Analysis of prototype foamy virus particle-host cell interaction with autofluorescent retroviral particles. Retrovirology 2010, 7, e45. [Google Scholar]
- Life, R.B.; Lee, E.G.; Eastman, S.W.; Linial, M.L. Mutations in the amino terminus of foamy virus Gag disrupt morphology and infectivity but do not target assembly. J. Virol. 2008, 82, 6109–6119. [Google Scholar] [CrossRef]
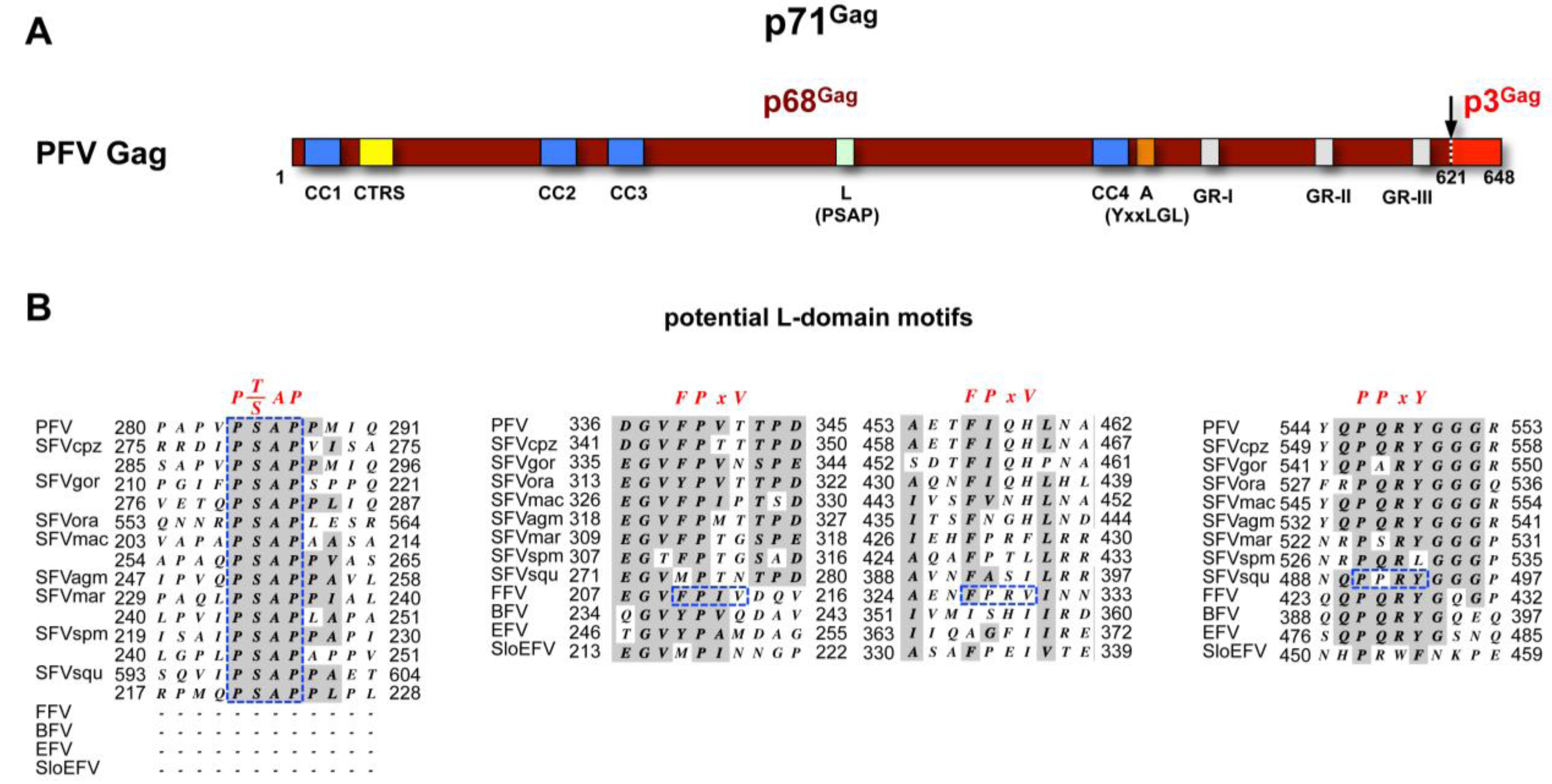
- Stange, A.; Mannigel, I.; Peters, K.; Heinkelein, M.; Stanke, N.; Cartellieri, M.; Göttlinger, H.; Rethwilm, A.; Zentgraf, H.; Lindemann, D. Characterization of prototype foamy virus gag late assembly domain motifs and their role in particle egress and infectivity. J. Virol. 2005, 79, 5466–5476. [Google Scholar] [CrossRef]
- Goff, S.P. Host factors exploited by retroviruses. Nat. Rev. Microbiol. 2007, 5, 253–263. [Google Scholar] [CrossRef]
- Xu, F.; Tan, J.; Liu, R.; Xu, D.; Li, Y.; Geng, Y.; Liang, C.; Qiao, W. Tetherin inhibits prototypic foamy virus release. Virol. J. 2011, 8, e198. [Google Scholar] [CrossRef]
- Jouvenet, N.; Neil, S.J.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009, 83, 1837–1844. [Google Scholar] [CrossRef]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Petit, C.; Giron, M.L.; Tobaly-Tapiero, J.; Bittoun, P.; Real, E.; Jacob, Y.; Tordo, N.; De The, H.; Saib, A. Targeting of incoming retroviral Gag to the centrosome involves a direct interaction with the dynein light chain 8. J. Cell Sci. 2003, 116, 3433–3442. [Google Scholar] [CrossRef]
- Saib, A.; Puvion Dutilleul, F.; Schmid, M.; Peries, J.; de The, H. Nuclear targeting of incoming human foamy virus Gag proteins involves a centriolar step. J. Virol. 1997, 71, 1155–1161. [Google Scholar]
- Zhadina, M.; Bieniasz, P.D. Functional interchangeability of late domains, late domain cofactors and ubiquitin in viral budding. PLoS Pathog. 2010, 6, e1001153. [Google Scholar] [CrossRef]
- Zhadina, M.; McClure, M.O.; Johnson, M.C.; Bieniasz, P.D. Ubiquitin-dependent virus particle budding without viral protein ubiquitination. Proc. Natl. Acad. Sci. USA 2007, 104, 20031–20036. [Google Scholar] [CrossRef]
- Patton, G.S.; Morris, S.A.; Chung, W.; Bieniasz, P.D.; McClure, M.O. Identification of domains in gag important for prototypic foamy virus egress. J. Virol. 2005, 79, 6392–6399. [Google Scholar] [CrossRef]
- Stanke, N.; Stange, A.; Lüftenegger, D.; Zentgraf, H.; Lindemann, D. Ubiquitination of the prototype foamy virus envelope glycoprotein leader peptide regulates subviral particle release. J. Virol. 2005, 79, 15074–15083. [Google Scholar] [CrossRef]
- Stange, A.; Luftenegger, D.; Reh, J.; Weissenhorn, W.; Lindemann, D. Subviral particle release determinants of prototype foamy virus. J. Virol. 2008, 82, 9858–9869. [Google Scholar] [CrossRef]
- Morita, E. Differential requirements of mammalian ESCRTs in multivesicular body formation, virus budding and cell division. FEBS J. 2012, 279, 1399–1406. [Google Scholar] [CrossRef]
- Demirov, D.G.; Freed, E.O. Retrovirus budding. Virus Res. 2004, 106, 87–102. [Google Scholar] [CrossRef]
- Bieniasz, P.D. Late budding domains and host proteins in enveloped virus release. Virology 2006, 344, 55–63. [Google Scholar] [CrossRef]
- Chen, B.J.; Lamb, R.A. Mechanisms for enveloped virus budding: Can some viruses do without an ESCRT? Virology 2008, 372, 221–232. [Google Scholar] [CrossRef]
- Schmitt, A.P.; Leser, G.P.; Morita, E.; Sundquist, W.I.; Lamb, R.A. Evidence for a new viral late-domain core sequence, FPIV, necessary for budding of a paramyxovirus. J. Virol. 2005, 79, 2988–2997. [Google Scholar] [CrossRef]
- Seo, E.J.; Leis, J. Budding of enveloped viruses: Interferon-induced ISG15-antivirus mechanisms targeting the release process. Adv. Virol. 2012, 2012, e532723. [Google Scholar]
- Mannigel, I.; Stange, A.; Zentgraf, H.; Lindemann, D. Correct capsid assembly mediated by a conserved YXXLGL motif in prototype foamy virus Gag is essential for infectivity and reverse transcription of the viral genome. J. Virol. 2007, 81, 3317–3326. [Google Scholar]
- Putterman, D.; Pepinsky, R.B.; Vogt, V.M. Ubiquitin in avian leukosis virus particles. Virology 1990, 176, 633–637. [Google Scholar] [CrossRef]
- Ott, D.E.; Coren, L.V.; Chertova, E.N.; Gagliardi, T.D.; Schubert, U. Ubiquitination of HIV-1 and MuLV Gag. Virology 2000, 278, 111–121. [Google Scholar] [CrossRef]
- Ott, D.E.; Coren, L.V.; Sowder, R.C., 2nd; Adams, J.; Nagashima, K.; Schubert, U. Equine infectious anemia virus and the ubiquitin-proteasome system. J. Virol. 2002, 76, 3038–3044. [Google Scholar] [CrossRef]
- Strack, B.; Calistri, A.; Accola, M.A.; Palu, G.; Gottlinger, H.G. A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. USA 2000, 97, 13063–13068. [Google Scholar] [CrossRef]
- Raiborg, C.; Rusten, T.E.; Stenmark, H. Protein sorting into multivesicular endosomes. Curr. Opin. Cell Biol. 2003, 15, 446–455. [Google Scholar] [CrossRef]
- Joshi, A.; Munshi, U.; Ablan, S.D.; Nagashima, K.; Freed, E.O. Functional replacement of a retroviral late domain by ubiquitin fusion. Traffic 2008, 9, 1972–1983. [Google Scholar] [CrossRef]
- Matthes, D.; Wiktorowicz, T.; Zahn, J.; Bodem, J.; Stanke, N.; Lindemann, D.; Rethwilm, A. Basic residues in the foamy virus gag protein. J. Virol. 2011, 85, 3986–3995. [Google Scholar] [CrossRef]
- Harris, R.S.; Hultquist, J.F.; Evans, D.T. The restriction factors of human immunodeficiency virus. J. Biol. Chem. 2012, 287, 40875–40883. [Google Scholar] [CrossRef]
- Biard-Piechaczyk, M.; Borel, S.; Espert, L.; de Bettignies, G.; Coux, O. HIV-1, ubiquitin and ubiquitin-like proteins: The dialectic interactions of a virus with a sophisticated network of post-translational modifications. Biol. Cell 2012, 104, 165–187. [Google Scholar] [CrossRef]
- Delebecque, F.; Suspene, R.; Calattini, S.; Casartelli, N.; Saib, A.; Froment, A.; Wain-Hobson, S.; Gessain, A.; Vartanian, J.P.; Schwartz, O. Restriction of foamy viruses by APOBEC cytidine deaminases. J. Virol. 2006, 80, 605–614. [Google Scholar] [CrossRef]
- Russell, R.A.; Wiegand, H.L.; Moore, M.D.; Schafer, A.; McClure, M.O.; Cullen, B.R. Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J. Virol. 2005, 79, 8724–8731. [Google Scholar] [CrossRef]
- Löchelt, M.; Romen, F.; Bastone, P.; Muckenfuss, H.; Kirchner, N.; Kim, Y.B.; Truyen, U.; Rosler, U.; Battenberg, M.; Saib, A.; et al. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7982–7987. [Google Scholar] [CrossRef]
- Chareza, S.; Slavkovic Lukic, D.; Liu, Y.; Rathe, A.M.; Munk, C.; Zabogli, E.; Pistello, M.; Lochelt, M. Molecular and functional interactions of cat APOBEC3 and feline foamy and immunodeficiency virus proteins: Different ways to counteract host-encoded restriction. Virology 2012, 424, 138–146. [Google Scholar] [CrossRef]
- Perkovic, M.; Schmidt, S.; Marino, D.; Russell, R.A.; Stauch, B.; Hofmann, H.; Kopietz, F.; Kloke, B.P.; Zielonka, J.; Strover, H.; et al. Species-specific inhibition of APOBEC3C by the prototype foamy virus protein bet. J. Biol. Chem. 2009, 284, 5819–5826. [Google Scholar]
- Prange, R. Host factors involved in hepatitis B virus maturation, assembly, and egress. Med. Microbiol. Immunol. 2012, 201, 449–461. [Google Scholar] [CrossRef]
- Patzer, E.J.; Nakamura, G.R.; Simonsen, C.C.; Levinson, A.D.; Brands, R. Intracellular assembly and packaging of hepatitis B surface antigen particles occur in the endoplasmic reticulum. J. Virol. 1986, 58, 884–892. [Google Scholar]
- Huovila, A.P.; Eder, A.M.; Fuller, S.D. Hepatitis B surface antigen assembles in a post-ER, pre-Golgi compartment. J. Cell Biol. 1992, 118, 1305–1320. [Google Scholar] [CrossRef]
- Ganem, D.; Prince, A.M. Hepatitis B virus infection—Natural history and clinical consequences. N. Engl. J. Med. 2004, 350, 1118–1129. [Google Scholar] [CrossRef]
- Chai, N.; Chang, H.E.; Nicolas, E.; Han, Z.; Jarnik, M.; Taylor, J. Properties of subviral particles of hepatitis B virus. J. Virol. 2008, 82, 7812–7817. [Google Scholar] [CrossRef]
- Ganem, D. Assembly of hepadnaviral virions and subviral particles. Curr. Top. Microbiol. Immunol. 1991, 168, 61–83. [Google Scholar] [CrossRef]
- Shaw, K.L.; Lindemann, D.; Mulligan, M.J.; Goepfert, P.A. Foamy virus envelope glycoprotein is sufficient for particle budding and release. J. Virol. 2003, 77, 2338–2348. [Google Scholar] [CrossRef]
- Pellman, D.; Garber, E.A.; Cross, F.R.; Hanafusa, H. An N-terminal peptide from p60src can direct myristylation and plasma membrane localization when fused to heterologous proteins. Nature 1985, 314, 374–377. [Google Scholar] [CrossRef]
- Wills, J.W.; Craven, R.C.; Weldon, R.A., Jr.; Nelle, T.D.; Erdie, C.R. Suppression of retroviral MA deletions by the amino-terminal membrane-binding domain of p60src. J. Virol. 1991, 65, 3804–3812. [Google Scholar]
- Verderame, M.F.; Nelle, T.D.; Wills, J.W. The membrane-binding domain of the Rous sarcoma virus Gag protein. J. Virol. 1996, 70, 2664–2668. [Google Scholar]
- Swiersy, A.; Wiek, C.; Zentgraf, H.; Lindemann, D. Characterization and manipulation of foamy virus membrane interactions. Cell. Microbiol. 2013, 15, 227–236. [Google Scholar] [CrossRef]
- Sanders, D.A. No false start for novel pseudotyped vectors. Curr. Opin. Biotechnol. 2002, 13, 437–442. [Google Scholar] [CrossRef]
- Cronin, J.; Zhang, X.Y.; Reiser, J. Altering the tropism of lentiviral vectors through pseudotyping. Curr. Gene. Ther. 2005, 5, 387–398. [Google Scholar] [CrossRef]
- Johnson, M.C. Mechanisms for Env glycoprotein acquisition by retroviruses. AIDS Res. Hum. Retrovir. 2011, 27, 239–247. [Google Scholar] [CrossRef]
- Ho, Y.P.; Schnabel, V.; Swiersy, A.; Stirnnagel, K.; Lindemann, D. A small-molecule-controlled system for efficient pseudotyping of prototype foamy virus vectors. Mol. Ther. 2012, 20, 1167–1176. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hütter, S.; Zurnic, I.; Lindemann, D. Foamy Virus Budding and Release. Viruses 2013, 5, 1075-1098. https://doi.org/10.3390/v5041075
Hütter S, Zurnic I, Lindemann D. Foamy Virus Budding and Release. Viruses. 2013; 5(4):1075-1098. https://doi.org/10.3390/v5041075
Chicago/Turabian StyleHütter, Sylvia, Irena Zurnic, and Dirk Lindemann. 2013. "Foamy Virus Budding and Release" Viruses 5, no. 4: 1075-1098. https://doi.org/10.3390/v5041075