Human Viruses and Cancer


Abstract
:1. Introduction: Historical and Epidemiological Aspects

{kind=link}
{kind=link}
{kind=link}
| Henle Koch’s Postulates [9] | Bradford Hill’s Causative Principles [10] |
|---|---|
|
|
2. General Principles of Viral Oncogenic Mechanisms
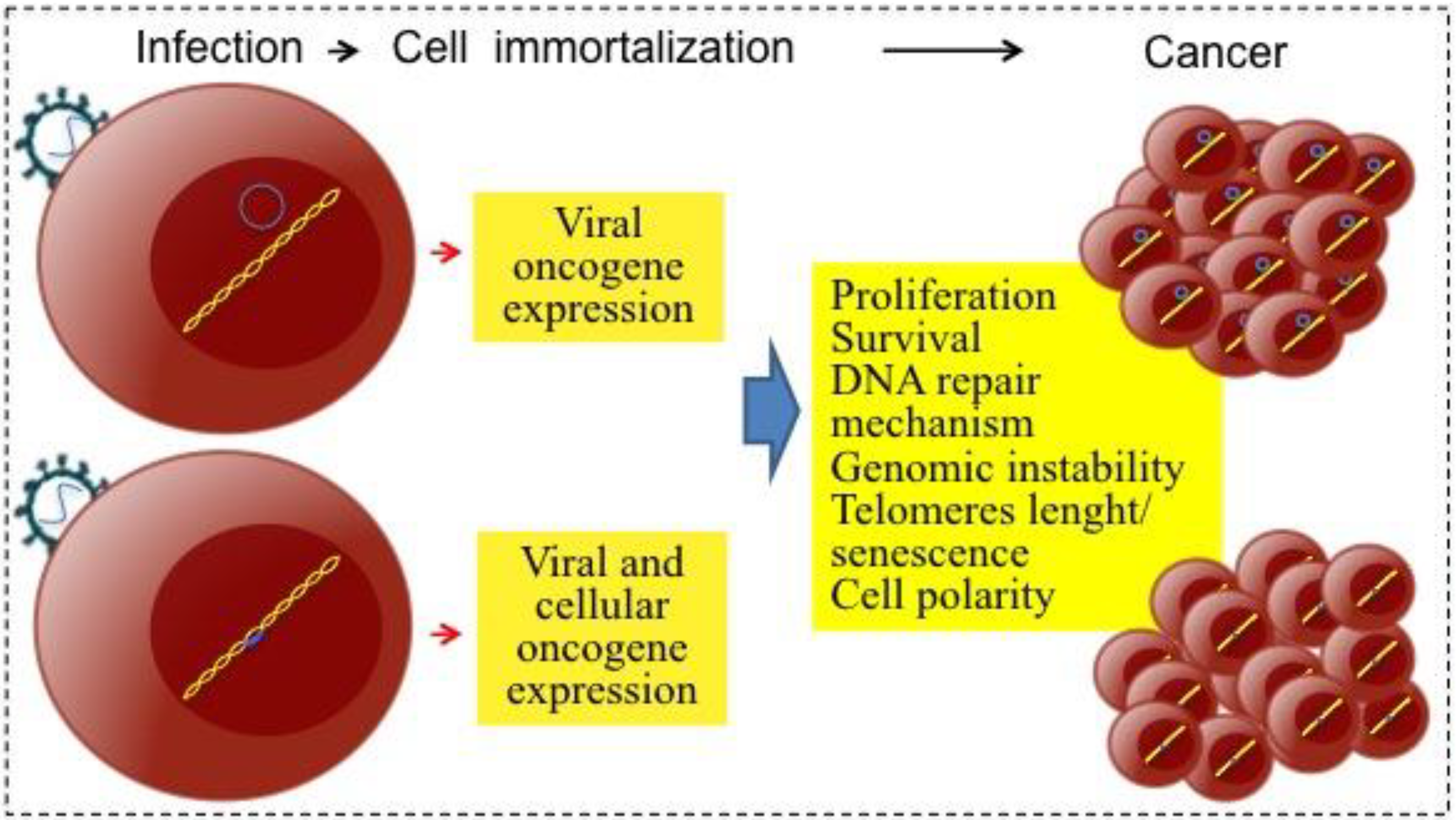
2.1. Direct and Indirect Viral Carcinogenesis


3. Human Oncogenic Viruses and Associated Cancers
| Human Virus | Associated Tumors | Reference |
|---|---|---|
| EBV | Burkitt’s lymphoma, Hodgkin’s lymphoma, immunosuppression-related lymphoma, T and NK cell lymphomas; nasopharyngeal and stomach carcinomas. | Reviewed in [11] |
| KSHV | Primary effusion lymphoma and Kaposi sarcoma | [20] |
| High-risk HPVs | Cervical, head and neck and anogenital tract carcinomas | Reviewed in [21] |
| MCPV | Merkel cell carcinoma | [22] |
| HBV | Hepatocellular carcinoma | [23] |
| HCV | Hepatocellular carcinoma | [24] |
| HTLV1 | Adult T-cell leukemia/lymphoma | [25] |
3.1. Herpesviruses: Epstein Barr Virus and Kaposi Sarcoma-Associated Herpesvirus
3.2. High-Risk Papillomaviruses
3.3. Merkel Cell Polyomavirus
3.4. Hepatitis B Virus
3.5. Hepatitis C Virus
3.6. Human T-Lymphotropic Virus Type 1
4. Common Mechanisms of Direct Carcinogenesis
4.1. Viral Oncogenes and Oncoproteins
4.1.1. p53 and pRb Inactivation and Other Targets of Increased Proliferation and Survival
4.1.2. Genomic Instability
4.1.3. Interfering with Telomere Shortening
4.1.4. Interfering with Cell Polarity
4.1.5. Viral miRNAs
4.2. Insertional Mutagenesis
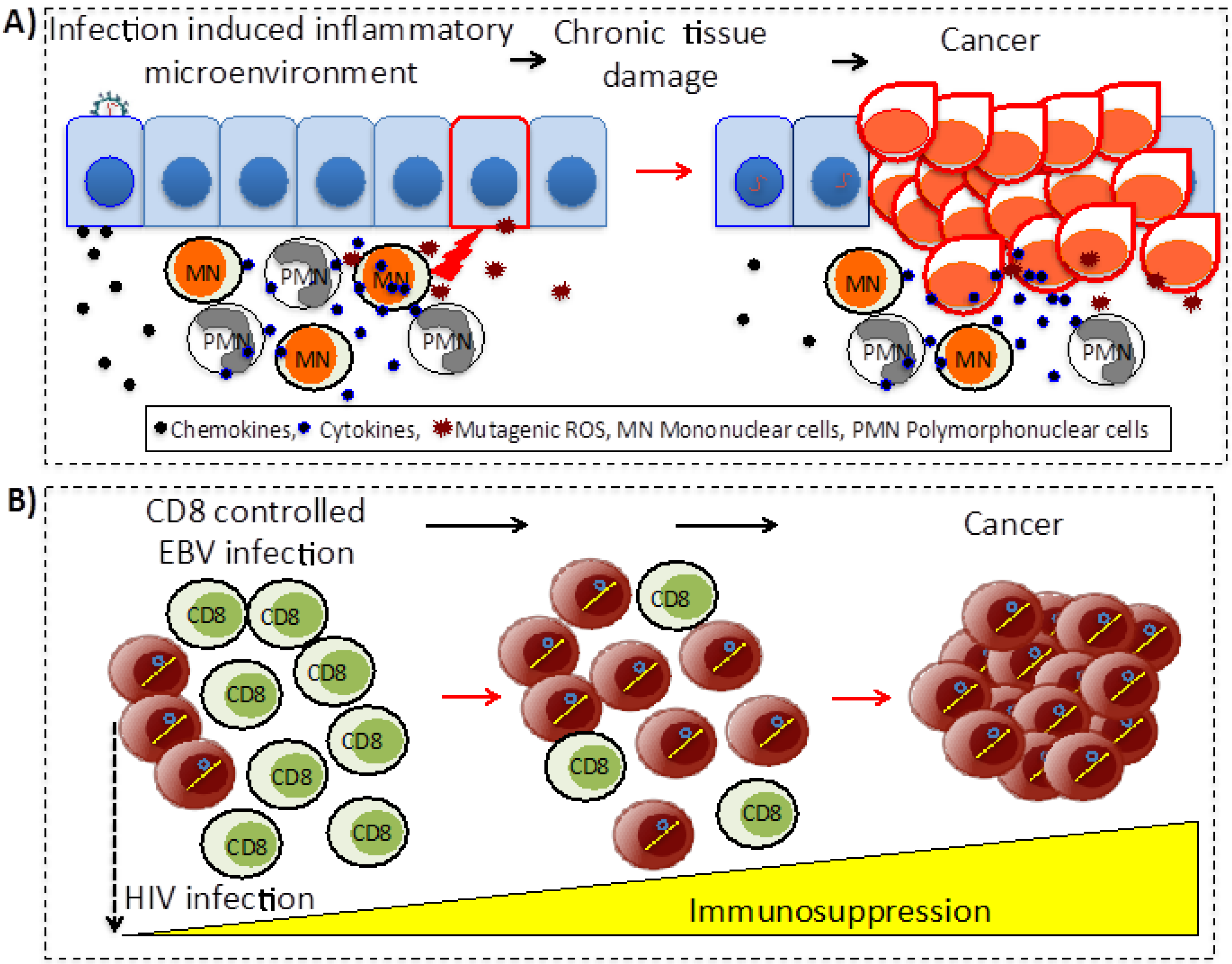
5. Common Mechanisms of Indirect Carcinogenesis

5.1. Chronic Inflammation
5.2. Immunosuppression
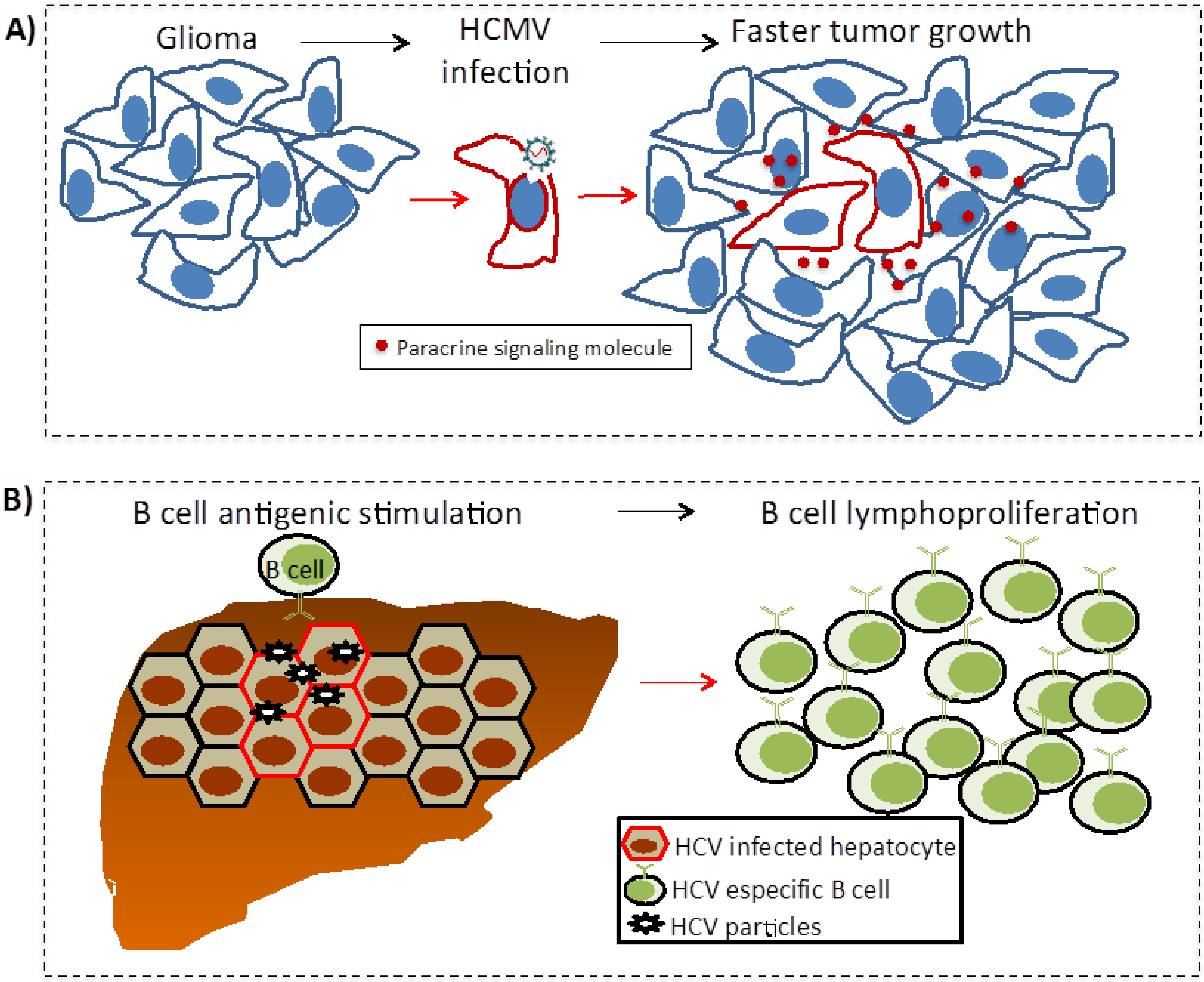
5.3. Oncomodulation
5.4. Chronic Antigen-Driven Lymphoproliferation
6. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Rous, P. A transmissible avian neoplasm. (sarcoma of the common fowl.). J. Exp. Med. 1910, 12, 696–705. [Google Scholar] [PubMed]
- Rous, P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J. Exp. Med. 1911, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Ellerman, V.; Bang, O. Experimentelle leukämie bei hühnern. Zent. Bakteriol. Parasitenkd. Infectionskr. Hyg. Abt. Orig. 1908, 46, 595–609. [Google Scholar]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef] [PubMed]
- Durst, M.; Gissmann, L.; Ikenberg, H.; zur Hausen, H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. USA 1983, 80, 3812–3815. [Google Scholar] [CrossRef] [PubMed]
- Boshart, M.; Gissmann, L.; Ikenberg, H.; Kleinheinz, A.; Scheurlen, W.; zur Hausen, H. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J. 1984, 3, 1151–1157. [Google Scholar] [PubMed]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [PubMed]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [PubMed]
- Koch, R. Untersuchungen über bakterien: V. Die ätiologie der milzbrand-krankheit, begründet auf die entwicklungsgeschichte des bacillus anthracis [investigations into bacteria: V. The etiology of anthrax, based on the ontogenesis of bacillus anthracis]. Cohns Beitr. Biol. Pflanz. 1876, 2, 277–310. [Google Scholar]
- Hill, A.B. The environment and disease: Association or causation? Proc. Royal Soc.Med. 1965, 58, 295–300. [Google Scholar]
- Thompson, M.P.; Kurzrock, R. Epstein-barr virus and cancer. Clin. Cancer Res. 2004, 10, 803–821. [Google Scholar] [CrossRef] [PubMed]
- Kulwichit, W.; Edwards, R.H.; Davenport, E.M.; Baskar, J.F.; Godfrey, V.; Raab-Traub, N. Expression of the epstein-barr virus latent membrane protein 1 induces b cell lymphoma in transgenic mice. Proc. Natl. Acad. Sci. USA 1998, 95, 11963–11968. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Springgate, C.F.; Battula, N. Errors in DNA replication as a basis of malignant changes. Cancer Res. 1974, 34, 2311–2321. [Google Scholar] [PubMed]
- Prindle, M.J.; Fox, E.J.; Loeb, L.A. The mutator phenotype in cancer: Molecular mechanisms and targeting strategies. Curr. Drug Targets 2010, 11, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Coleman, W.B. Mechanisms of human hepatocarcinogenesis. Curr. Mol. Med. 2003, 3, 573–588. [Google Scholar]
- Zucman-Rossi, J.; Laurent-Puig, P. Genetic diversity of hepatocellular carcinomas and its potential impact on targeted therapies. Pharmacogenomics 2007, 8, 997–1003. [Google Scholar] [PubMed]
- Chadburn, A.; Abdul-Nabi, A.M.; Teruya, B.S.; Lo, A.A. Lymphoid proliferations associated with human immunodeficiency virus infection. Arch. Pathol. Lab. Med. 2013, 137, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.; Piazuelo, M.B. The gastric precancerous cascade. J. Dig. Dis. 2012, 13, 2–9. [Google Scholar] [PubMed]
- Hatakeyama, M.; Higashi, H. Helicobacter pylori caga: A new paradigm for bacterial carcinogenesis. Cancer Sci. 2005, 96, 835–843. [Google Scholar] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in aids-associated kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [PubMed]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Beasley, R.P.; Hwang, L.Y.; Lin, C.C.; Chien, C.S. Hepatocellular carcinoma and hepatitis b virus. A prospective study of 22,707 men in taiwan. Lancet 1981, 2, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-a, non-b viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [PubMed]
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type c retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous t-cell lymphoma. Proc. Natl. Acad. Sci. USA 1980, 77, 7415–7419. [Google Scholar] [CrossRef] [PubMed]
- Rickinson, A.B.; Kieff, E. Epstein-barr virus. In Fields Virology; Lippincott-Raven Publishers: Philadelphia, PA, USA, 1996; Volume 2, pp. 2397–2446. [Google Scholar]
- Henle, G.; Henle, W.; Clifford, P.; Diehl, V.; Kafuko, G.W.; Kirya, B.G.; Klein, G.; Morrow, R.H.; Munube, G.M.; Pike, P.; et al. Antibodies to epstein-barr virus in burkittʼs lymphoma and control groups. J. Natl. Cancer Inst. 1969, 43, 1147–1157. [Google Scholar]
- Gerber, P.; Walsh, J.H.; Rosenblum, E.N.; Purcell, R.H. Association of eb-virus infection with the post-perfusion syndrome. Lancet 1969, 1, 593–595. [Google Scholar] [PubMed]
- Hoagland, R.J. The transmission of infectious mononucleosis. Am. J. Med. Sci. 1955, 229, 262–272. [Google Scholar] [PubMed]
- Goldberg, G.N.; Fulginiti, V.A.; Ray, C.G.; Ferry, P.; Jones, J.F.; Cross, H.; Minnich, L. In utero epstein-barr virus (infectious mononucleosis) infection. JAMA: J. Am. Med. Assoc. 1981, 246, 1579–1581. [Google Scholar] [CrossRef]
- Meyohas, M.C.; Marechal, V.; Desire, N.; Bouillie, J.; Frottier, J.; Nicolas, J.C. Study of mother-to-child epstein-barr virus transmission by means of nested pcrs. J. Virol. 1996, 70, 6816–6819. [Google Scholar]
- Stock, I. Infectious mononucleosis—A “childhood disease” of great medical concern. Med. Mon. Pharma. 2013, 36, 364–368. [Google Scholar]
- Hjalgrim, H. On the aetiology of hodgkin lymphoma. Dan. Med. J. 2012, 59, B4485. [Google Scholar] [PubMed]
- Klein, G.; Klein, E.; Kashuba, E. Interaction of epstein-barr virus (ebv) with human b-lymphocytes. Biochem. Biophys. Res. Commun. 2010, 396, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.G.; Niederman, J.C.; Miller, G.; Smith, H.W.; Dowaliby, J.M. Site of epstein-barr virus replication in the oropharynx. Lancet 1979, 2, 1154–1157. [Google Scholar] [PubMed]
- Shair, K.H.; Schnegg, C.I.; Raab-Traub, N. Ebv latent membrane protein 1 effects on plakoglobin, cell growth, and migration. Cancer Res. 2008, 68, 6997–7005. [Google Scholar] [PubMed]
- Caldwell, R.G.; Wilson, J.B.; Anderson, S.J.; Longnecker, R. Epstein-barr virus lmp2a drives b cell development and survival in the absence of normal b cell receptor signals. Immunity 1998, 9, 405–411. [Google Scholar] [PubMed]
- Scholle, F.; Bendt, K.M.; Raab-Traub, N. Epstein-barr virus lmp2a transforms epithelial cells, inhibits cell differentiation, and activates akt. J. Virol. 2000, 74, 10681–10689. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.L.; Warren, N.; Sugden, B. Stable replication of plasmids derived from epstein-barr virus in various mammalian cells. Nature 1985, 313, 812–815. [Google Scholar] [PubMed]
- Dukers, N.H.; Rezza, G. Human herpesvirus 8 epidemiology: What we do and do not know. Aids 2003, 17, 1717–1730. [Google Scholar] [PubMed]
- Cai, Q.; Verma, S.C.; Lu, J.; Robertson, E.S. Molecular biology of kaposi’s sarcoma-associated herpesvirus and related oncogenesis. Adv. Virus Res. 2010, 78, 87–142. [Google Scholar] [PubMed]
- Viejo-Borbolla, A.; Ottinger, M.; Schulz, T.F. Human herpesvirus 8: Biology and role in the pathogenesis of kaposi’s sarcoma and other aids-related malignancies. Curr. HIV/AIDS Rep. 2004, 1, 5–11. [Google Scholar] [PubMed]
- Beral, V.; Peterman, T.A.; Berkelman, R.L.; Jaffe, H.W. Kaposi’s sarcoma among persons with aids: A sexually transmitted infection? Lancet 1990, 335, 123–128. [Google Scholar] [CrossRef]
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of kaposi sarcoma-associated herpesvirus targets the retinoblastoma-e2f pathway and with the oncogene hras transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127. [Google Scholar] [PubMed]
- Chugh, P.; Matta, H.; Schamus, S.; Zachariah, S.; Kumar, A.; Richardson, J.A.; Smith, A.L.; Chaudhary, P.M. Constitutive nf-kappab activation, normal fas-induced apoptosis, and increased incidence of lymphoma in human herpes virus 8 k13 transgenic mice. Proc. Natl. Acad. Sci. USA 2005, 102, 12885–12890. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zachariah, S.; Chaudhary, P.M. The human herpes virus 8-encoded viral flice-inhibitory protein induces cellular transformation via nf-kappab activation. J. Biol. Chem. 2003, 278, 52437–52445. [Google Scholar] [PubMed]
- Wang, L.; Dittmer, D.P.; Tomlinson, C.C.; Fakhari, F.D.; Damania, B. Immortalization of primary endothelial cells by the k1 protein of kaposi’s sarcoma-associated herpesvirus. Cancer Res. 2006, 66, 3658–3666. [Google Scholar] [PubMed]
- Staskus, K.A.; Zhong, W.; Gebhard, K.; Herndier, B.; Wang, H.; Renne, R.; Beneke, J.; Pudney, J.; Anderson, D.J.; Ganem, D.; et al. Kaposi’s sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 1997, 71, 715–719. [Google Scholar] [PubMed]
- Xie, J.; Pan, H.; Yoo, S.; Gao, S.J. Kaposi’s sarcoma-associated herpesvirus induction of ap-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J. Virol. 2005, 79, 15027–15037. [Google Scholar] [CrossRef] [PubMed]
- Naranatt, P.P.; Krishnan, H.H.; Svojanovsky, S.R.; Bloomer, C.; Mathur, S.; Chandran, B. Host gene induction and transcriptional reprogramming in kaposi’s sarcoma-associated herpesvirus (kshv/hhv-8)-infected endothelial, fibroblast, and b cells: Insights into modulation events early during infection. Cancer Res. 2004, 64, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Masood, R.; Cesarman, E.; Smith, D.L.; Gill, P.S.; Flore, O. Human herpesvirus-8-transformed endothelial cells have functionally activated vascular endothelial growth factor/vascular endothelial growth factor receptor. Am. J. Pathol. 2002, 160, 23–29. [Google Scholar] [PubMed]
- Cerimele, F.; Curreli, F.; Ely, S.; Friedman-Kien, A.E.; Cesarman, E.; Flore, O. Kaposi’s sarcoma-associated herpesvirus can productively infect primary human keratinocytes and alter their growth properties. J. Virol. 2001, 75, 2435–2443. [Google Scholar] [PubMed]
- Hong, G.K.; Gulley, M.L.; Feng, W.H.; Delecluse, H.J.; Holley-Guthrie, E.; Kenney, S.C. Epstein-barr virus lytic infection contributes to lymphoproliferative disease in a scid mouse model. J. Virol. 2005, 79, 13993–14003. [Google Scholar] [CrossRef] [PubMed]
- Martel-Renoir, D.; Grunewald, V.; Touitou, R.; Schwaab, G.; Joab, I. Qualitative analysis of the expression of epstein-barr virus lytic genes in nasopharyngeal carcinoma biopsies. J. Gen. Virol. 1995, 76, 1401–1408. [Google Scholar] [PubMed]
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. International Agency for Research on Cancer Multicenter Cervical Cancer Study, G. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Clifford, G.M.; Smith, J.S.; Plummer, M.; Munoz, N.; Franceschi, S. Human papillomavirus types in invasive cervical cancer worldwide: A meta-analysis. Br. J. Cancer 2003, 88, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.T.; Broker, T.R. Human papillomavirus infections: Warts or cancer? Cold Spring Harbor Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Woodman, C.B.; Collins, S.I.; Young, L.S. The natural history of cervical hpv infection: Unresolved issues. Nat. Rev. Cancer 2007, 7, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses—To vaccination and beyond. Biochem. Biokhimiia 2008, 73, 498–503. [Google Scholar] [CrossRef]
- Ghittoni, R.; Accardi, R.; Hasan, U.; Gheit, T.; Sylla, B.; Tommasino, M. The biological properties of e6 and e7 oncoproteins from human papillomaviruses. Virus Genes 2010, 40, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Houben, R.; Ugurel, S.; Trefzer, U.; Pfohler, C.; Schrama, D. Mc polyomavirus is frequently present in merkel cell carcinoma of european patients. J. Investig. Dermatol. 2009, 129, 248–250. [Google Scholar] [CrossRef]
- Carter, J.J.; Paulson, K.G.; Wipf, G.C.; Miranda, D.; Madeleine, M.M.; Johnson, L.G.; Lemos, B.D.; Lee, S.; Warcola, A.H.; Iyer, J.G.; et al. Association of merkel cell polyomavirus-specific antibodies with merkel cell carcinoma. J. Natl. Cancer Inst. 2009, 101, 1510–1522. [Google Scholar] [CrossRef] [PubMed]
- Duncavage, E.J.; Zehnbauer, B.A.; Pfeifer, J.D. Prevalence of merkel cell polyomavirus in merkel cell carcinoma. Mod. Pathol. 2009, 22, 516–521. [Google Scholar] [PubMed]
- Agelli, M.; Clegg, L.X. Epidemiology of primary merkel cell carcinoma in the United States. J. Am. Acad. Dermatol. 2003, 49, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, N.C. Merkel cell carcinoma: Changing incidence trends. J. Surg. Oncol. 2005, 89, 1–4. [Google Scholar] [PubMed]
- Kassem, A.; Schopflin, A.; Diaz, C.; Weyers, W.; Stickeler, E.; Werner, M.; Zur Hausen, A. Frequent detection of merkel cell polyomavirus in human merkel cell carcinomas and identification of a unique deletion in the vp1 gene. Cancer Res. 2008, 68, 5009–5013. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Guastafierro, A.; Shuda, M.; Meinke, G.; Bohm, A.; Moore, P.S.; Chang, Y. The minimum replication origin of merkel cell polyomavirus has a unique large t-antigen loading architecture and requires small t-antigen expression for optimal replication. J. Virol. 2009, 83, 12118–12128. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; van Ghelue, M.; Johannessen, M. Oncogenic potentials of the human polyomavirus regulatory proteins. Cell. Mol. Life Sci. 2007, 64, 1656–1678. [Google Scholar] [PubMed]
- Shuda, M.; Arora, R.; Kwun, H.J.; Feng, H.; Sarid, R.; Fernandez-Figueras, M.T.; Tolstov, Y.; Gjoerup, O.; Mansukhani, M.M.; Swerdlow, S.H.; et al. Human merkel cell polyomavirus infection i. Mcv t antigen expression in merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int. J. Cancer 2009, 125, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [PubMed]
- Houben, R.; Adam, C.; Baeurle, A.; Hesbacher, S.; Grimm, J.; Angermeyer, S.; Henzel, K.; Hauser, S.; Elling, R.; Brocker, E.B.; et al. An intact retinoblastoma protein-binding site in merkel cell polyomavirus large t antigen is required for promoting growth of merkel cell carcinoma cells. Int. J. Cancer 2012, 130, 847–856. [Google Scholar] [PubMed]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel cell polyomavirus-infected merkel cell carcinoma cells require expression of viral t antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human merkel cell polyomavirus small t antigen is an oncoprotein targeting the 4e-bp1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [PubMed]
- Kew, M.C. Epidemiology of chronic hepatitis b virus infection, hepatocellular carcinoma, and hepatitis b virus-induced hepatocellular carcinoma. Pathol.-Biol. 2010, 58, 273–277. [Google Scholar] [CrossRef]
- Tarocchi, M.; Polvani, S.; Marroncini, G.; Galli, A. Molecular mechanism of hepatitis b virus-induced hepatocarcinogenesis. World J. Gastroenterol. 2014, 20, 11630–11640. [Google Scholar] [PubMed]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent hbv integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Golubkov, V.S.; Strongin, A.Y.; Jiang, W.; Reed, J.C. Interaction of hepatitis b viral oncoprotein with cellular target hbxip dysregulates centrosome dynamics and mitotic spindle formation. J. Biol. Chem. 2008, 283, 2793–2803. [Google Scholar] [PubMed]
- Kim, C.M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. Hbx gene of hepatitis b virus induces liver cancer in transgenic mice. Nature 1991, 351, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Bukh, J.; Combet, C.; Deleage, G.; Enomoto, N.; Feinstone, S.; Halfon, P.; Inchauspe, G.; Kuiken, C.; Maertens, G.; et al. Consensus proposals for a unified system of nomenclature of hepatitis c virus genotypes. Hepatology 2005, 42, 962–973. [Google Scholar] [PubMed]
- Tang, H.; Grise, H. Cellular and molecular biology of hcv infection and hepatitis. Clin. Sci. 2009, 117, 49–65. [Google Scholar] [PubMed]
- Morgan, R.L.; Baack, B.; Smith, B.D.; Yartel, A.; Pitasi, M.; Falck-Ytter, Y. Eradication of hepatitis c virus infection and the development of hepatocellular carcinoma: A meta-analysis of observational studies. Ann. Intern. Med. 2013, 158, 329–337. [Google Scholar] [PubMed]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis c virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [PubMed]
- Moriya, K.; Yotsuyanagi, H.; Shintani, Y.; Fujie, H.; Ishibashi, K.; Matsuura, Y.; Miyamura, T.; Koike, K. Hepatitis c virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 1997, 78 Pt 7, 1527–1531. [Google Scholar] [PubMed]
- Pei, Y.; Zhang, T.; Renault, V.; Zhang, X. An overview of hepatocellular carcinoma study by omics-based methods. Acta Biochim. Biophys. Sin. 2009, 41, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Nunez, O.; Fernandez-Martinez, A.; Majano, P.L.; Apolinario, A.; Gomez-Gonzalo, M.; Benedicto, I.; Lopez-Cabrera, M.; Bosca, L.; Clemente, G.; Garcia-Monzon, C.; et al. Increased intrahepatic cyclooxygenase 2, matrix metalloproteinase 2, and matrix metalloproteinase 9 expression is associated with progressive liver disease in chronic hepatitis c virus infection: Role of viral core and ns5a proteins. Gut 2004, 53, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A.; Ohigashi, H. Targeting nox, inos and cox-2 in inflammatory cells: Chemoprevention using food phytochemicals. Int. J. Cancer 2007, 121, 2357–2363. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, H.; Tazawa, H.; Sylla, B.S.; Sawa, T. Prevention of human cancer by modulation of chronic inflammatory processes. Mutat. Res. 2005, 591, 110–122. [Google Scholar] [PubMed]
- Fujinaga, H.; Tsutsumi, T.; Yotsuyanagi, H.; Moriya, K.; Koike, K. Hepatocarcinogenesis in hepatitis c: Hcv shrewdly exacerbates oxidative stress by modulating both production and scavenging of reactive oxygen species. Oncology 2011, 81 (Suppl. S1), 11–17. [Google Scholar]
- Kannian, P.; Green, P.L. Human t lymphotropic virus type 1 (htlv-1): Molecular biology and oncogenesis. Viruses 2010, 2, 2037–2077. [Google Scholar] [CrossRef] [PubMed]
- Proietti, F.A.; Carneiro-Proietti, A.B.; Catalan-Soares, B.C.; Murphy, E.L. Global epidemiology of htlv-i infection and associated diseases. Oncogene 2005, 24, 6058–6068. [Google Scholar] [PubMed]
- Grassmann, R.; Dengler, C.; Muller-Fleckenstein, I.; Fleckenstein, B.; McGuire, K.; Dokhelar, M.C.; Sodroski, J.G.; Haseltine, W.A. Transformation to continuous growth of primary human t lymphocytes by human t-cell leukemia virus type i x-region genes transduced by a herpesvirus saimiri vector. Proc. Natl. Acad. Sci. USA 1989, 86, 3351–3355. [Google Scholar] [PubMed]
- Grassmann, R.; Berchtold, S.; Radant, I.; Alt, M.; Fleckenstein, B.; Sodroski, J.G.; Haseltine, W.A.; Ramstedt, U. Role of human t-cell leukemia virus type 1 x region proteins in immortalization of primary human lymphocytes in culture. J. Virol. 1992, 66, 4570–4575. [Google Scholar] [PubMed]
- Akagi, T.; Shimotohno, K. Proliferative response of tax1-transduced primary human t cells to anti-cd3 antibody stimulation by an interleukin-2-independent pathway. J. Virol. 1993, 67, 1211–1217. [Google Scholar] [PubMed]
- Hasegawa, H.; Sawa, H.; Lewis, M.J.; Orba, Y.; Sheehy, N.; Yamamoto, Y.; Ichinohe, T.; Tsunetsugu-Yokota, Y.; Katano, H.; Takahashi, H.; et al. Thymus-derived leukemia-lymphoma in mice transgenic for the tax gene of human t-lymphotropic virus type i. Nat. Med. 2006, 12, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Benvenisty, N.; Ornitz, D.M.; Bennett, G.L.; Sahagan, B.G.; Kuo, A.; Cardiff, R.D.; Leder, P. Brain tumours and lymphomas in transgenic mice that carry htlv-i ltr/c-myc and ig/tax genes. Oncogene 1992, 7, 2399–2405. [Google Scholar] [PubMed]
- Azran, I.; Schavinsky-Khrapunsky, Y.; Aboud, M. Role of tax protein in human t-cell leukemia virus type-i leukemogenicity. Retrovirology 2004, 1, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.H.; Gaynor, R.B. Mechanisms of nf-kappab activation by the htlv type 1 tax protein. AIDS Res. Hum. Retrovir. 2000, 16, 1583–1590. [Google Scholar] [PubMed]
- Harhaj, E.W.; Harhaj, N.S.; Grant, C.; Mostoller, K.; Alefantis, T.; Sun, S.C.; Wigdahl, B. Human t cell leukemia virus type i tax activates cd40 gene expression via the nf-kappa b pathway. Virology 2005, 333, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Fujii, M.; Cheng, G.; Ikeda, S.; Yamasaki, Y.; Yamada, Y.; Tomonaga, M.; Yamamoto, N. Human t-cell leukemia virus type i tax protein induces the expression of anti-apoptotic gene bcl-xl in human t-cells through nuclear factor-kappab and c-amp responsive element binding protein pathways. Virus Genes 2001, 22, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. P53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Momand, J.; Finlay, C.A. The p53 tumour suppressor gene. Nature 1991, 351, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Khidr, L.; Chen, P.L. Rb, the conductor that orchestrates life, death and differentiation. Oncogene 2006, 25, 5210–5219. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Bernstein, A. Apoptosis, cancer and the p53 tumour suppressor gene. Cancer Metastasis Rev. 1995, 14, 149–161. [Google Scholar] [PubMed]
- Amundson, S.A.; Myers, T.G.; Fornace, A.J., Jr. Roles for p53 in growth arrest and apoptosis: Putting on the brakes after genotoxic stress. Oncogene 1998, 17, 3287–3299. [Google Scholar] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The e6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The hpv-16 e6 and e6-ap complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Reznikoff, C.A.; Yeager, T.R.; Belair, C.D.; Savelieva, E.; Puthenveettil, J.A.; Stadler, W.M. Elevated p16 at senescence and loss of p16 at immortalization in human papillomavirus 16 e6, but not e7, transformed human uroepithelial cells. Cancer Res. 1996, 56, 2886–2890. [Google Scholar] [PubMed]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 e7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [PubMed]
- Giam, C.Z.; Jeang, K.T. Htlv-1 tax and adult t-cell leukemia. Front. Biosci.: J. Virtual Libr. 2007, 12, 1496–1507. [Google Scholar] [CrossRef]
- Pise-Masison, C.A.; Brady, J.N. Setting the stage for transformation: Htlv-1 tax inhibition of p53 function. Front. Biosci. 2005, 10, 919–930. [Google Scholar] [PubMed]
- Haller, K.; Wu, Y.; Derow, E.; Schmitt, I.; Jeang, K.T.; Grassmann, R. Physical interaction of human t-cell leukemia virus type 1 tax with cyclin-dependent kinase 4 stimulates the phosphorylation of retinoblastoma protein. Mol. Cell. Biol. 2002, 22, 3327–3338. [Google Scholar] [PubMed]
- Suzuki, T.; Narita, T.; Uchida-Toita, M.; Yoshida, M. Down-regulation of the ink4 family of cyclin-dependent kinase inhibitors by tax protein of htlv-1 through two distinct mechanisms. Virology 1999, 259, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kitao, S.; Matsushime, H.; Yoshida, M. Htlv-1 tax protein interacts with cyclin-dependent kinase inhibitor p16ink4a and counteracts its inhibitory activity towards cdk4. EMBO J. 1996, 15, 1607–1614. [Google Scholar] [PubMed]
- Satou, Y.; Yasunaga, J.; Yoshida, M.; Matsuoka, M. Htlv-i basic leucine zipper factor gene mrna supports proliferation of adult t cell leukemia cells. Proc. Natl. Acad. Sci. USA 2006, 103, 720–725. [Google Scholar] [PubMed]
- Cai, Q.L.; Knight, J.S.; Verma, S.C.; Zald, P.; Robertson, E.S. Ec5s ubiquitin complex is recruited by kshv latent antigen lana for degradation of the vhl and p53 tumor suppressors. PLoS Pathog. 2006, 2, e116. [Google Scholar] [PubMed]
- Friborg, J., Jr.; Kong, W.; Hottiger, M.O.; Nabel, G.J. P53 inhibition by the lana protein of kshv protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [PubMed]
- Si, H.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J. Virol. 2006, 80, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Spender, L.C.; Cannell, E.J.; Hollyoake, M.; Wensing, B.; Gawn, J.M.; Brimmell, M.; Packham, G.; Farrell, P.J. Control of cell cycle entry and apoptosis in b lymphocytes infected by epstein-barr virus. J. Virol. 1999, 73, 4678–4688. [Google Scholar] [PubMed]
- Mei, Y.P.; Zhou, J.M.; Wang, Y.; Huang, H.; Deng, R.; Feng, G.K.; Zeng, Y.X.; Zhu, X.F. Silencing of lmp1 induces cell cycle arrest and enhances chemosensitivity through inhibition of akt signaling pathway in ebv-positive nasopharyngeal carcinoma cells. Cell Cycle 2007, 6, 1379–1385. [Google Scholar]
- Portis, T.; Longnecker, R. Epstein-barr virus (ebv) lmp2a mediates b-lymphocyte survival through constitutive activation of the ras/pi3k/akt pathway. Oncogene 2004, 23, 8619–8628. [Google Scholar] [PubMed]
- Desbien, A.L.; Kappler, J.W.; Marrack, P. The epstein-barr virus bcl-2 homolog, bhrf1, blocks apoptosis by binding to a limited amount of bim. Proc. Natl. Acad. Sci. USA 2009, 106, 5663–5668. [Google Scholar] [CrossRef] [PubMed]
- Saggioro, D.; Silic-Benussi, M.; Biasiotto, R.; D’Agostino, D.M.; Ciminale, V. Control of cell death pathways by htlv-1 proteins. Front. Biosci. 2009, 14, 3338–3351. [Google Scholar]
- Underbrink, M.P.; Howie, H.L.; Bedard, K.M.; Koop, J.I.; Galloway, D.A. E6 proteins from multiple human betapapillomavirus types degrade bak and protect keratinocytes from apoptosis after uvb irradiation. J. Virol. 2008, 82, 10408–10417. [Google Scholar]
- Garnett, T.O.; Filippova, M.; Duerksen-Hughes, P.J. Accelerated degradation of fadd and procaspase 8 in cells expressing human papilloma virus 16 e6 impairs trail-mediated apoptosis. Cell Death Differ. 2006, 13, 1915–1926. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Fu, F.; Zhuo, J.; Wang, W.; Nishitani, J.; An, D.S.; Chen, I.S.; Liu, X. Human papillomavirus type 16 e6 and e7 oncoproteins upregulate c-iap2 gene expression and confer resistance to apoptosis. Oncogene 2005, 24, 5069–5078. [Google Scholar] [CrossRef]
- Filippova, M.; Parkhurst, L.; Duerksen-Hughes, P.J. The human papillomavirus 16 e6 protein binds to fas-associated death domain and protects cells from fas-triggered apoptosis. J. Biol. Chem. 2004, 279, 25729–25744. [Google Scholar] [PubMed]
- Du, J.; Chen, G.G.; Vlantis, A.C.; Chan, P.K.; Tsang, R.K.; van Hasselt, C.A. Resistance to apoptosis of hpv 16-infected laryngeal cancer cells is associated with decreased bak and increased bcl-2 expression. Cancer Lett. 2004, 205, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.A.; Lee, T.H.; Butel, J.S.; Slagle, B.L. Hepatitis b virus x protein interferes with cellular DNA repair. J. Virol. 1998, 72, 266–272. [Google Scholar] [PubMed]
- Aweya, J.J.; Tan, Y.J. Modulation of programmed cell death pathways by the hepatitis c virus. Front. Biosci. 2011, 16, 608–618. [Google Scholar] [CrossRef] [Green Version]
- Plug-DeMaggio, A.W.; Sundsvold, T.; Wurscher, M.A.; Koop, J.I.; Klingelhutz, A.J.; McDougall, J.K. Telomere erosion and chromosomal instability in cells expressing the hpv oncogene 16e6. Oncogene 2004, 23, 3561–3571. [Google Scholar] [PubMed]
- Duensing, S.; Munger, K. The human papillomavirus type 16 e6 and e7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002, 62, 7075–7082. [Google Scholar] [PubMed]
- Duensing, S.; Duensing, A.; Crum, C.P.; Munger, K. Human papillomavirus type 16 e7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. 2001, 61, 2356–2360. [Google Scholar] [PubMed]
- Duensing, S.; Lee, L.Y.; Duensing, A.; Basile, J.; Piboonniyom, S.; Gonzalez, S.; Crum, C.P.; Munger, K. The human papillomavirus type 16 e6 and e7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc. Natl. Acad. Sci. USA 2000, 97, 10002–10007. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J. Genomic instability induced by human papillomavirus oncogenes. North Am. J. Med. Sci. 2010, 3, 43–47. [Google Scholar]
- Bornkamm, G.W. Epstein-barr virus and its role in the pathogenesis of burkitt’s lymphoma: An unresolved issue. Semin. Cancer Biol. 2009, 19, 351–365. [Google Scholar] [PubMed]
- Heath, E.; Begue-Pastor, N.; Chaganti, S.; Croom-Carter, D.; Shannon-Lowe, C.; Kube, D.; Feederle, R.; Delecluse, H.J.; Rickinson, A.B.; Bell, A.I. Epstein-barr virus infection of naive b cells in vitro frequently selects clones with mutated immunoglobulin genotypes: Implications for virus biology. PLoS Pathog. 2012, 8, e1002697. [Google Scholar] [CrossRef] [PubMed]
- Marriott, S.J.; Lemoine, F.J.; Jeang, K.T. Damaged DNA and miscounted chromosomes: Human t cell leukemia virus type i tax oncoprotein and genetic lesions in transformed cells. J. Biomed. Sci. 2002, 9, 292–298. [Google Scholar] [PubMed]
- Lemoine, F.J.; Marriott, S.J. Genomic instability driven by the human t-cell leukemia virus type i (htlv-i) oncoprotein, tax. Oncogene 2002, 21, 7230–7234. [Google Scholar] [PubMed]
- Chandhasin, C.; Ducu, R.I.; Berkovich, E.; Kastan, M.B.; Marriott, S.J. Human t-cell leukemia virus type 1 tax attenuates the atm-mediated cellular DNA damage response. J. Virol. 2008, 82, 6952–6961. [Google Scholar] [CrossRef] [PubMed]
- Kao, S.Y.; Lemoine, F.J.; Marriott, S.J. P53-independent induction of apoptosis by the htlv-i tax protein following uv irradiation. Virology 2001, 291, 292–298. [Google Scholar] [PubMed]
- Jin, D.Y.; Spencer, F.; Jeang, K.T. Human t cell leukemia virus type 1 oncoprotein tax targets the human mitotic checkpoint protein mad1. Cell 1998, 93, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Hong, S.; Tang, Z.; Yu, H.; Giam, C.Z. Htlv-i tax directly binds the cdc20-associated anaphase-promoting complex and activates it ahead of schedule. Proc. Natl. Acad. Sci. USA 2005, 102, 63–68. [Google Scholar] [PubMed]
- Cohen, S.B.; Graham, M.E.; Lovrecz, G.O.; Bache, N.; Robinson, P.J.; Reddel, R.R. Protein composition of catalytically active human telomerase from immortal cells. Science 2007, 315, 1850–1853. [Google Scholar] [PubMed]
- Terrin, L.; dal Col, J.; Rampazzo, E.; Zancai, P.; Pedrotti, M.; Ammirabile, G.; Bergamin, S.; Rizzo, S.; Dolcetti, R.; de Rossi, A. Latent membrane protein 1 of epstein-barr virus activates the htert promoter and enhances telomerase activity in b lymphocytes. J. Virol. 2008, 82, 10175–10187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, N.; Zhang, H.; You, J.; Wang, H.; Ye, L. Effects of hepatitis b virus x protein on human telomerase reverse transcriptase expression and activity in hepatoma cells. J. Lab. Clin. Med. 2005, 145, 98–104. [Google Scholar] [PubMed]
- Verma, S.C.; Borah, S.; Robertson, E.S. Latency-associated nuclear antigen of kaposi’s sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor sp1. J. Virol. 2004, 78, 10348–10359. [Google Scholar] [PubMed]
- Gewin, L.; Myers, H.; Kiyono, T.; Galloway, D.A. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 e6/e6-ap complex. Genes Dev. 2004, 18, 2269–2282. [Google Scholar] [PubMed]
- Chen, X.; Kamranvar, S.A.; Masucci, M.G. Tumor viruses and replicative immortality—Avoiding the telomere hurdle. Semin. Cancer Biol. 2014, 26, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Sakurai, M.; Higuchi, M.; Mori, N.; Fukushi, M.; Oie, M.; Coffey, R.J.; Yoshiura, K.; Tanaka, Y.; Uchiyama, M.; et al. Human t-cell leukemia virus type 1 tax oncoprotein induces and interacts with a multi-pdz domain protein, magi-3. Virology 2004, 320, 52–62. [Google Scholar] [PubMed]
- Glaunsinger, B.A.; Lee, S.S.; Thomas, M.; Banks, L.; Javier, R. Interactions of the pdz-protein magi-1 with adenovirus e4-orf1 and high-risk papillomavirus e6 oncoproteins. Oncogene 2000, 19, 5270–5280. [Google Scholar] [CrossRef] [PubMed]
- Rousset, R.; Fabre, S.; Desbois, C.; Bantignies, F.; Jalinot, P. The c-terminus of the htlv-1 tax oncoprotein mediates interaction with the pdz domain of cellular proteins. Oncogene 1998, 16, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Weiss, R.S.; Javier, R.T. Binding of human virus oncoproteins to hdlg/sap97, a mammalian homolog of the drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 6670–6675. [Google Scholar] [PubMed]
- Spanos, W.C.; Hoover, A.; Harris, G.F.; Wu, S.; Strand, G.L.; Anderson, M.E.; Klingelhutz, A.J.; Hendriks, W.; Bossler, A.D.; Lee, J.H. The pdz binding motif of human papillomavirus type 16 e6 induces ptpn13 loss, which allows anchorage-independent growth and synergizes with ras for invasive growth. J. Virol. 2008, 82, 2493–2500. [Google Scholar] [PubMed]
- Spanos, W.C.; Geiger, J.; Anderson, M.E.; Harris, G.F.; Bossler, A.D.; Smith, R.B.; Klingelhutz, A.J.; Lee, J.H. Deletion of the pdz motif of hpv16 e6 preventing immortalization and anchorage-independent growth in human tonsil epithelial cells. Head Neck 2008, 30, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Yamamoto, B.; Haoudi, A.; Semmes, O.J.; Green, P.L. Pdz binding motif of htlv-1 tax promotes virus-mediated t-cell proliferation in vitro and persistence in vivo. Blood 2006, 107, 1980–1988. [Google Scholar] [PubMed]
- Hirata, A.; Higuchi, M.; Niinuma, A.; Ohashi, M.; Fukushi, M.; Oie, M.; Akiyama, T.; Tanaka, Y.; Gejyo, F.; Fujii, M. Pdz domain-binding motif of human t-cell leukemia virus type 1 tax oncoprotein augments the transforming activity in a rat fibroblast cell line. Virology 2004, 318, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microrna targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. Microrna signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. Microrna expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Schafer, A.; Lu, S.; Bilello, J.P.; Desrosiers, R.C.; Edwards, R.; Raab-Traub, N.; Cullen, B.R. Epstein-barr virus micrornas are evolutionarily conserved and differentially expressed. PLoS Pathog. 2006, 2, e23. [Google Scholar]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of virus-encoded micrornas. Science 2004, 304, 734–736. [Google Scholar]
- Choy, E.Y.; Siu, K.L.; Kok, K.H.; Lung, R.W.; Tsang, C.M.; To, K.F.; Kwong, D.L.; Tsao, S.W.; Jin, D.Y. An epstein-barr virus-encoded microrna targets puma to promote host cell survival. J. Exp. Med. 2008, 205, 2551–2560. [Google Scholar] [PubMed]
- Dolken, L.; Malterer, G.; Erhard, F.; Kothe, S.; Friedel, C.C.; Suffert, G.; Marcinowski, L.; Motsch, N.; Barth, S.; Beitzinger, M.; et al. Systematic analysis of viral and cellular microrna targets in cells latently infected with human gamma-herpesviruses by risc immunoprecipitation assay. Cell Host Microbe 2010, 7, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Marquitz, A.R.; Mathur, A.; Nam, C.S.; Raab-Traub, N. The epstein-barr virus bart micrornas target the pro-apoptotic protein bim. Virology 2011, 412, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Marquitz, A.R.; Raab-Traub, N. The role of mirnas and ebv barts in npc. Semin. Cancer Biol. 2012, 22, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Marquitz, A.R.; Mathur, A.; Shair, K.H.; Raab-Traub, N. Infection of epstein-barr virus in a gastric carcinoma cell line induces anchorage independence and global changes in gene expression. Proc. Natl. Acad. Sci. USA 2012, 109, 9593–9598. [Google Scholar] [PubMed]
- Blattner, W.A. Human retroviruses: Their role in cancer. Proc. Assoc. Am. Phys. 1999, 111, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.H.; van den Heuvel, A.P.; Schmidt, J.W.; Ross, S.R. Novel common integration sites targeted by mouse mammary tumor virus insertion in mammary tumors have oncogenic activity. PLoS One 2011, 6, e27425. [Google Scholar] [CrossRef] [PubMed]
- Sourvinos, G.; Tsatsanis, C.; Spandidos, D.A. Mechanisms of retrovirus-induced oncogenesis. Folia Biol. 2000, 46, 226–232. [Google Scholar]
- Chimal-Ramirez, G.K.; Espinoza-Sanchez, N.A.; Fuentes-Panana, E.M. Protumor activities of the immune response: Insights in the mechanisms of immunological shift, oncotraining, and oncopromotion. J. Oncol. 2013, 2013, 835956. [Google Scholar] [PubMed]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Schistosomes, Liver Flukes and Helicobacter Pylori. Iarc working group on the evaluation of carcinogenic risks to humans. Lyon, 7–14 June 1994. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 61, 1–241.
- Fuentes-Panana, E.; Camorlinga-Ponce, M.; Maldonado-Bernal, C. Infection, inflammation and gastric cancer. Salud Publica Mex. 2009, 51, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Kusters, J.G.; van Vliet, A.H.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yin, Z.; Cao, S.; Gao, W.; Liu, L.; Yin, Y.; Liu, P.; Shu, Y. Systematic review and meta-analysis on the association between il-1b polymorphisms and cancer risk. PLoS One 2013, 8, e63654. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Lin, B.; Ni, P.; Xu, H.; Huang, G. Interleukin-1b and interleukin-1 rn polymorphisms and gastric carcinoma risk: A meta-analysis. J. Gastroenterol. Hepatol. 2010, 25, 1604–1617. [Google Scholar] [PubMed]
- Crusius, J.B.; Canzian, F.; Capella, G.; Pena, A.S.; Pera, G.; Sala, N.; Agudo, A.; Rico, F.; Del Giudice, G.; Palli, D.; et al. Cytokine gene polymorphisms and the risk of adenocarcinoma of the stomach in the european prospective investigation into cancer and nutrition (epic-eurgast). Ann. Oncol. 2008, 19, 1894–1902. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Hao, Y.; Zhou, W.; Ma, Y. Positive association between interleukin-8 -251a>t polymorphism and susceptibility to gastric carcinogenesis: A meta-analysis. Cancer Cell Int. 2013, 13, 100. [Google Scholar] [PubMed]
- Poli, G. Pathogenesis of liver fibrosis: Role of oxidative stress. Mol. Asp. Med. 2000, 21, 49–98. [Google Scholar]
- Cardenas-Mondragon, M.G.; Carreon-Talavera, R.; Camorlinga-Ponce, M.; Gomez-Delgado, A.; Torres, J.; Fuentes-Panana, E.M. Epstein barr virus and helicobacter pylori co-infection are positively associated with severe gastritis in pediatric patients. PLoS One 2013, 8, e62850. [Google Scholar] [PubMed]
- Cárdenas-Mondragón, M.; Carreón-Talavera, R.; Flores-Luna, L.; Camorlinga-Ponce, M.; Gómez-Delgado, A.; Torres, J.; Fuentes-Pananá, E. Epstein barr virus reactivation is an important trigger of gastric inflammation and progression to intestinal type gastric cancer. In Proceedings of the Epstein Barr Virus 50th Anniversary Conference, Keble College, Oxford, UK, 23–25 March, 2014.
- Bernstein, W.B.; Little, R.F.; Wilson, W.H.; Yarchoan, R. Acquired immunodeficiency syndrome-related malignancies in the era of highly active antiretroviral therapy. Int. J. Hematol. 2006, 84, 3–11. [Google Scholar] [PubMed]
- Young, L.S.; Murray, P.G. Epstein-barr virus and oncogenesis: From latent genes to tumours. Oncogene 2003, 22, 5108–5121. [Google Scholar] [PubMed]
- Young, L.; Alfieri, C.; Hennessy, K.; Evans, H.; O’Hara, C.; Anderson, K.C.; Ritz, J.; Shapiro, R.S.; Rickinson, A.; Kieff, E.; et al. Expression of epstein-barr virus transformation-associated genes in tissues of patients with ebv lymphoproliferative disease. N. Engl. J. Med. 1989, 321, 1080–1085. [Google Scholar] [PubMed]
- Nourse, J.P.; Jones, K.; Gandhi, M.K. Epstein-barr virus-related post-transplant lymphoproliferative disorders: Pathogenetic insights for targeted therapy. Am. J. Transplant. 2011, 11, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.K.; Wilkie, G.M.; Dua, U.; Mollee, P.N.; Grimmett, K.; Williams, T.; Whitaker, N.; Gill, D.; Crawford, D.H. Immunity, homing and efficacy of allogeneic adoptive immunotherapy for posttransplant lymphoproliferative disorders. Am. J. Transplant. 2007, 7, 1293–1299. [Google Scholar] [PubMed]
- Haque, T.; Wilkie, G.M.; Jones, M.M.; Higgins, C.D.; Urquhart, G.; Wingate, P.; Burns, D.; McAulay, K.; Turner, M.; Bellamy, C.; et al. Allogeneic cytotoxic t-cell therapy for ebv-positive posttransplantation lymphoproliferative disease: Results of a phase 2 multicenter clinical trial. Blood 2007, 110, 1123–1131. [Google Scholar] [PubMed]
- Michaelis, M.; Doerr, H.W.; Cinatl, J. The story of human cytomegalovirus and cancer: Increasing evidence and open questions. Neoplasia 2009, 11, 1–9. [Google Scholar] [PubMed]
- Boldogh, I.; Huang, E.S.; Rady, P.; Arany, I.; Tyring, S.; Albrecht, T. Alteration in the coding potential and expression of h-ras in human cytomegalovirus-transformed cells. Intervirology 1994, 37, 321–329. [Google Scholar] [PubMed]
- Geder, K.M.; Lausch, R.; O’Neill, F.; Rapp, F. Oncogenic transformation of human embryo lung cells by human cytomegalovirus. Science 1976, 192, 1134–1137. [Google Scholar] [CrossRef] [PubMed]
- Geder, L.; Kreider, J.; Rapp, F. Human cells transformed in vitro by human cytomegalovirus: Tumorigenicity in athymic nude mice. J. Natl. Cancer Inst. 1977, 58, 1003–1009. [Google Scholar] [PubMed]
- Geder, L.; Laychock, A.M.; Gorodecki, J.; Rapp, F. Alterations in biological properties of different lines of cytomegalorivus-transformed human embryo lung cells following in vitro cultivation. IARC Sci. Publ. 1978, 24, 591–601. [Google Scholar] [PubMed]
- Cinatl, J.; Scholz, M.; Kotchetkov, R.; Vogel, J.U.; Doerr, H.W. Molecular mechanisms of the modulatory effects of hcmv infection in tumor cell biology. Trends Mol. Med. 2004, 10, 19–23. [Google Scholar] [PubMed]
- Scheurer, M.E.; Bondy, M.L.; Aldape, K.D.; Albrecht, T.; El-Zein, R. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathol. 2008, 116, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Cobbs, C.S.; Soroceanu, L.; Denham, S.; Zhang, W.; Kraus, M.H. Modulation of oncogenic phenotype in human glioma cells by cytomegalovirus ie1-mediated mitogenicity. Cancer Res. 2008, 68, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Maussang, D.; Verzijl, D.; van Walsum, M.; Leurs, R.; Holl, J.; Pleskoff, O.; Michel, D.; van Dongen, G.A.; Smit, M.J. Human cytomegalovirus-encoded chemokine receptor us28 promotes tumorigenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 13068–13073. [Google Scholar] [CrossRef] [PubMed]
- Vallat, L.; Benhamou, Y.; Gutierrez, M.; Ghillani, P.; Hercher, C.; Thibault, V.; Charlotte, F.; Piette, J.C.; Poynard, T.; Merle-Beral, H.; et al. Clonal b cell populations in the blood and liver of patients with chronic hepatitis c virus infection. Arthritis Rheum. 2004, 50, 3668–3678. [Google Scholar] [PubMed]
- Quinn, E.R.; Chan, C.H.; Hadlock, K.G.; Foung, S.K.; Flint, M.; Levy, S. The b-cell receptor of a hepatitis c virus (hcv)-associated non-hodgkin lymphoma binds the viral e2 envelope protein, implicating hcv in lymphomagenesis. Blood 2001, 98, 3745–3749. [Google Scholar] [CrossRef]
- Chan, C.H.; Hadlock, K.G.; Foung, S.K.; Levy, S. V(h)1–69 gene is preferentially used by hepatitis c virus-associated b cell lymphomas and by normal b cells responding to the e2 viral antigen. Blood 2001, 97, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Hermine, O.; Lefrere, F.; Bronowicki, J.P.; Mariette, X.; Jondeau, K.; Eclache-Saudreau, V.; Delmas, B.; Valensi, F.; Cacoub, P.; Brechot, C.; et al. Regression of splenic lymphoma with villous lymphocytes after treatment of hepatitis c virus infection. N. Engl. J. Med. 2002, 347, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Zullo, A.; Hassan, C.; Cristofari, F.; Andriani, A.; de Francesco, V.; Ierardi, E.; Tomao, S.; Stolte, M.; Morini, S.; Vaira, D. Effects of helicobacter pylori eradication on early stage gastric mucosa-associated lymphoid tissue lymphoma. Clin. Gastroenterol. Hepatol. 2010, 8, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Skinner, G.R. Transformation of primary hamster embryo fibroblasts by type 2 simplex virus: Evidence for a “hit and run” mechanism. Br. J. Exp. Pathol. 1976, 57, 361–376. [Google Scholar] [PubMed]
- Birdwell, C.E.; Queen, K.J.; Kilgore, P.C.; Rollyson, P.; Trutschl, M.; Cvek, U.; Scott, R.S. Genome-wide DNA methylation as an epigenetic consequence of epstein-barr virus infection of immortalized keratinocytes. J. Virol. 2014, 88, 11442–11458. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for apobec3b mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Sánchez, A.; Fuentes-Pananá, E.M. Human Viruses and Cancer. Viruses 2014, 6, 4047-4079. https://doi.org/10.3390/v6104047
Morales-Sánchez A, Fuentes-Pananá EM. Human Viruses and Cancer. Viruses. 2014; 6(10):4047-4079. https://doi.org/10.3390/v6104047
Chicago/Turabian StyleMorales-Sánchez, Abigail, and Ezequiel M. Fuentes-Pananá. 2014. "Human Viruses and Cancer" Viruses 6, no. 10: 4047-4079. https://doi.org/10.3390/v6104047
APA StyleMorales-Sánchez, A., & Fuentes-Pananá, E. M. (2014). Human Viruses and Cancer. Viruses, 6(10), 4047-4079. https://doi.org/10.3390/v6104047