Epigenetic Control of Cytomegalovirus Latency and Reactivation

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. CMV Latency
2.1. Cellular Sites of Latency
2.2. Viral Gene Expression Is Repressed in Latency
2.3. Latent Viral Genomes Are Heterochromatinized
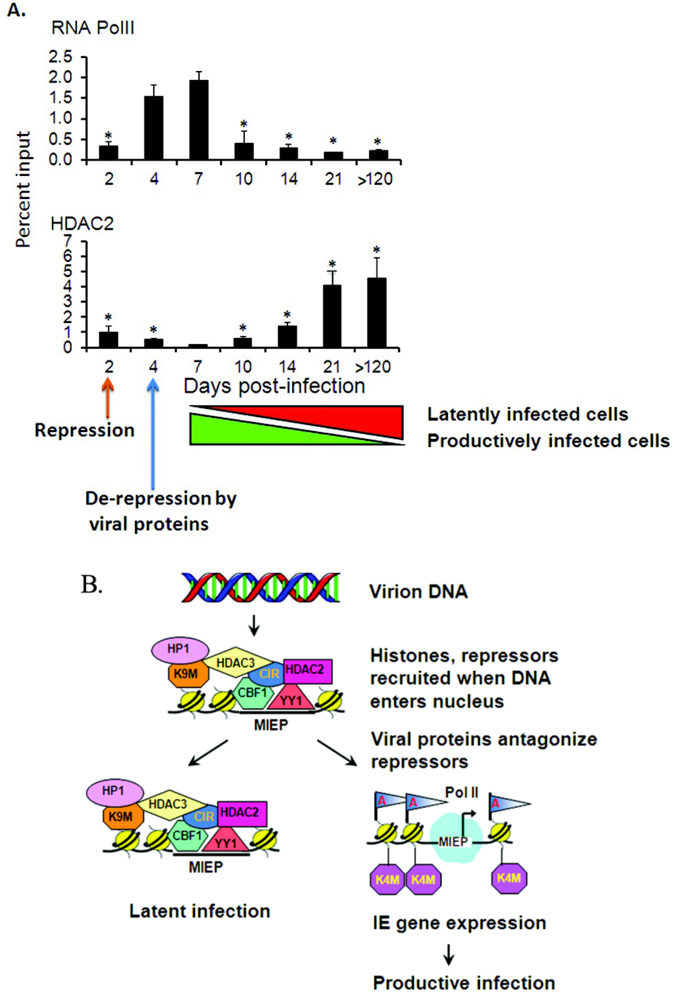
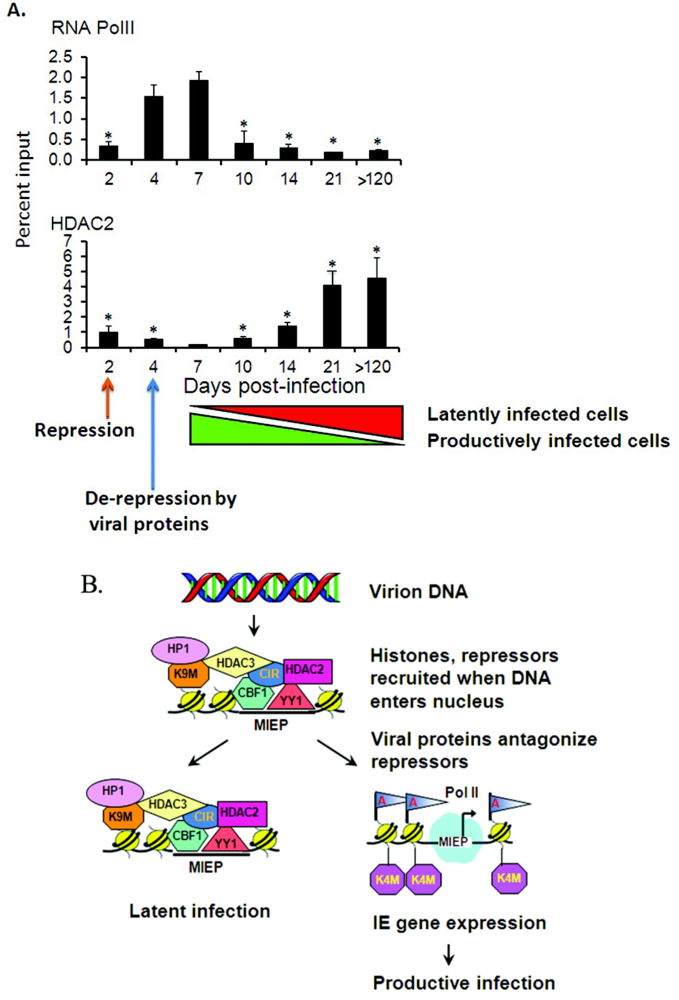
2.4. Transcriptional Repressors Are Recruited onto Viral Genomes in Latency
2.5. Latency May Be the Default State in Viral Infection
3. Reactivation of Latent CMV
3.1. Organ Transplantation Induces Reactivation of Viral Gene Expression
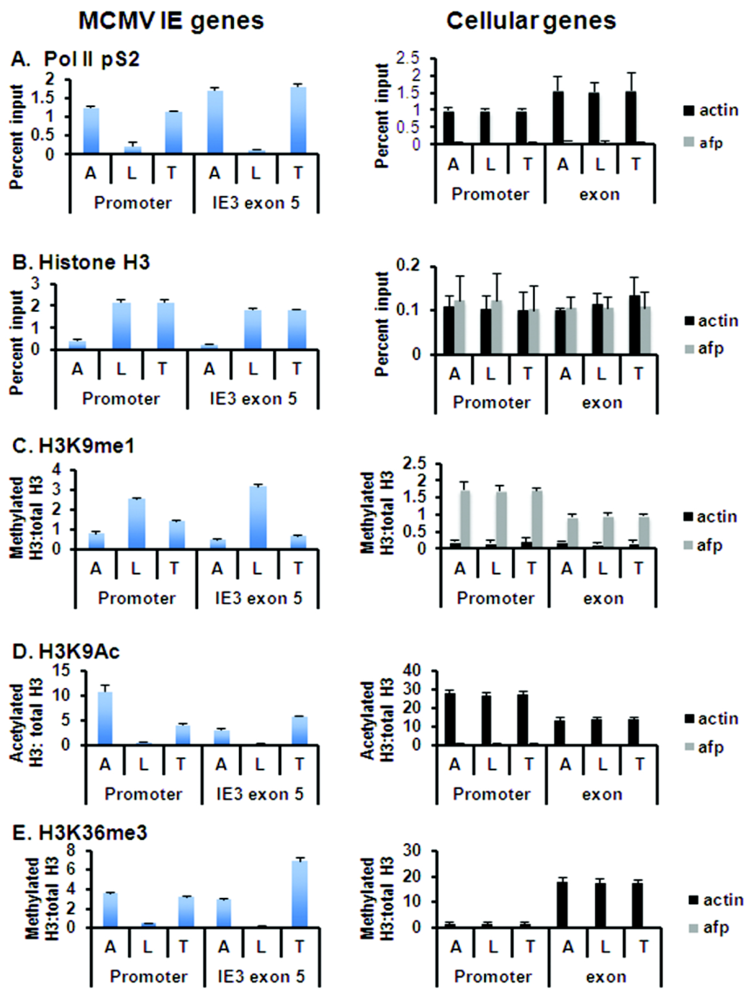
3.2. Epigenetic Reprogramming of Viral Chromatin in the ie Region Is Induced by Transplantation
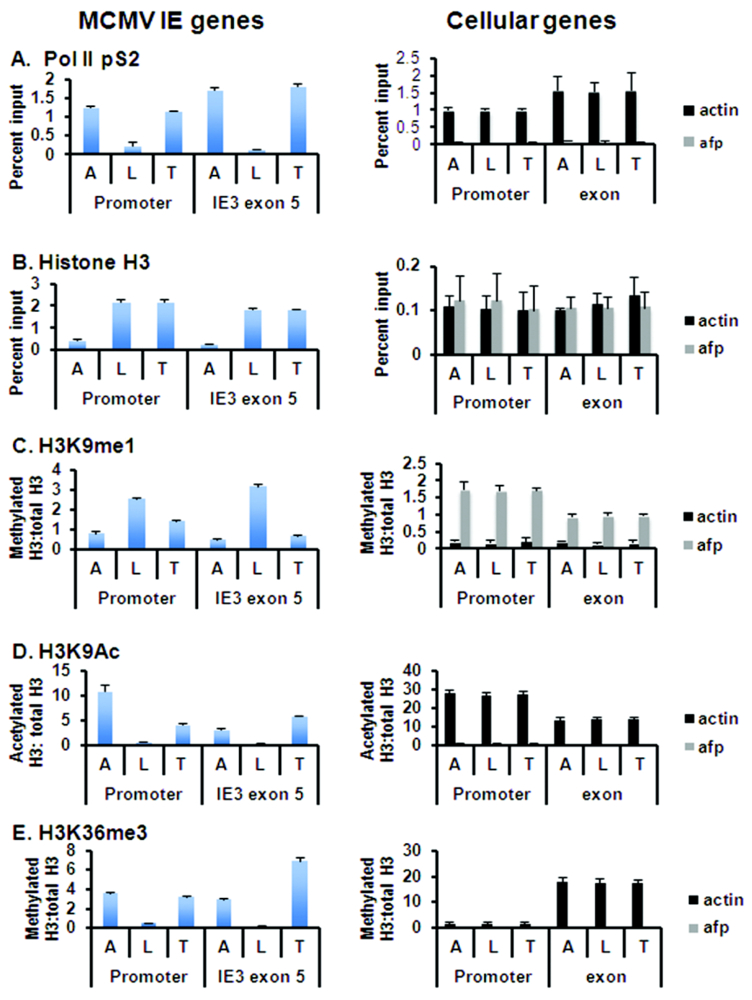
3.3. Global Changes in Viral Chromatin Are Likely Required for Reactivation of Virus
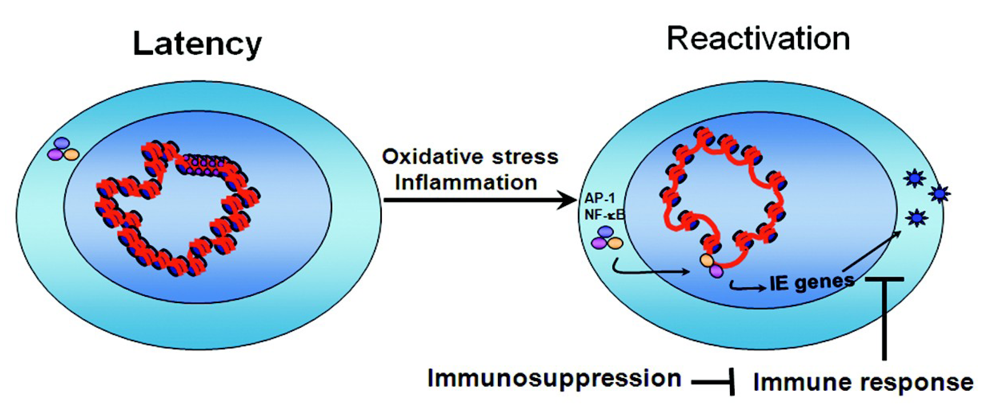
3.4. Mechanisms of Chromatin Remodeling Induced by Transplantation
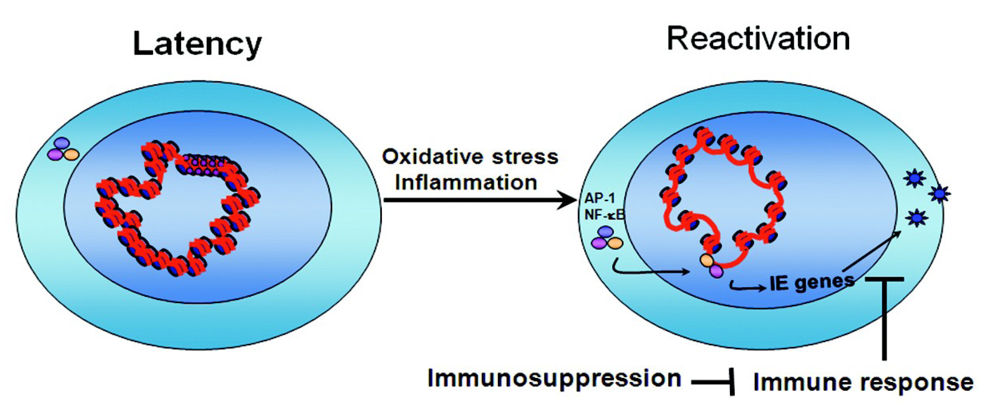
4. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Britt, W. Manifestations of human cytomegalovirus infection: Proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 2008, 325, 417–470. [Google Scholar] [CrossRef]
- Freeman, R.B. The ‘indirect’ effects of cytomegalovirus infection. Am. J. Transplant. 2009, 9, 2453–2458. [Google Scholar] [CrossRef]
- Fishman, J.A.; Emery, V.; Freeman, R.; Pascual, M.; Rostaing, L.; Schlitt, H.J.; Sgarabotto, D.; Torre-Cisneros, J.; Uknis, M.E. Cytomegalovirus in transplantation—Challenging the status quo. Clin. Transplant. 2007, 21, 149–158. [Google Scholar] [CrossRef]
- Singh, N. Late-onset cytomegalovirus disease as a significant complication in solid organ transplant recipients receiving antiviral prophylaxis: A call to heed the mounting evidence. Clin. Infect. Dis. 2005, 40, 704–708. [Google Scholar] [CrossRef]
- Pereira, L.; Maidji, E. Cytomegalovirus infection in the human placenta: Maternal immunity and developmentally regulated receptors on trophoblasts converge. Curr. Top. Microbiol. Immunol. 2008, 325, 383–395. [Google Scholar] [CrossRef]
- Gaytant, M.A.; Steegers, E.A.; Semmekrot, B.A.; Merkus, H.M.; Galama, J.M. Congenital cytomegalovirus infection: Review of the epidemiology and outcome. Obstet. Gynecol. Surv. 2002, 57, 245–256. [Google Scholar] [CrossRef]
- Sinzger, C.; Digel, M.; Jahn, G. Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 2008, 325, 63–83. [Google Scholar] [CrossRef]
- Mocarski, E.S.; Shenk, T.; Pass, R.F. Cytomegaloviruses. In Fields Virology, 5th; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 2702–2772. [Google Scholar]
- Reddehase, M.J.; Simon, C.O.; Seckert, C.K.; Lemmermann, N.; Grzimek, N.K. Murine model of cytomegalovirus latency and reactivation. Curr. Top. Microbiol. Immunol. 2008, 325, 315–331. [Google Scholar]
- Reeves, M.; Sinclair, J. Aspects of human cytomegalovirus latency and reactivation. Curr. Top. Microbiol. Immunol. 2008, 325, 297–313. [Google Scholar] [CrossRef]
- Seckert, C.K.; Griebl, M.; Buttner, J.; Freitag, K.; Lemmermann, N.; Hummel, M.; Liu, X.-F.; Abecassis, M.; Angulo, A.; Messerle, M.; et al. Immune surveillance of cytomegalovirus latency and reactivation in murine models: Link to "memory inflation". In Cytomegaloviruses: From Molecular Pathogenesis to Therapy; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK; Volume 1, in press.
- Meier, J.L.; Stinski, M.F. Major Immediate-early Enhancer and its Gene Products. In Cytomegaloviruses Molecular Biology and Immunology; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2006; pp. 151–166. [Google Scholar]
- Hummel, M.; Abecassis, M.M. A model for reactivation of CMV from latency. J. Clin. Virol. 2002, 25, S123–S136. [Google Scholar] [CrossRef]
- Hummel, M.; Zhang, Z.; Yan, S.; DePlaen, I.; Golia, P.; Varghese, T.; Thomas, G.; Abecassis, M.I. Allogeneic transplantation induces expression of cytomegalovirus immediate-early genes in vivo: A model for reactivation from latency. J. Virol. 2001, 75, 4814–4822. [Google Scholar]
- Hendrix, R.M.; Wagenaar, M.; Slobbe, R.L.; Bruggeman, C.A. Widespread presence of cytomegalovirus DNA in tissues of healthy trauma victims. J. Clin. Pathol. 1997, 50, 59–63. [Google Scholar]
- Koffron, A.J.; Patterson, B.K.; Yan, S.; Kaufman, D.B.; Fryer, J.P.; Stuart, F.P.; Abecassis, M.I. Latent human cytomegalovirus: A functional study. Transplant. Proc. 1997, 29, 793–795. [Google Scholar]
- Toorkey, C.B.; Carrigan, D.R. Immunohistochemical detection of an immediate early antigen of human cytomegalovirus in normal tissues. J. Infect. Dis. 1989, 160, 741–751. [Google Scholar]
- Reeves, M.B.; Coleman, H.; Chadderton, J.; Goddard, M.; Sissons, J.G.; Sinclair, J.H. Vascular endothelial and smooth muscle cells are unlikely to be major sites of latency of human cytomegalovirus in vivo. J. Gen. Virol. 2004, 85, 3337–3341. [Google Scholar] [CrossRef]
- Chang, H.H.; Ganem, D. A unique herpesviral transcriptional program in KSHV-infected lymphatic endothelial cells leads to mTORC1 activation and rapamycin sensitivity. Cell Host Microbe 2013, 13, 429–440. [Google Scholar] [CrossRef]
- Koffron, A.J.; Hummel, M.; Patterson, B.K.; Yan, S.; Kaufman, D.B.; Fryer, J.P.; Stuart, F.P.; Abecassis, M.I. Cellular localization of latent murine cytomegalovirus. J. Virol. 1998, 72, 95–103. [Google Scholar]
- Seckert, C.K.; Renzaho, A.; Tervo, H.M.; Krause, C.; Deegen, P.; Kuhnapfel, B.; Reddehase, M.J.; Grzimek, N.K. Liver sinusoidal endothelial cells are a site of murine cytomegalovirus latency and reactivation. J. Virol. 2009, 83, 8869–8884. [Google Scholar] [CrossRef]
- Pollock, J.L.; Presti, R.M.; Paetzold, S.; Virgin, H.W., IVth. Latent murine cytomegalovirus infection in macrophages. Virology 1997, 227, 168–179. [Google Scholar]
- Seckert, C.K.; Renzaho, A.; Reddehase, M.J.; Grzimek, N.K. Hematopoietic stem cell transplantation with latently infected donors does not transmit virus to immunocompromised recipients in the murine model of cytomegalovirus infection. Med. Microbiol. Immunol. 2008, 197, 251–259. [Google Scholar] [CrossRef]
- Reeves, M.; Sinclair, J. Regulation of human cytomegalovirus transcription in latency: Beyond the major immediate-early promoter. Viruses 2013. submitted for publication. [Google Scholar]
- Weekes, M.P.; Tan, S.Y.; Poole, E.; Talbot, S.; Antrobus, R.; Smith, D.L.; Montag, C.; Gygi, S.P.; Sinclair, J.H.; Lehner, P.J. Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science 2013, 340, 199–202. [Google Scholar]
- Keyes, L.R.; Hargett, D.; Soland, M.; Bego, M.G.; Rossetto, C.C.; Almeida-Porada, G.; St Jeor, S. HCMV protein LUNA is required for viral reactivation from latently infected primary CD14(+) cells. PLoS One 2012, 7, e52827. [Google Scholar]
- Grzimek, N.K.; Dreis, D.; Schmalz, S.; Reddehase, M.J. Random, Asynchronous, and asymmetric transcriptional activity of enhancer-flanking major immediate-early genes ie1/3 and ie2 during murine cytomegalovirus latency in the lungs. J. Virol. 2001, 75, 2692–2705. [Google Scholar]
- Kurz, S.; Steffens, H.P.; Mayer, A.; Harris, J.R.; Reddehase, M.J. Latency versus persistence or intermittent recurrences: Evidence for a latent state of murine cytomegalovirus in the lungs. J. Virol. 1997, 71, 2980–2987. [Google Scholar]
- Kurz, S.K.; Rapp, M.; Steffens, H.P.; Grzimek, N.K.; Schmalz, S.; Reddehase, M.J. Focal transcriptional activity of murine cytomegalovirus during latency in the lungs. J. Virol. 1999, 73, 482–494. [Google Scholar]
- Simon, C.O.; Seckert, C.K.; Dreis, D.; Reddehase, M.J.; Grzimek, N.K. Role for tumor necrosis factor alpha in murine cytomegalovirus transcriptional reactivation in latently infected lungs. J. Virol. 2005, 79, 326–340. [Google Scholar] [CrossRef]
- Liu, X.F.; Yan, S.; Abecassis, M.; Hummel, M. Establishment of murine cytomegalovirus latency in vivo is associated with changes in histone modifications and recruitment of transcriptional repressors to the major immediate-early promoter. J. Virol. 2008, 82, 10922–10931. [Google Scholar] [CrossRef]
- Liu, X.F.; Yan, S.; Abecassis, M.; Hummel, M. Biphasic recruitment of transcriptional repressors to the murine cytomegalovirus major immediate-early promoter during the course of infection in vivo. J. Virol. 2010, 84, 3631–3643. [Google Scholar] [CrossRef]
- Henry, S.C.; Hamilton, J.D. Detection of murine cytomegalovirus immediate early 1 transcripts in the spleens of latently infected mice. J. Infec. Dis. 1993, 167, 950–954. [Google Scholar] [CrossRef]
- Yu, Y.; Henry, S.C.; Xu, F.; Hamilton, J.D. Expression of a murine cytomegalovirus early-late protein in “latently” infected mice. J. Infec. Dis. 1995, 172, 371–379. [Google Scholar] [CrossRef]
- Yuhasz, S.A.; Dissette, V.B.; Cook, M.L.; Stevens, J.G. Murine cytomegalovirus is present in both chronic active and latent states in persistently infected mice. Virology 1994, 202, 272–280. [Google Scholar] [CrossRef]
- Zaidi, S.K.; Young, D.W.; Montecino, M.; van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Stein, G.S. Bookmarking the genome: Maintenance of epigenetic information. J. Biol. Chem. 2011, 286, 18355–18361. [Google Scholar]
- Hummel, M.; Yan, S.; Li, Z.; Varghese, T.K.; Abecassis, M. Transcriptional reactivation of murine cytomegalovirus ie gene expression by 5-aza-2'-deoxycytidine and trichostatin A in latently infected cells despite lack of methylation of the major immediate-early promoter. J. Gen. Virol. 2007, 88, 1097–1102. [Google Scholar] [CrossRef]
- Kubat, N.J.; Tran, R.K.; McAnany, P.; Bloom, D.C. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J. Virol. 2004, 78, 1139–1149. [Google Scholar] [CrossRef]
- Streblow, D.N.; Varnum, S.M.; Smith, R.D.; Nelson, J.A. A proteomics analysis of human cytomegalovirus particles. In Cytomegaloviruses: Molecular Biology and Immunology; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2006; pp. 91–110. [Google Scholar]
- Groves, I.J.; Reeves, M.B.; Sinclair, J.H. Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic 'pre-immediate-early' repression of viral gene expression mediated by histone post-translational modification. J. Gen. Virol. 2009, 90, 2364–2374. [Google Scholar] [CrossRef]
- Liashkovich, I.; Hafezi, W.; Kuhn, J.M.; Oberleithner, H.; Shahin, V. Nuclear delivery mechanism of herpes simplex virus type 1 genome. J. Mol. Recognit. 2011, 24, 414–421. [Google Scholar]
- Nitzsche, A.; Paulus, C.; Nevels, M. Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J. Virol. 2008, 82, 11167–11180. [Google Scholar] [CrossRef]
- Reeves, M.B.; MacAry, P.A.; Lehner, P.J.; Sissons, J.G.; Sinclair, J.H. Latency, Chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. USA 2005, 102, 4140–4145. [Google Scholar]
- Reeves, M.B.; Sinclair, J.H. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J. Gen. Virol. 2010, 91, 599–604. [Google Scholar] [CrossRef]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef]
- Knipe, D.M.; Lieberman, P.M.; Jung, J.U.; McBride, A.A.; Morris, K.V.; Ott, M.; Margolis, D.; Nieto, A.; Nevels, M.; Parks, R.J.; et al. Snapshots: Chromatin control of viral infection. Virology 2013, 435, 141–156. [Google Scholar] [CrossRef]
- Nevels, M.; Nitzsche, A.; Paulus, C. How to control an infectious bead string: Nucleosome-based regulation and targeting of herpesvirus chromatin. Rev. Med. Virol. 2011, 21, 154–180. [Google Scholar] [CrossRef]
- Toth, Z.; Maglinte, D.T.; Lee, S.H.; Lee, H.R.; Wong, L.Y.; Brulois, K.F.; Lee, S.; Buckley, J.D.; Laird, P.W.; Marquez, V.E.; et al. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 2010, 6, e1001013. [Google Scholar] [CrossRef]
- Tempera, I.; Lieberman, P.M. Chromatin organization of gammaherpesvirus latent genomes. Biochim. Biophys. Acta 2010, 1799, 236–245. [Google Scholar] [CrossRef]
- Trojer, P.; Reinberg, D. Facultative heterochromatin: Is there a distinctive molecular signature? Mol. Cell 2007, 28, 1–13. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef]
- Reeves, M.B. Chromatin-mediated regulation of cytomegalovirus gene expression. Virus Res. 2011, 157, 134–143. [Google Scholar] [CrossRef]
- Corpet, A.; Almouzni, G. Making copies of chromatin: The challenge of nucleosomal organization and epigenetic information. Trends Cell Biol. 2009, 19, 29–41. [Google Scholar] [CrossRef]
- Drane, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Gene. Dev. 2010, 24, 1253–1265. [Google Scholar] [CrossRef]
- Meier, J.L. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: Role of trichostatin A, Retinoic acid, and deletion of the 21-base-pair repeats and modulator. J. Virol. 2001, 75, 1581–1593. [Google Scholar] [CrossRef]
- Lu, F.; Zhou, J.; Wiedmer, A.; Madden, K.; Yuan, Y.; Lieberman, P.M. Chromatin remodeling of the Kaposi’s sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J. Virol. 2003, 77, 11425–11435. [Google Scholar] [CrossRef]
- Lai, A.Y.; Wade, P.A. Cancer biology and NuRD: A multifaceted chromatin remodelling complex. Nat. Rev. Cancer 2011, 11, 588–596. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Bray, S.; Bernard, F. Notch targets and their regulation. Curr. Top. Dev. Biol. 2010, 92, 253–275. [Google Scholar] [CrossRef]
- Hertzog, P. A notch in the toll belt. Immunity 2008, 29, 663–665. [Google Scholar] [CrossRef]
- Minter, L.M.; Osborne, B.A. Canonical and non-canonical Notch signaling in CD4(+) T cells. Curr. Top. Microbiol. Immunol. 2012, 360, 99–114. [Google Scholar] [CrossRef]
- Radtke, F.; Fasnacht, N.; Macdonald, H.R. Notch signaling in the immune system. Immunity 2010, 32, 14–27. [Google Scholar] [CrossRef]
- Hayward, S.D.; Liu, J.; Fujimuro, M. Notch and Wnt signaling: Mimicry and manipulation by gamma herpesviruses. Sci. STKE 2006, 2006, re4. [Google Scholar]
- Tyagi, M.; Karn, J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007, 26, 4985–4995. [Google Scholar] [CrossRef]
- Gordon, S.; Akopyan, G.; Garban, H.; Bonavida, B. Transcription factor YY1: Structure, Function, And therapeutic implications in cancer biology. Oncogene 2006, 25, 1125–1142. [Google Scholar] [CrossRef]
- Liu, R.; Baillie, J.; Sissons, J.G.; Sinclair, J.H. The transcription factor YY1 binds to negative regulatory elements in the human cytomegalovirus major immediate early enhancer/promoter and mediates repression in non-permissive cells. Nucleic Acids Res. 1994, 22, 2453–2459. [Google Scholar] [CrossRef]
- Coull, J.J.; Romerio, F.; Sun, J.M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar]
- Huang, N.E.; Lin, C.H.; Lin, Y.S.; Yu, W.C. Modulation of YY1 activity by SAP30. Biochem. Biophys. Res. Commun. 2003, 306, 267–275. [Google Scholar] [CrossRef]
- Le May, N.; Mansuroglu, Z.; Leger, P.; Josse, T.; Blot, G.; Billecocq, A.; Flick, R.; Jacob, Y.; Bonnefoy, E.; Bouloy, M. A SAP30 complex inhibits IFN-beta expression in Rift Valley fever virus infected cells. PLoS Pathog. 2008, 4, e13. [Google Scholar] [CrossRef]
- Yang, W.M.; Inouye, C.; Zeng, Y.; Bearss, D.; Seto, E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl. Acad. Sci. USA 1996, 93, 12845–12850. [Google Scholar]
- Kassardjian, A.; Rizkallah, R.; Riman, S.; Renfro, S.H.; Alexander, K.E.; Hurt, M.M. The transcription factor YY1 is a novel substrate for Aurora B kinase at G2/M transition of the cell cycle. PLoS One 2012, 7, e50645. [Google Scholar]
- Riman, S.; Rizkallah, R.; Kassardjian, A.; Alexander, K.E.; Luscher, B.; Hurt, M.M. Phosphorylation of the transcription factor YY1 by CK2alpha prevents cleavage by caspase 7 during apoptosis. Mol. Cell Biol. 2012, 32, 797–807. [Google Scholar]
- Rizkallah, R.; Hurt, M.M. Regulation of the transcription factor YY1 in mitosis through phosphorylation of its DNA-binding domain. Mol. Biol. Cell 2009, 20, 4766–4776. [Google Scholar]
- Yao, Y.L.; Yang, W.M.; Seto, E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol. Cell Biol. 2001, 21, 5979–5991. [Google Scholar]
- Everett, R.D. The use of fluorescence microscopy to study the association between herpesviruses and intrinsic resistance factors. Viruses 2011, 3, 2412–2424. [Google Scholar] [CrossRef]
- Maul, G.G. Initiation of cytomegalovirus infection at ND10. Curr. Top. Microbiol. Immunol. 2008, 325, 117–132. [Google Scholar]
- Kalejta, R.F. Functions of human cytomegalovirus tegument proteins prior to immediate early gene expression. Curr. Top. Microbiol. Immunol. 2008, 325, 101–115. [Google Scholar]
- Cantrell, S.R.; Bresnahan, W.A. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 2006, 80, 6188–6191. [Google Scholar] [CrossRef]
- Saffert, R.T.; Kalejta, R.F. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 2006, 80, 3863–3871. [Google Scholar] [CrossRef]
- Saffert, R.T.; Kalejta, R.F. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 2007, 81, 9109–9120. [Google Scholar] [CrossRef]
- Woodhall, D.L.; Groves, I.J.; Reeves, M.B.; Wilkinson, G.; Sinclair, J.H. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 2006, 281, 37652–37660. [Google Scholar]
- Everett, R.D.; Murray, J. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 2005, 79, 5078–5089. [Google Scholar] [CrossRef]
- Dimitropoulou, P.; Caswell, R.; McSharry, B.P.; Greaves, R.F.; Spandidos, D.A.; Wilkinson, G.W.; Sourvinos, G. Differential relocation and stability of PML-body components during productive human cytomegalovirus infection: Detailed characterization by live-cell imaging. Eur. J. Cell Biol. 2010, 89, 757–768. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Li, Z.; Wang, X.; Yan, S.; Zhang, Z.; Jie, C.; Sustento-Reodica, N.; Hummel, M.; Abecassis, M. A mouse model of CMV transmission following kidney transplantation. Am. J. Transplant. 2012, 12, 1024–1028. [Google Scholar] [CrossRef]
- Saunders, A.; Core, L.J.; Lis, J.T. Breaking barriers to transcription elongation. Nat. Rev. Mol. Cell. Biol. 2006, 7, 557–567. [Google Scholar] [CrossRef]
- Messerle, M.; Buhler, B.; Keil, G.M.; Koszinowski, U.H. Structural organization, Expression, And functional characterization of the murine cytomegalovirus immediate-early gene 3. J. Virol. 1992, 66, 27–36. [Google Scholar]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef]
- Busche, A.; Angulo, A.; Kay-Jackson, P.; Ghazal, P.; Messerle, M. Phenotypes of major immediate-early gene mutants of mouse cytomegalovirus. Med. Microbiol. Immunol. 2008, 197, 233–240. [Google Scholar] [CrossRef]
- Nevels, M.; Paulus, C.; Shenk, T. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc. Natl. Acad. Sci. USA 2004, 101, 17234–17239. [Google Scholar] [CrossRef]
- Tang, Q.; Maul, G. Immediate-Early Interactions and Epigenetic Defense Mechanisms. In Cytomegaloviruses: Molecular Biology and Immunology; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2006; pp. 131–150. [Google Scholar]
- Tang, Q.; Maul, G.G. Mouse cytomegalovirus immediate-early protein 1 binds with host cell repressors to relieve suppressive effects on viral transcription and replication during lytic infection. J. Virol. 2003, 77, 1357–1367. [Google Scholar] [CrossRef]
- Buhler, B.; Keil, G.M.; Weiland, F.; Koszinowski, U.H. Characterization of the murine cytomegalovirus early transcription unit e1 that is induced by immediate-early proteins. J. Virol. 1990, 64, 1907–1919. [Google Scholar]
- Angulo, A.; Ghazal, P.; Messerle, M. The major immediate-early gene ie3 of mouse cytomegalovirus is essential for viral growth. J. Virol. 2000, 74, 11129–11136. [Google Scholar] [CrossRef]
- Martinez, F.P.; Cosme, R.S.; Tang, Q. Murine cytomegalovirus major immediate-early protein 3 interacts with cellular and viral proteins in viral DNA replication compartments and is important for early gene activation. J. Gen. Virol. 2010, 91, 2664–2676. [Google Scholar] [CrossRef]
- Stinski, M.F.; Isomura, H. Role of the cytomegalovirus major immediate early enhancer in acute infection and reactivation from latency. Med. Microbiol. Immunol. 2008, 197, 223–231. [Google Scholar] [CrossRef]
- Keller, M.J.; Wu, A.W.; Andrews, J.I.; McGonagill, P.W.; Tibesar, E.E.; Meier, J.L. Reversal of human cytomegalovirus major immediate-early enhancer/promoter silencing in quiescently infected cells via the cyclic AMP signaling pathway. J. Virol. 2007, 81, 6669–6681. [Google Scholar]
- Lashmit, P.; Wang, S.; Li, H.; Isomura, H.; Stinski, M.F. The CREB site in the proximal enhancer is critical for cooperative interaction with the other transcription factor binding sites to enhance transcription of the major intermediate-early genes in human cytomegalovirus-infected cells. J. Virol. 2009, 83, 8893–8904. [Google Scholar] [CrossRef]
- Gustems, M.; Busche, A.; Messerle, M.; Ghazal, P.; Angulo, A. In vivo competence of murine cytomegalovirus under the control of the human cytomegalovirus major immediate-early enhancer in the establishment of latency and reactivation. J. Virol. 2008, 82, 10302–10307. [Google Scholar]
- Isomura, H.; Stinski, M.F.; Kudoh, A.; Daikoku, T.; Shirata, N.; Tsurumi, T. Two Sp1/Sp3 binding sites in the major immediate-early proximal enhancer of human cytomegalovirus have a significant role in viral replication. J. Virol. 2005, 79, 9597–9607. [Google Scholar]
- Isomura, H.; Stinski, M.F.; Kudoh, A.; Nakayama, S.; Murata, T.; Sato, Y.; Iwahori, S.; Tsurumi, T. A cis element between the TATA Box and the transcription start site of the major immediate-early promoter of human cytomegalovirus determines efficiency of viral replication. J. Virol. 2008, 82, 849–858. [Google Scholar] [CrossRef]
- Netterwald, J.; Yang, S.; Wang, W.; Ghanny, S.; Cody, M.; Soteropoulos, P.; Tian, B.; Dunn, W.; Liu, F.; Zhu, H. Two gamma interferon-activated site-like elements in the human cytomegalovirus major immediate-early promoter/enhancer are important for viral replication. J. Virol. 2005, 79, 5035–5046. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-kappaB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef]
- Gerald, D.; Berra, E.; Frapart, Y.M.; Chan, D.A.; Giaccia, A.J.; Mansuy, D.; Pouyssegur, J.; Yaniv, M.; Mechta-Grigoriou, F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell 2004, 118, 781–794. [Google Scholar] [CrossRef]
- Hernandez, J.M.; Floyd, D.H.; Weilbaecher, K.N.; Green, P.L.; Boris-Lawrie, K. Multiple facets of junD gene expression are atypical among AP-1 family members. Oncogene 2008, 27, 4757–4767. [Google Scholar] [CrossRef]
- Lamb, J.A.; Ventura, J.J.; Hess, P.; Flavell, R.A.; Davis, R.J. JunD mediates survival signaling by the JNK signal transduction pathway. Mol. Cell 2003, 11, 1479–1489. [Google Scholar] [CrossRef]
- Tsuji, Y. JunD activates transcription of the human ferritin H gene through an antioxidant response element during oxidative stress. Oncogene 2005, 24, 7567–7578. [Google Scholar] [CrossRef]
- Hummel, M.; Kurian, S.M.; Lin, S.; Borodyanskiy, A.; Zhang, Z.; Li, Z.; Kim, S.J.; Salomon, D.R.; Abecassis, M. Intragraft TNF receptor signaling contributes to activation of innate and adaptive immunity in a renal allograft model. Transplantation 2009, 87, 178–188. [Google Scholar] [CrossRef]
- Cook, C.H.; Trgovcich, J.; Zimmerman, P.D.; Zhang, Y.; Sedmak, D.D. Lipopolysaccharide, Tumor necrosis factor alpha, Or interleukin-1beta triggers reactivation of latent cytomegalovirus in immunocompetent mice. J. Virol. 2006, 80, 9151–9158. [Google Scholar] [CrossRef]
- Koskinen, P.K.; Kallio, E.A.; Tikkanen, J.M.; Sihvola, R.K.; Hayry, P.J.; Lemstrom, K.B. Cytomegalovirus infection and cardiac allograft vasculopathy. Transplant. Infect. Dis. 1999, 1, 115–126. [Google Scholar] [CrossRef]
- Reinke, P.; Prosch, S.; Kern, F.; Volk, H.D. Mechanisms of human cytomegalovirus (HCMV) (re)activation and its impact on organ transplant patients. Transplant. Infect. Dis. 1999, 1, 157–164. [Google Scholar] [CrossRef]
- Powers, C.; DeFilippis, V.; Malouli, D.; Fruh, K. Cytomegalovirus immune evasion. Curr. Top. Microbiol. Immunol. 2008, 325, 333–359. [Google Scholar]
- Hargett, D.; Shenk, T.E. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 20039–20044. [Google Scholar] [CrossRef]
- Soderberg-Naucler, C.; Fish, K.N.; Nelson, J.A. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 1997, 91, 119–126. [Google Scholar] [CrossRef]
- Reeves, M.B.; Compton, T. Inhibition of inflammatory interleukin-6 activity via extracellular signal-regulated kinase-mitogen-activated protein kinase signaling antagonizes human cytomegalovirus reactivation from dendritic cells. J. Virol. 2011, 85, 12750–12758. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, X.-f.; Wang, X.; Yan, S.; Zhang, Z.; Abecassis, M.; Hummel, M. Epigenetic Control of Cytomegalovirus Latency and Reactivation. Viruses 2013, 5, 1325-1345. https://doi.org/10.3390/v5051325
Liu X-f, Wang X, Yan S, Zhang Z, Abecassis M, Hummel M. Epigenetic Control of Cytomegalovirus Latency and Reactivation. Viruses. 2013; 5(5):1325-1345. https://doi.org/10.3390/v5051325
Chicago/Turabian StyleLiu, Xue-feng, Xueqiong Wang, Shixian Yan, Zheng Zhang, Michael Abecassis, and Mary Hummel. 2013. "Epigenetic Control of Cytomegalovirus Latency and Reactivation" Viruses 5, no. 5: 1325-1345. https://doi.org/10.3390/v5051325