KSHV Targeted Therapy: An Update on Inhibitors of Viral Lytic Replication


Abstract
:1. Introduction
2. Management of KSHV-Associated Diseases

{kind=link}
{kind=link}
{kind=link}
| Treatment | KSHV-related Diseases | |
|---|---|---|
| Intensification of HAART | AIDS-KS | |
| Surgical excision | KS (single skin lesion) | |
| Radiotherapy | KS | |
| Immunotherapy | Reduction of immunosuppressive therapy | KS, PEL and MCD |
| Anti-CD20 (Rituximab) Anti-human IL-6 receptor (Tocilizumab) Anti-IL6 chimeric monoclonal antibody (Siltuximab) | MCD | |
| Chemotherapy | Liposomal anthracyclines | KS |
| CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) | PEL and MCD | |
| Antiviral drugs | (Val)ganciclovir, foscarnet | KS, PEL and MCD |
| Intracavity cidofovir | PEL | |
| Others | mTOR inhibitor (Rapamycin) Proteasome inhibitor (Bortezomib) | KS, PEL PEL |
| Paclitaxel, anti-angiogenic agents, matrix metalloproteinase inhibitors | KS |
4. Inhibitors of KSHV Lytic Replication under Investigation
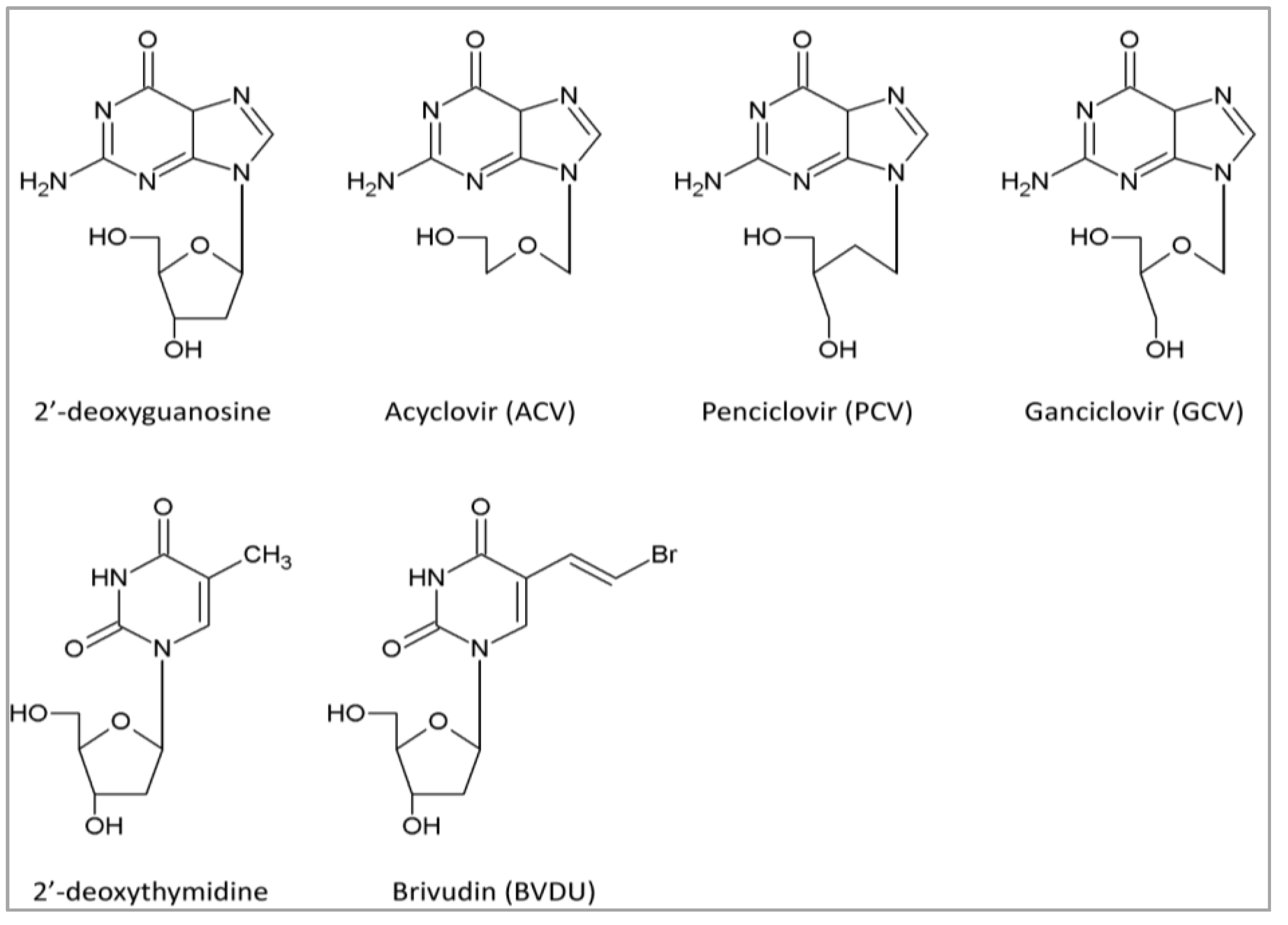
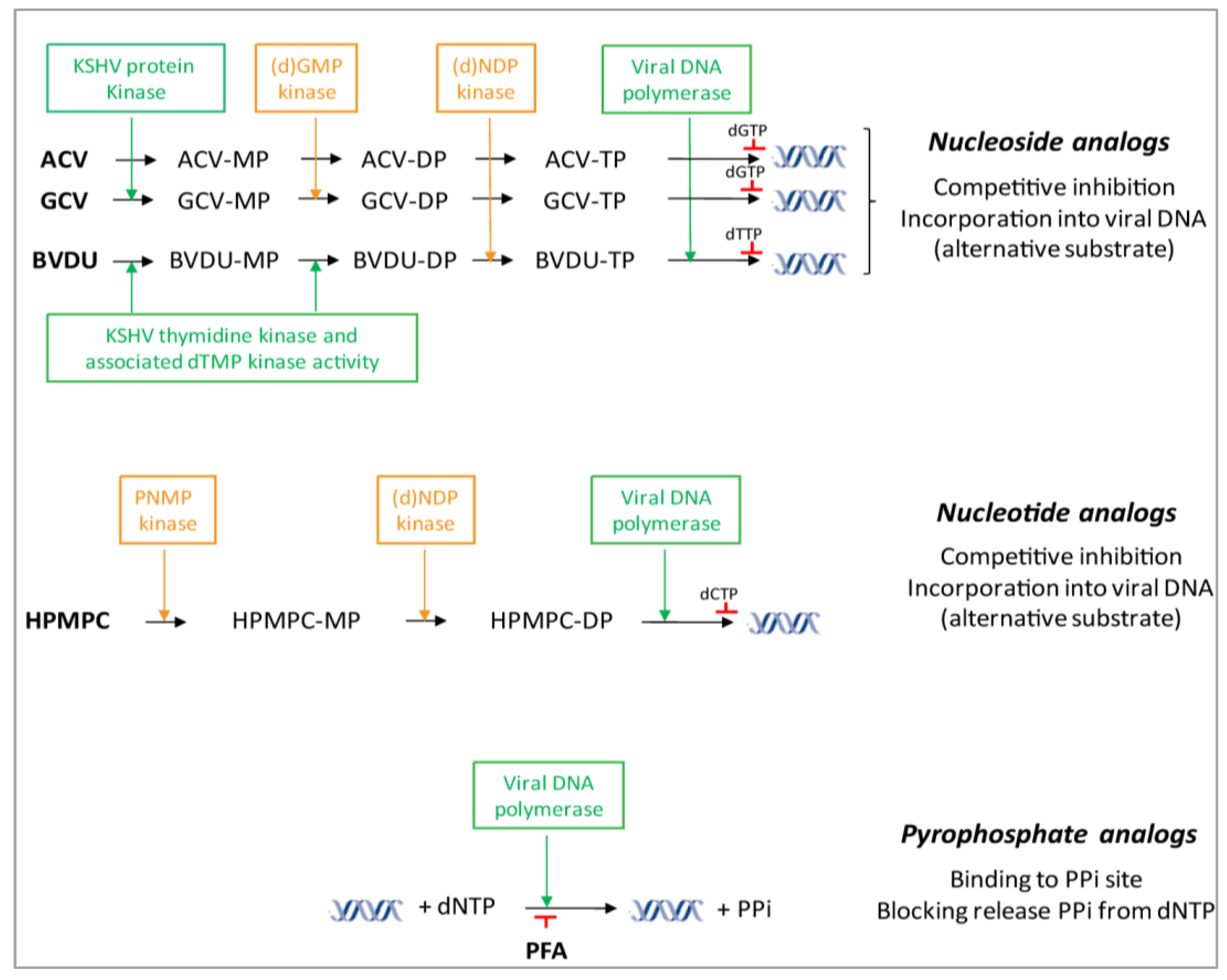
4.1. DNA Polymerase Inhibitors: Nucleoside Analogs


| Class | Subclass | Abbreviation | Drug Name | EC50 Range (µM) a | Stage of Development d | Refs. |
|---|---|---|---|---|---|---|
| Nucleoside analogs | Purine analogs | ACV | Acyclovir | 26–138 | Cohort study | [35,36,72,73,74,75,90] |
| PCV | Penciclovir | 43 | In vitro | [72] | ||
| A-5021 | (1S,2R)-9-[[1,2-bis(hydroxymethyl) cycloprop-1yl]methyl]guanine | 75 | In vitro | [79] | ||
| H2G | Omaciclovir | 42 | In vitro | [72] | ||
| GCV | Ganciclovir | 1.0–10 | Randomized, controlled trial (with VGCV) | [31,38,72,73,74,75,90] | ||
| S2242 | 2-Amino-7-[(1,3-dihydroxy-2-propoxy)-methyl]purine | 0.1 | In vitro | [72] | ||
| Methylenecyclopropane nucleosides | CPV | Cyclopropavir | 3.8 b | In vitro | [89] | |
| 6-Alkoxy-substituted derivatives | 1.8–3.5 b | In vitro | [89] | |||
| 6-Alkylthio-substituted derivatives | 1.9–7.3 b | In vitro | [89] | |||
| Pyrimidine analogs | AZT | Zidovudine | Randomized trial | [17] | ||
| BVDU | Brivudine | 0.9–24 | In vivo | [72,73,90] | ||
| N-MCT | (North)-methanocarbathymidine | 0.08 | In vitro | [96] | ||
| L-dioxolane uracil analog | HDVD | 1-[(2S,4S-2-(hydroxymethyl)-1,3-dioxolan-4-yl]5-vinylpyrimidine-2,4(1H,3H)-dione | 0.09 | In vivo | [90] | |
| Thiothymidine analogs | KAY-2-41 | 1-methyl substituted 4-thiothymidine | ≥130 | In vivo | [97] | |
| KAH-39-149 | 4-azido substituted 4-thiothymidine | >200 | In vivo | [97] | ||
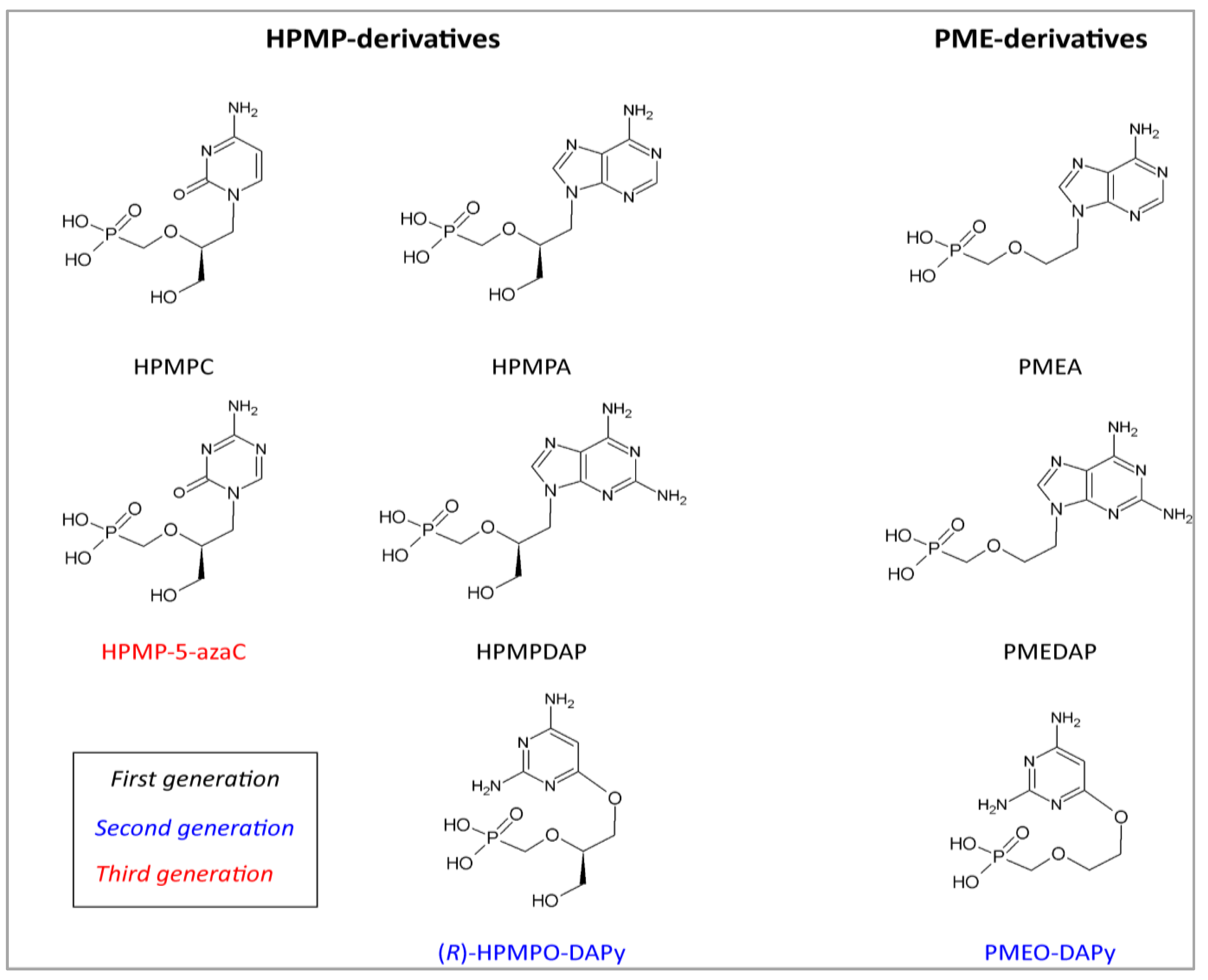
| Acyclic nucleoside phosphonates | HPMP derivatives | HPMPC, CDV | Cidofovir | 0.3–6.3 | Pilot study | [39,46,72,73,74,75,90] |
| CMX001 | Brincidofovir | 0.7 | In vitro | [98] | ||
| HPMP-5-azaC | 1-(S)-[3-hydroxy-2-(phosphonomethoxy)-propyl]-5-azacytosine | 0.7 | In vivo | [98] | ||
| HPMPA | (S)-9-[3-hydroxy-2-(phosphonomethoxy)-propyl]adenine | 0.7 | In vitro | [72,98] | ||
| HPMPDAP | (S)-9-[3-hydroxy-2-(phosphonomethoxy)-propyl]-2,6-diaminopurine | 0.9 | In vitro | [98] | ||
| HPMPO-DAPy | (R)-(2,4-diamino-3-hydroxy-6-[2-(phosphono-methoxy)propoxy])- pyrimidine | 5.1 | In vitro | [98] | ||
| PMEderivatives | PMEA | Adefovir | 18–44 | In vitro | [72,73,75,98] | |
| PMEDAP | (9-[2-(phosphonomethoxy)ethyl]-2,6-diamino-purine | 16 | In vitro | [98] | ||
| PMEO-DAPy | 2,4-diamino-6-[2-(phosphono-methoxy)ethoxy]-pyrimidine | 12 | In vitro | [98] | ||
| PMPderivatives | PMPA | Tenofovir | >150 | In vitro | Our unpublished data | |
| Pyrophosphate analog | PFA | Foscarnet sodium | 34–39 | Cohort study | [35,36,74,75] | |
| Non-nucleoside inhibitors | 4-oxo-dihydroquinolines | 183792, 529311, 568561, 570886 | 1.9–11.1 c | In vitro | [99] | |
| Pyrimidoquinoline analog | NSC 373989 | (5-((3-(dimethylamino)propyl)amino) -3,10-dimethy-lpyrimido[4,5-b] quinoline-2,4(3H,-10H)-dione) | 1.9 | In vitro | [100] |
4.2. DNA Polymerase Inhibitors: Acyclic Nucleoside Phosphonates

4.3. DNA Polymerase Inhibitors: Pyrophosphate Analogs
4.4. DNA Polymerase Inhibitors: Non-Nucleoside Analogs
4.5. Non-DNA Polymerase Inhibitors that Target Viral DNA Synthesis
4.6. Other Inhibitors of KSHV Replication
4.7. Potential Novel Drug Targets in KSHV
5. Cellular Targets
7. Conclusions
Acknowledgements
Conflict of Interest
References and Notes
- Stanescu, L.; Foarfa, C.; Georgescu, A.C.; Georgescu, I. Kaposi’s sarcoma associated with AIDS. Rom. J. Morphol. Embryol. 2007, 4, 181–187. [Google Scholar]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Ablashi, D.V.; Chatlynne, L.G.; Whitman, J.E., Jr.; Cesarman, E. Spectrum of Kaposi’s sarcoma-associated herpesvirus, or human herpesvirus 8, diseases. Clin. Microbiol. Rev. 2002, 15, 439–464. [Google Scholar] [CrossRef] [PubMed]
- Dourmishev, L.A.; Dourmishev, A.L.; Palmeri, D.; Schwartz, R.A.; Lukac, D.M. Molecular genetics of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 2003, 67, 175–212. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, V.; Lavergne, A.; de Thoisy, B.; Pouliquen, J.F.; Gessain, A. Genetic diversity and molecular evolution of human and non-human primate Gammaherpesvirinae. Infect. Genet. Evol. 2010, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Meggetto, F.; Brousset, P. Human herpesvirus 8 infections in patients with immunodeficiencies. Hum. Pathol. 2008, 39, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.J.; Pantanowitz, L.; Casper, C.; Stebbing, J.; Dezube, B.J. HIV/AIDS: Epidemiology, pathophysiology, and treatment of Kaposi sarcoma-associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clin. Infect. Dis. 2008, 47, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Fatahzadeh, M. Kaposi sarcoma: Review and medical management update. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2012, 113, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.A. Kaposi’s sarcoma: An update. J. Surg. Oncol. 2004, 87, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Hengge, U.R.; Ruzicka, T.; Tyring, S.K.; Stuschke, M.; Roggendorf, M.; Schwartz, R.A.; Seeber, S. Update on Kaposi’s sarcoma and other HHV8 associated diseases. Part 1: Epidemiology, environmental predispositions, clinical manifestations, and therapy. Lancet Infect. Dis. 2002, 2, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Szajerka, T.; Jablecki, J. Kaposi’s sarcoma revisited. AIDS Rev. 2007, 9, 230–236. [Google Scholar] [PubMed]
- Martorelli, D.; Muraro, E.; Merlo, A.; Turrini, R.; Fae, D.A.; Rosato, A.; Dolcetti, R. Exploiting the interplay between innate and adaptive immunity to improve immunotherapeutic strategies for Epstein-Barr-virus-driven disorders. Clin. Dev. Immunol. 2012, 2012, 931952. [Google Scholar] [CrossRef] [PubMed]
- Von Roenn, J.H. Clinical presentations and standard therapy of AIDS-associated Kaposi’s sarcoma. Hematol. Oncol. Clin. North Am. 2003, 17, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Vanni, T.; Sprinz, E.; Machado, M.W.; Santana, R.C.; Fonseca, B.A.; Schwartsmann, G. Systemic treatment of AIDS-related Kaposi sarcoma: Current status and perspectives. Cancer Treat. Rev. 2006, 32, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Uldrick, T.S.; Whitby, D. Update on KSHV epidemiology, Kaposi Sarcoma pathogenesis, and treatment of Kaposi Sarcoma. Cancer Lett. 2011, 305, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Casper, C.; Wald, A. The use of antiviral drugs in the prevention and treatment of Kaposi sarcoma, multicentric Castleman disease and primary effusion lymphoma. Curr. Top. Microbiol. Immunol. 2007, 312, 289–307. [Google Scholar] [PubMed]
- Joffe, M.M.; Hoover, D.R.; Jacobson, L.P.; Kingsley, L.; Chmiel, J.S.; Visscher, B.R. Effect of treatment with zidovudine on subsequent incidence of Kaposi’s sarcoma. Clin. Infect. Dis. 1997, 25, 1125–1133. [Google Scholar] [CrossRef]
- Sgadari, C.; Monini, P.; Barillari, G.; Ensoli, B. Use of HIV protease inhibitors to block Kaposi’s sarcoma and tumour growth. Lancet Oncol. 2003, 4, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Lock, M.J.; Thorley, N.; Teo, J.; Emery, V.C. Azidodeoxythymidine and didehydrodeoxythymidine as inhibitors and substrates of the human herpesvirus 8 thymidine kinase. J. Antimicrob. Chemother. 2002, 49, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Gantt, S.; Casper, C.; Ambinder, R.F. Insights into the broad cellular effects of nelfinavir and the HIV protease inhibitors supporting their role in cancer treatment and prevention. Curr. Opin. Oncol. 2013, 25, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Gantt, S.; Carlsson, J.; Ikoma, M.; Gachelet, E.; Gray, M.; Geballe, A.P.; Corey, L.; Casper, C.; Lagunoff, M.; Vieira, J. The HIV protease inhibitor nelfinavir inhibits Kaposi’s sarcoma-associated herpesvirus replication in vitro. Antimicrob. Agents Chemother. 2011, 55, 2696–2703. [Google Scholar]
- Ruocco, E.; Ruocco, V.; Tornesello, M.L.; Gambardella, A.; Wolf, R.; Buonaguro, F.M. Kaposi’s sarcoma: Etiology and pathogenesis, inducing factors, causal associations, and treatments: Facts and controversies. Clin. Dermatol. 2013, 31, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Simonelli, C.; Spina, M.; Cinelli, R.; Talamini, R.; Tedeschi, R.; Gloghini, A.; Vaccher, E.; Carbone, A.; Tirelli, U. Clinical features and outcome of primary effusion lymphoma in HIV-infected patients: A single-institution study. J. Clin. Oncol. 2003, 21, 3948–3954. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.P.; Richards, K.L.; Damania, B. Treatment of Kaposi sarcoma-associated herpesvirus-associated cancers. Front Microbiol. 2012, 3, 141. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.T.; Rubin, N.; Said, J.; Levine, A.M. Primary effusion lymphoma: Successful treatment with highly active antiretroviral therapy and rituximab. Ann. Hematol. 2005, 84, 551–552. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.J.; Pantanowitz, L.; Dezube, B.J. Targeted therapy for Kaposi sarcoma. BioDrugs 2009, 23, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Song, S.N.; Tomosugi, N.; Kawabata, H.; Ishikawa, T.; Nishikawa, T.; Yoshizaki, K. Down-regulation of hepcidin resulting from long-term treatment with an anti-IL-6 receptor antibody (tocilizumab) improves anemia of inflammation in multicentric Castleman disease. Blood 2010, 116, 3627–3634. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.C. First IL-6-blocking drug nears approval for rare blood disorder. Nat. Med. 2013, 19, 1193. [Google Scholar] [CrossRef] [PubMed]
- Marcelin, A.G.; Aaron, L.; Mateus, C.; Gyan, E.; Gorin, I.; Viard, J.P.; Calvez, V.; Dupin, N. Rituximab therapy for HIV-associated Castleman disease. Blood 2003, 102, 2786–2788. [Google Scholar] [CrossRef] [PubMed]
- Ocio, E.M.; Sanchez-Guijo, F.M.; Diez-Campelo, M.; Castilla, C.; Blanco, O.J.; Caballero, D.; San Miguel, J.F. Efficacy of rituximab in an aggressive form of multicentric Castleman disease associated with immune phenomena. Am. J. Hematol. 2005, 78, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Casper, C.; Nichols, W.G.; Huang, M.L.; Corey, L.; Wald, A. Remission of HHV-8 and HIV-associated multicentric Castleman disease with ganciclovir treatment. Blood 2004, 103, 1632–1634. [Google Scholar] [CrossRef] [PubMed]
- Gessain, A.; Duprez, R. Spindle cells and their role in Kaposi’s sarcoma. Int. J. Biochem. Cell Biol. 2005, 37, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Gantt, S.; Casper, C. Human herpesvirus 8-associated neoplasms: The roles of viral replication and antiviral treatment. Curr. Opin. Infect. Dis. 2011, 24, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Asahi-Ozaki, Y.; Sato, Y.; Kanno, T.; Sata, T.; Katano, H. Quantitative analysis of Kaposi sarcoma-associated herpesvirus (KSHV) in KSHV-associated diseases. J. Infect. Dis. 2006, 193, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Glesby, M.J.; Hoover, D.R.; Weng, S.; Graham, N.M.; Phair, J.P.; Detels, R.; Ho, M.; Saah, A.J. Use of antiherpes drugs and the risk of Kaposi’s sarcoma: Data from the Multicenter AIDS Cohort Study. J. Infect. Dis. 1996, 173, 1477–1480. [Google Scholar] [CrossRef] [PubMed]
- Mocroft, A.; Youle, M.; Gazzard, B.; Morcinek, J.; Halai, R.; Phillips, A.N. Anti-herpesvirus treatment and risk of Kaposi’s sarcoma in HIV infection. Royal Free/Chelsea and Westminster Hospitals Collaborative Group. AIDS 1996, 10, 1101–1105. [Google Scholar] [PubMed]
- Spector, S.A.; McKinley, G.F.; Lalezari, J.P.; Samo, T.; Andruczk, R.; Follansbee, S.; Sparti, P.D.; Havlir, D.V.; Simpson, G.; Buhles, W.; Wong, R.; Stempien, M. Oral ganciclovir for the prevention of cytomegalovirus disease in persons with AIDS. Roche Cooperative Oral Ganciclovir Study Group. N. Engl. J. Med. 1996, 334, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Casper, C.; Krantz, E.M.; Corey, L.; Kuntz, S.R.; Wang, J.; Selke, S.; Hamilton, S.; Huang, M.L.; Wald, A. Valganciclovir for suppression of human herpesvirus-8 replication: A randomized, double-blind, placebo-controlled, crossover trial. J. Infect. Dis. 2008, 198, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Little, R.F.; Merced-Galindez, F.; Staskus, K.; Whitby, D.; Aoki, Y.; Humphrey, R.; Pluda, J.M.; Marshall, V.; Walters, M.; Welles, L.; Rodriguez-Chavez, I.R.; Pittaluga, S.; Tosato, G.; Yarchoan, R. A pilot study of cidofovir in patients with kaposi sarcoma. J. Infect Dis. 2003, 187, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Mazzi, R.; Parisi, S.G.; Sarmati, L.; Uccella, I.; Nicastri, E.; Carolo, G.; Gatti, F.; Concia, E.; Andreoni, M. Efficacy of cidofovir on human herpesvirus 8 viraemia and Kaposi’s sarcoma progression in two patients with AIDS. AIDS 2001, 15, 2061–2062. [Google Scholar] [CrossRef] [PubMed]
- Simonart, T.; Noel, J.C.; De Dobbeleer, G.; Parent, D.; Van Vooren, J.P.; De Clercq, E.; Snoeck, R. Treatment of classical Kaposi’s sarcoma with intralesional injections of cidofovir: Report of a case. J. Med. Virol. 1998, 55, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Suen, J.; Frias, C.; Pfeiffer, R.; Tsai, M.H.; Chuang, E.; Zeichner, S.L. Dissection of the Kaposi’s sarcoma-associated herpesvirus gene expression program by using the viral DNA replication inhibitor cidofovir. J. Virol. 2004, 78, 13637–13652. [Google Scholar] [CrossRef] [PubMed]
- Crum-Cianflone, N.F.; Wallace, M.R.; Looney, D. Successful secondary prophylaxis for primary effusion lymphoma with human herpesvirus 8 therapy. AIDS 2006, 20, 1567–1569. [Google Scholar] [CrossRef] [PubMed]
- Pastore, R.D.; Chadburn, A.; Kripas, C.; Schattner, E.J. Novel association of haemophagocytic syndrome with Kaposi’s sarcoma-associated herpesvirus-related primary effusion lymphoma. Br. J. Haematol. 2000, 111, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Hocqueloux, L.; Agbalika, F.; Oksenhendler, E.; Molina, J.M. Long-term remission of an AIDS-related primary effusion lymphoma with antiviral therapy. AIDS 2001, 15, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Luppi, M.; Trovato, R.; Barozzi, P.; Vallisa, D.; Rossi, G.; Re, A.; Ravazzini, L.; Potenza, L.; Riva, G.; Morselli, M.; et al. Treatment of herpesvirus associated primary effusion lymphoma with intracavity cidofovir. Leukemia 2005, 19, 473–476. [Google Scholar] [CrossRef]
- Boulanger, E. Human herpesvirus 8 (HHV8). II. Pathogenic role and sensitivity to antiviral drugs. Ann. Biol. Clin. (Paris) 1999, 57, 19–28. [Google Scholar]
- Moyo, T.K.; Richards, K.L.; Damania, B. Use of cidofovir for the treatment of HIV-negative human herpes virus-8-associated primary effusion lymphoma. Clin. Adv. Hematol. Oncol. 2010, 8, 372–374. [Google Scholar] [PubMed]
- Valencia, M.E.; Moreno, V.; Martinez, P.; Casado, I. Favorable outcome of Castleman’s disease treated with oral valganciclovir. Med. Clin. (Barc.) 2005, 125, 399. [Google Scholar] [CrossRef]
- Berezne, A.; Agbalika, F.; Oksenhendler, E. Failure of cidofovir in HIV-associated multicentric Castleman disease. Blood 2004, 103, 4368–4369. [Google Scholar] [CrossRef] [PubMed]
- Uldrick, T.S.; Polizzotto, M.N.; Aleman, K.; O’Mahony, D.; Wyvill, K.M.; Wang, V.; Marshall, V.; Pittaluga, S.; Steinberg, S.M.; Tosato, G.; et al. High-dose zidovudine plus valganciclovir for Kaposi sarcoma herpesvirus-associated multicentric Castleman disease: A pilot study of virus-activated cytotoxic therapy. Blood 2011, 117, 6977–6986. [Google Scholar]
- Kenney, S.C.; Fingeroth, J.D. The Molecular Basis of Lytic Induction Therapy in Relation to Gamma herpesvirus (KSHV, EBV)-Associated, AIDS-Related Tumors. In Molecular Basis for Therapy if AIDS-defining Cancers; Dittmer, D., Krown, S.E., Eds.; Springer: New York, NY, USA, 2010; pp. 111–135. [Google Scholar]
- Klass, C.M.; Offermann, M.K. Targeting human herpesvirus-8 for treatment of Kaposi’s sarcoma and primary effusion lymphoma. Curr. Opin. Oncol. 2005, 17, 447–455. [Google Scholar] [CrossRef]
- Klass, C.M.; Krug, L.T.; Pozharskaya, V.P.; Offermann, M.K. The targeting of primary effusion lymphoma cells for apoptosis by inducing lytic replication of human herpesvirus 8 while blocking virus production. Blood 2005, 105, 4028–4034. [Google Scholar] [CrossRef] [PubMed]
- Lechowicz, M.; Dittmer, D.P.; Lee, J.Y.; Krown, S.E.; Wachsman, W.; Aboulafia, D.; Dezube, B.J.; Ratner, L.; Said, J.; Ambinder, R.F. Molecular and clinical assessment in the treatment of AIDS Kaposi sarcoma with valproic Acid. Clin. Infec. Dis. 2009, 49, 1946–1949. [Google Scholar] [CrossRef]
- Fu, D.X.; Tanhehco, Y.; Chen, J.; Foss, C.A.; Fox, J.J.; Chong, J.M.; Hobbs, R.F.; Fukayama, M.; Sgouros, G.; Kowalski, J.; Pomper, M.G.; Ambinder, R.F. Bortezomib-induced enzyme-targeted radiation therapy in herpesvirus-associated tumors. Nature Med. 2008, 14, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Reid, E.G. Bortezomib-induced Epstein-Barr virus and Kaposi sarcoma herpesvirus lytic gene expression: Oncolytic strategies. Curr. Opin. Oncol. 2011, 23, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.J.; McBride, W.H.; Zack, J.A.; Sun, R. Prostratin and bortezomib are novel inducers of latent Kaposi’s sarcoma-associated herpesvirus. Antivir. Ther. 2005, 10, 745–751. [Google Scholar] [PubMed]
- Bhatt, S.; Ashlock, B.M.; Toomey, N.L.; Diaz, L.A.; Mesri, E.A.; Lossos, I.S.; Ramos, J.C. Efficacious proteasome/HDAC inhibitor combination therapy for primary effusion lymphoma. J. Clin. Invest 2013, 123, 2616–2628. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, A.T.; Gentry, G.A.; Subak-Sharpe, J.H. Induction of both thymidine and deoxycytidine kinase activity by herpes viruses. J. Gen. Virol. 1974, 24, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.H.; Miller, R.L. Phosphorylation of acyclovir diphosphate by cellular enzymes. Biochem. Pharmacol. 1982, 31, 3879–3884. [Google Scholar] [CrossRef] [PubMed]
- Andrei, G.; De Clercq, E.; Snoeck, R. Viral DNA Polymerase Inhibitors. In Viral Genome Replication; Cameron, C.E., Gotte, M., Raney, K., Eds.; Springer: New York, NY, USA, 2009; pp. 481–526. [Google Scholar]
- Gustafson, E.A.; Schinazi, R.F.; Fingeroth, J.D. Human herpesvirus 8 open reading frame 21 is a thymidine and thymidylate kinase of narrow substrate specificity that efficiently phosphorylates zidovudine but not ganciclovir. J. Virol 2000, 74, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.S.; Hamzeh, F.; Moore, S.; Nicholas, J.; Ambinder, R.F. Human herpesvirus 8-encoded thymidine kinase and phosphotransferase homologues confer sensitivity to ganciclovir. J. Virol 1999, 73, 4786–4793. [Google Scholar] [PubMed]
- Lurain, N.S.; Chou, S. Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 2010, 23, 689–712. [Google Scholar] [CrossRef]
- De Clercq, E. Discovery and development of BVDU (brivudin) as a therapeutic for the treatment of herpes zoster. Biochem. Pharmacol. 2004, 68, 2301–2315. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, E.A.; Chillemi, A.C.; Sage, D.R.; Fingeroth, J.D. The Epstein-Barr virus thymidine kinase does not phosphorylate ganciclovir or acyclovir and demonstrates a narrow substrate specificity compared to the herpes simplex virus type 1 thymidine kinase. Antimicrob. Agents Chemother. 1998, 42, 2923–2931. [Google Scholar] [PubMed]
- De Clercq, E. Antiviral drug discovery and development: Where chemistry meets with biomedicine. Antivir. Res. 2005, 67, 56–75. [Google Scholar] [CrossRef] [PubMed]
- Reardon, J.E.; Spector, T. Herpes simplex virus type 1 DNA polymerase. Mechanism of inhibition by acyclovir triphosphate. J. Biol. Chem. 1989, 264, 7405–7411. [Google Scholar] [PubMed]
- Sellar, R.S.; Peggs, K.S. Management of multidrug-resistant viruses in the immunocompromised host. Br. J. Hematol. 2012, 156, 559–572. [Google Scholar] [CrossRef]
- De Clercq, E.; Naesens, L.; De Bolle, L.; Schols, D.; Zhang, Y.; Neyts, J. Antiviral agents active against human herpesviruses HHV-6, HHV-7 and HHV-8. Rev. Med. Virol 2001, 11, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Neyts, J.; De Clercq, E. Antiviral drug susceptibility of human herpesvirus 8. Antimicrob. Agents Chemother. 1997, 41, 2754–2756. [Google Scholar]
- Friedrichs, C.; Neyts, J.; Gaspar, G.; De Clercq, E.; Wutzler, P. Evaluation of antiviral activity against human herpesvirus 8 (HHV-8) and Epstein-Barr virus (EBV) by a quantitative real-time PCR assay. Antivir. Res. 2004, 62, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Bounaadja, L.; Piret, J.; Goyette, N.; Boivin, G. Evaluation of Epstein-Barr virus, human herpesvirus 6 (HHV-6), and HHV-8 antiviral drug susceptibilities by use of real-time-PCR-based assays. J. Clin. Microbiol. 2013, 51, 1244–1246. [Google Scholar] [CrossRef] [PubMed]
- Sergerie, Y.; Boivin, G. Evaluation of susceptibility of human herpesvirus 8 to antiviral drugs by quantitative real-time PCR. J. Clin. Microbiol. 2003, 41, 3897–3900. [Google Scholar] [CrossRef] [PubMed]
- Medveczky, M.M.; Horvath, E.; Lund, T.; Medveczky, P.G. In vitro antiviral drug sensitivity of the Kaposi’s sarcoma-associated herpesvirus. AIDS 1997, 11, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Bacon, T.H.; Boyd, M.R. Activity of penciclovir against Epstein-Barr virus. Antimicrob. Agents Chemother. 1995, 39, 1599–1602. [Google Scholar] [CrossRef] [PubMed]
- Bacon, T.H.; Howard, B.A.; Spender, L.C.; Boyd, M.R. Activity of penciclovir in antiviral assays against herpes simplex virus. J. Antimicrob. Chemother. 1996, 37, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Neyts, J.; Naesens, L.; Ying, C.; De, B.L.; De, C.E. Anti-herpesvirus activity of (1’S,2’R)-9-[[1’,2’-bis(hydroxymethyl)-cycloprop-1’-yl]methyl] x guanine (A-5021) in vitro and in vivo. Antivir. Res. 2001, 49, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Faulds, D.; Heel, R.C. Ganciclovir. A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in cytomegalovirus infections. Drugs 1990, 39, 597–638. [Google Scholar] [CrossRef]
- De Clercq, E. Antiviral agents: Characteristic activity spectrum depending on the molecular target with which they interact. Adv. Virus Res. 1993, 42, 1–55. [Google Scholar] [PubMed]
- Biron, K.K. Antiviral drugs for cytomegalovirus diseases. Antiviral Res. 2006, 71, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Neyts, J.; Balzarini, J.; Andrei, G.; Chaoyong, Z.; Snoeck, R.; Zimmermann, A.; Mertens, T.; Karlsson, A.; De Clercq, E. Intracellular metabolism of the N7-substituted acyclic nucleoside analog 2-amino-7-(1,3-dihydroxy-2-propoxymethyl)purine, a potent inhibitor of herpesvirus replication. Mol. Pharmacol. 1998, 53, 157–165. [Google Scholar] [PubMed]
- Zimmermann, A.; Michel, D.; Pavic, I.; Hampl, W.; Luske, A.; Neyts, J.; De Clercq, E.; Mertens, T. Phosphorylation of aciclovir, ganciclovir, penciclovir and S2242 by the cytomegalovirus UL97 protein: A quantitative analysis using recombinant vaccinia viruses. Antiviral Res. 1997, 36, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Neyts, J.; Andrei, G.; Snoeck, R.; Jahne, G.; Winkler, I.; Helsberg, M.; Balzarini, J.; De Clercq, E. The N-7-substituted acyclic nucleoside analog 2-amino-7-[(1,3-dihydroxy-2-propoxy)methyl]purine is a potent and selective inhibitor of herpesvirus replication. Antimicrob. Agents Chemother. 1994, 38, 2710–2716. [Google Scholar] [CrossRef] [PubMed]
- Andrei, G.; De Clercq, E.; Snoeck, R. Drug targets in cytomegalovirus infection. Infec. Dis. Drug Targets 2009, 9, 201–222. [Google Scholar] [CrossRef]
- Gentry, B.G.; Kamil, J.P.; Coen, D.M.; Zemlicka, J.; Drach, J.C. Stereoselective phosphorylation of cyclopropavir by pUL97 and competitive inhibition by maribavir. Antimicrob. Agents Chemother. 2010, 54, 3093–3098. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Quenelle, D.C.; Prichard, M.N.; Drach, J.C.; Zemlicka, J. Synthesis and antiviral activity of 6-deoxycyclopropavir, a new prodrug of cyclopropavir. Bioorg. Med. Chem. 2012, 20, 2669–2674. [Google Scholar] [CrossRef] [PubMed]
- Prichard, M.N.; Williams, J.D.; Komazin-Meredith, G.; Khan, A.R.; Price, N.B.; Jefferson, G.M.; Harden, E.A.; Hartline, C.B.; Peet, N.P.; Bowlin, T.L. Synthesis and antiviral activities of methylenecyclopropane analogs with 6-alkoxy and 6-alkylthio substitutions that exhibit broad-spectrum antiviral activity against human herpesviruses. Antimicrob. Agents Chemother. 2013, 57, 3518–3527. [Google Scholar] [CrossRef] [PubMed]
- Coen, N.; Singh, U.; Vuyyuru, V.; van den Oord, J.J.; Balzarini, J.; Duraffour, S.; Snoeck, R.; Cheng, Y.C.; Chu, C.K.; Andrei, G. Activity and mechanism of action of HDVD, a novel pyrimidine nucleoside derivative with high levels of selectivity and potency against gammaherpesviruses. J. Virol. 2013, 87, 3839–3851. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Machida, H. Comparison of two bromovinyl nucleoside analogs, 1-beta-D-arabinofuranosyl-E-5-(2-bromovinyl)uracil and E-5-(2-bromovinyl)-2’-deoxyuridine, with acyclovir in inhibition of Epstein-Barr virus replication. Antimicrob. Agents Chemother. 1988, 32, 1068–1072. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Li, L.; Grill, S.; Gullen, E.; Lee, C.S.; Gumina, G.; Tsujii, E.; Cheng, Y.C.; Chu, C.K. Structure-activity relationships of (E)-5-(2-bromovinyl)uracil and related pyrimidine nucleosides as antiviral agents for herpes viruses. J. Med. Chem. 2000, 43, 2538–2546. [Google Scholar] [CrossRef] [PubMed]
- Neyts, J.; De Clercq, E. In vitro and in vivo inhibition of murine gamma herpesvirus 68 replication by selected antiviral agents. Antimicrob. Agents Chemother. 1998, 42, 170–172. [Google Scholar]
- De Clercq, E.; Zhang, Z.X.; Sim, I.S. Treatment of experimental herpes simplex virus encephalitis with (E)-5-(2-bromovinyl)-2’-deoxyuridine in mice. Antimicrob. Agents Chemother. 1982, 22, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Weber, O. Novel mouse models for the investigation of experimental drugs with activity against human varicella-zoster virus. Antiviral Chem. Chemother. 2000, 11, 283–290. [Google Scholar]
- Zhu, W.; Burnette, A.; Dorjsuren, D.; Roberts, P.E.; Huleihel, M.; Shoemaker, R.H.; Marquez, V.E.; Agbaria, R.; Sei, S. Potent antiviral activity of north-methanocarbathymidine against Kaposi’s sarcoma-associated herpesvirus. Antimicrob. Agents Chemother. 2005, 49, 4965–4973. [Google Scholar] [CrossRef] [PubMed]
- Coen, N.; Duraffour, S.; Haraguchi, K.; Balzarini, J.; van den Oord, J.J.; Snoeck, R.; Andrei, G. Anti-herpesvirus Activity of Two Novel 4’-Thiothymidine Derivatives KAY-2-41 and KAH-39-149 is dependent on viral and cellular thymidine kinases. Antimicrob. Agents Chemother. 2014, 58, 4328–4340. [Google Scholar] [CrossRef] [PubMed]
- Coen, N.; Duraffour, S.; Naesens, L.; Krecmerova, M.; Van den Oord, J.; Snoeck, R.; Andrei, G. Evaluation of Novel Acyclic Nucleoside Phosphonates against Human and Animal Gammaherpesviruses Revealed an Altered Metabolism of Cyclic Prodrugs upon Epstein-Barr Virus Reactivation in P3HR-1 Cells. J. Virol. 2013, 87, 12422–12432. [Google Scholar] [CrossRef] [PubMed]
- Hartline, C.B.; Harden, E.A.; Williams-Aziz, S.L.; Kushner, N.L.; Brideau, R.J.; Kern, E.R. Inhibition of herpesvirus replication by a series of 4-oxo-dihydroquinolines with viral polymerase activity. Antivir. Res. 2005, 65, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Dorjsuren, D.; Burnette, A.; Gray, G.N.; Chen, X.; Zhu, W.; Roberts, P.E.; Currens, M.J.; Shoemaker, R.H.; Ricciardi, R.P.; Sei, S. Chemical library screen for novel inhibitors of Kaposi’s sarcoma-associated herpesvirus processive DNA synthesis. Antivir. Res. 2006, 69, 9–23. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Holy, A.; Rosenberg, I.; Sakuma, T.; Balzarini, J.; Maudgal, P.C. A novel selective broad-spectrum anti-DNA virus agent. Nature 1986, 323, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.A.; Martin, J.C. Perspectives on the development of acyclic nucleotide analogs as antiviral drugs. Antivir. Res. 2006, 71, 254–259. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Field, H.J. Antiviral prodrugs—The development of successful prodrug strategies for antiviral chemotherapy. Br. J. Pharmacol 2006, 147, 1–11. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Holy, A. Acyclic nucleoside phosphonates: A key class of antiviral drugs. Nat. Rev. Drug Discov. 2005, 4, 928–940. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2004, 2, 704–720. [Google Scholar] [CrossRef] [PubMed]
- Neyts, J.; Snoeck, R.; Balzarini, J.; De Clercq, E. Particular characteristics of the anti-human cytomegalovirus activity of (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine (HPMPC) in vitro. Antivir. Res. 1991, 16, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.T.; Woods, K.L.; Bronson, J.J.; De, B.H.; Martin, J.C.; Hitchcock, M.J. Intracellular metabolism of the antiherpes agent (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine. Mol. Pharmacol. 1992, 41, 197–202. [Google Scholar] [PubMed]
- De Clercq, E. The clinical potential of the acyclic (and cyclic) nucleoside phosphonates. The magic of the phosphonate bond. Biochem. Pharmacol. 2011, 2, 99–109. [Google Scholar] [CrossRef]
- Krecmerova, M. Nucleoside and Nucleotide Analogues for the Treatment of Herpesvirus Infections: Current Stage and New Prospects in the Field of Acyclic Nucleoside Phosphonates. In Herpesviridae—A Look Into This Unique Family of Viruses; Magel, G., Tyring, S., Eds.; InTech: Rijeka, Croatia, 2012; pp. 245–270. [Google Scholar]
- Holy, A. Phosphonomethoxyalkyl analogs of nucleotides. Cur. Pharmac. Des. 2003, 9, 2567–2592. [Google Scholar] [CrossRef]
- Krecmerova, M.; Holy, A.; Piskala, A.; Masojidkova, M.; Andrei, G.; Naesens, L.; Neyts, J.; Balzarini, J.; De, C.E.; Snoeck, R. Antiviral activity of triazine analogues of 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine (cidofovir) and related compounds. J. Med. Chem. 2007, 50, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Tichy, T.; Andrei, G.; Dracinsky, M.; Holy, A.; Balzarini, J.; Snoeck, R.; Krecmerova, M. New prodrugs of Adefovir and Cidofovir. Bioorg. Med. Chem. 2011, 19, 3527–3539. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.D.; Adams, M.M.; Wallace, G.; Burrage, A.M.; Lindsey, S.F.; Smith, A.J.; Swetnam, D.; Manning, B.R.; Gray, S.A.; Lampert, B.; et al. Efficacy of CMX001 as a post exposure antiviral in New Zealand White rabbits infected with rabbitpox virus, a model for orthopoxvirus infections of humans. Viruses 2011, 3, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Painter, W.; Robertson, A.; Trost, L.C.; Godkin, S.; Lampert, B.; Painter, G. First pharmacokinetic and safety study in humans of the novel lipid antiviral conjugate CMX001, a broad-spectrum oral drug active against double-stranded DNA viruses. Antimicrob. Agents Chemother. 2012, 56, 2726–2734. [Google Scholar] [CrossRef] [PubMed]
- Marty, F.M.; Winston, D.J.; Rowley, S.D.; Vance, E.; Papanicolaou, G.A.; Mullane, K.M.; Brundage, T.M.; Robertson, A.T.; Godkin, S.; Mommeja-Marin, H.; et al. CMX001 to prevent cytomegalovirus disease in hematopoietic-cell transplantation. N. Engl. J. Med. 2013, 369, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Crumpacker, C.S. Mechanism of action of foscarnet against viral polymerases. Am. J. Med. 1992, 92, 3S–7S. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Ricciardi, R.P. A rapid plate assay for the screening of inhibitors against herpesvirus DNA polymerases and processivity factors. J. Virol. Methods 2000, 88, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Wang, L.; Gonzalez-Molleda, L.; Wang, Y.; Xu, J.; Yuan, Y. Antiviral activity of (+)-rutamarin against Kaposi’s sarcoma-associated herpesvirus by inhibition of the catalytic activity of human topoisomerase II. Antimicrob. Agents Chemother. 2014, 58, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Jeong, S.G.; Park, J.E.; Han, J.A.; Kang, H.R.; Lee, D.; Song, M.J. Antiviral activity of angelicin against gammaherpesviruses. Antivir. Res. 2013, 100, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Dyson, O.F.; Walker, L.R.; Whitehouse, A.; Cook, P.P.; Akula, S.M. Resveratrol inhibits KSHV reactivation by lowering the levels of cellular EGR-1. PLoS One 2012, 7, e33364. [Google Scholar] [CrossRef] [PubMed]
- Medveczky, M.M.; Sherwood, T.A.; Klein, T.W.; Friedman, H.; Medveczky, P.G. Delta-9 tetrahydrocannabinol (THC) inhibits lytic replication of gamma oncogenic herpesviruses in vitro. BMC. Med. 2004, 2, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.J.; Stein, D.A.; Fan, S.M.; Wang, K.Y.; Kroeker, A.D.; Meng, X.J.; Iversen, P.L.; Matson, D.O. Suppression of porcine reproductive and respiratory syndrome virus replication by morpholino antisense oligomers. Vet. Microbiol. 2006, 117, 117–129. [Google Scholar] [CrossRef]
- Skorenski, M.; Sienczyk, M. Anti-herpesvirus agents: A patent and literature review (2003 to present). Expert Opin. Ther. Pat. 2014, 24, 925–941. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, A.D.; Davies, D.E.; Ertl, P.F.; Exall, A.M.; Haley, T.M.; Hart, G.J.; Jackson, D.L.; Parry, N.R.; Patikis, A.; Trivedi, N.; et al. Design and synthesis of pyrrolidine-5,5’-trans-lactams (5-oxo-hexahydropyrrolo[3,2-b]pyrroles) as novel mechanism-based inhibitors of human cytomegalovirus protease. 4. Antiviral activity and plasma stability. J. Med. Chem. 2003, 46, 4428–4449. [Google Scholar] [CrossRef] [PubMed]
- Gopalsamy, A.; Lim, K.; Ellingboe, J.W.; Mitsner, B.; Nikitenko, A.; Upeslacis, J.; Mansour, T.S.; Olson, M.W.; Bebernitz, G.A.; Grinberg, D.; et al. Design and syntheses of 1,6-naphthalene derivatives as selective HCMV protease inhibitors. J. Med. Chem. 2004, 47, 1893–1899. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, A.D. Design of translactam HCMV protease inhibitors as potent antivirals. Med. Res. Rev. 2005, 25, 427–452. [Google Scholar] [CrossRef] [PubMed]
- Shahian, T.; Lee, G.M.; Lazic, A.; Arnold, L.A.; Velusamy, P.; Roels, C.M.; Guy, R.K.; Craik, C.S. Inhibition of a viral enzyme by a small-molecule dimer disruptor. Nat. Chem. Biol. 2009, 5, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.M.; Shahian, T.; Baharuddin, A.; Gable, J.E.; Craik, C.S. Enzyme inhibition by allosteric capture of an inactive conformation. J Mol. Biol. 2011, 411, 999–1016. [Google Scholar] [CrossRef] [PubMed]
- Gable, J.E.; Lee, G.M.; Jaishankar, P.; Hearn, B.R.; Waddling, C.A.; Renslo, A.R.; Craik, C.S. Broad-spectrum allosteric inhibition of herpesvirus proteases. Biochemistry 2014, 53, 4648–4660. [Google Scholar] [CrossRef] [PubMed]
- Field, H.J.; Mickleburgh, I. The helicase-primase complex as a target for effective herpesvirus antivirals. Adv. Exp. Med. Biol. 2013, 767, 145–159. [Google Scholar] [PubMed]
- Underwood, M.R.; Ferris, R.G.; Selleseth, D.W.; Davis, M.G.; Drach, J.C.; Townsend, L.B.; Biron, K.K.; Boyd, F.L. Mechanism of action of the ribopyranoside benzimidazole GW275175X against human cytomegalovirus. Antimicrobial. Agents Chemother. 2004, 48, 1647–1651. [Google Scholar] [CrossRef]
- Reefschlaeger, J.; Bender, W.; Hallenberger, S.; Weber, O.; Eckenberg, P.; Goldmann, S.; Haerter, M.; Buerger, I.; Trappe, J.; Herrington, J.A.; et al. Novel non-nucleoside inhibitors of cytomegaloviruses (BAY 38–4766): In vitro and in vivo antiviral activity and mechanism of action. J. Antimicrob. Chemother. 2001, 48, 757–767. [Google Scholar] [PubMed]
- Li, R.; Hayward, S.D. Potential of protein kinase inhibitors for treating herpesvirus-associated disease. Trends Microbiol. 2013, 21, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Andrei, G.; Snoeck, R. Advances in the treatment of varicella-zoster virus infections. Adv. Pharmacol. 2013, 67, 107–168. [Google Scholar] [PubMed]
- Nichols, L.A.; Adang, L.A.; Kedes, D.H. Rapamycin blocks production of KSHV/HHV8: insights into the anti-tumor activity of an immunosuppressant drug. PLoS One 2011, 6, e14535. [Google Scholar] [CrossRef]
- Nash, A.A.; Dutia, B.M.; Stewart, J.P.; Davison, A.J. Natural history of murine gamma-herpesvirus infection. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2001, 356, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Simas, J.P.; Efstathiou, S. Murine gammaherpesvirus 68: A model for the study of gammaherpesvirus pathogenesis. Trends Microbiol. 1998, 6, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Speck, S.H.; Virgin, H.W. Host and viral genetics of chronic infection: A mouse model of gamma-herpesvirus pathogenesis. Curr. Opin. Microbiol. 1999, 2, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Cipkova-Jarcuskova, J.; Chalupkova, A.; Hrabovska, Z.; Wagnerova, M.; Mistrikova, J. Biological and pathogenetic characterization of different isolates of murine gammaherpesvirus 68 (MHV-68) in the context of study of human oncogenic gammaherpesviruses. Acta Virol. 2013, 57, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Arico, E.; Robertson, K.A.; Belardelli, F.; Ferrantini, M.; Nash, A.A. Vaccination with inactivated murine gammaherpesvirus 68 strongly limits viral replication and latency and protects type I IFN receptor knockout mice from a lethal infection. Vaccine 2004, 22, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Boname, J.M.; Coleman, H.M.; May, J.S.; Stevenson, P.G. Protection against wild-type murine gammaherpesvirus-68 latency by a latency-deficient mutant. J. Gen. Virol. 2004, 85, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Fowler, P.; Efstathiou, S. Vaccine potential of a murine gammaherpesvirus-68 mutant deficient for ORF73. J. Gen. Virol. 2004, 85, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Freeman, M.L.; Yager, E.J.; McHardy, I.; Tong, L.; Martinez-Guzman, D.; Rickabaugh, T.; Hwang, S.; Blackman, M.A.; Sun, R.; et al. Induction of protective immunity against murine gammaherpesvirus 68 infection in the absence of viral latency. J. Virol. 2010, 84, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Kayhan, B.; Yager, E.J.; Lanzer, K.; Cookenham, T.; Jia, Q.; Wu, T.T.; Woodland, D.L.; Sun, R.; Blackman, M.A. A replication-deficient murine gamma-herpesvirus blocked in late viral gene expression can establish latency and elicit protective cellular immunity. J. Immunol. 2007, 179, 8392–8402. [Google Scholar] [CrossRef] [PubMed]
- Obar, J.J.; Donovan, D.C.; Crist, S.G.; Silvia, O.; Stewart, J.P.; Usherwood, E.J. T-cell responses to the M3 immune evasion protein of murid gammaherpesvirus 68 are partially protective and induced with lytic antigen kinetics. J. Virol. 2004, 78, 10829–10832. [Google Scholar] [CrossRef] [PubMed]
- Rickabaugh, T.M.; Brown, H.J.; Martinez-Guzman, D.; Wu, T.T.; Tong, L.; Yu, F.; Cole, S.; Sun, R. Generation of a latency-deficient gammaherpesvirus that is protective against secondary infection. J. Virol. 2004, 78, 9215–9223. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, P.G.; Doherty, P.C. Kinetic analysis of the specific host response to a murine gammaherpesvirus. J. Virol. 1998, 72, 943–949. [Google Scholar] [PubMed]
- Stewart, J.P.; Micali, N.; Usherwood, E.J.; Bonina, L.; Nash, A.A. Murine gamma-herpesvirus 68 glycoprotein 150 protects against virus-induced mononucleosis: A model system for gamma-herpesvirus vaccination. Vaccine 1999, 17, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, S.A.; McClellan, J.S.; Gangappa, S.; Speck, S.H.; Virgin, H.W. Effective vaccination against long-term gammaherpesvirus latency. J. Virol. 2003, 77, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Usherwood, E.J.; Ward, K.A.; Blackman, M.A.; Stewart, J.P.; Woodland, D.L. Latent antigen vaccination in a model gammaherpesvirus infection. J. Virol. 2001, 75, 8283–8288. [Google Scholar] [CrossRef] [PubMed]
- Woodland, D.L.; Usherwood, E.J.; Liu, L.; Flano, E.; Kim, I.J.; Blackman, M.A. Vaccination against murine gamma-herpesvirus infection. Viral Immunol. 2001, 14, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.T.; Qian, J.; Ang, J.; Sun, R. Vaccine prospect of Kaposi sarcoma-associated herpesvirus. Cur. Opin. Virol. 2012, 2, 482–488. [Google Scholar] [CrossRef]
- Wu, T.T.; Blackman, M.A.; Sun, R. Prospects of a novel vaccination strategy for human gamma-herpesviruses. Immunol. Res. 2010, 48, 122–146. [Google Scholar] [CrossRef] [PubMed]
- Barnes, A.; Dyson, H.; Sunil-Chandra, N.P.; Collins, P.; Nash, A.A. 2’-Deoxy-5-ethyl-beta-4’-thiouridine inhibits replication of murine gammaherpesvirus and delays the onset of virus latency. Antivir. Chem. Chemother. 1999, 10, 321–326. [Google Scholar] [PubMed]
- Parsons, C.H.; Adang, L.A.; Overdevest, J.; O’Connor, C.M.; Taylor, J.R., Jr.; Camerini, D.; Kedes, D.H. KSHV targets multiple leukocyte lineages during long-term productive infection in NOD/SCID mice. J. Clin. Invest. 2006, 116, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Vieira, J.; Fiore, N.; Banerjee, P.; Sieburg, M.; Rochford, R.; Harrington, W., Jr.; Feuer, G. KSHV/HHV-8 infection of human hematopoietic progenitor (CD34+) cells: Persistence of infection during hematopoiesis in vitro and in vivo. Blood 2006, 108, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Murakami, M.; Bajaj, B.; Kaul, R.; He, Z.; Gan, R.; Feldman, M.; Robertson, E.S. Inhibition of KSHV-infected primary effusion lymphomas in NOD/SCID mice by gamma-secretase inhibitor. Cancer Biol. Ther. 2009, 8, 2136–2143. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, C.; Tanaka, J.; Katano, H.; Senba, M.; Mori, N. Hippuristanol reduces the viability of primary effusion lymphoma cells both in vitro and in vivo. Mar. Drugs 2013, 11, 3410–3424. [Google Scholar] [CrossRef] [PubMed]
- Kariya, R.; Taura, M.; Suzu, S.; Kai, H.; Katano, H.; Okada, S. HIV protease inhibitor Lopinavir induces apoptosis of primary effusion lymphoma cells via suppression of NF-kappaB pathway. Cancer Lett. 2014, 342, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Rochford, R.; Toomey, L.; Harrington, W.J.; Feuer, G. Inhibition of HHV-8/KSHV infected primary effusion lymphomas in NOD/SCID mice by azidothymidine and interferon-alpha. Leukemia Res. 2005, 29, 545–555. [Google Scholar] [CrossRef]
- Dai, L.; Trillo-Tinoco, J.; Bai, L.; Kang, B.; Xu, Z.; Wen, X.; Del Valle, L.; Qin, Z. Systematic analysis of a xenograft mice model for KSHV+ primary effusion lymphoma (PEL). PLoS One 2014, 9, e90349. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coen, N.; Duraffour, S.; Snoeck, R.; Andrei, G. KSHV Targeted Therapy: An Update on Inhibitors of Viral Lytic Replication. Viruses 2014, 6, 4731-4759. https://doi.org/10.3390/v6114731
Coen N, Duraffour S, Snoeck R, Andrei G. KSHV Targeted Therapy: An Update on Inhibitors of Viral Lytic Replication. Viruses. 2014; 6(11):4731-4759. https://doi.org/10.3390/v6114731
Chicago/Turabian StyleCoen, Natacha, Sophie Duraffour, Robert Snoeck, and Graciela Andrei. 2014. "KSHV Targeted Therapy: An Update on Inhibitors of Viral Lytic Replication" Viruses 6, no. 11: 4731-4759. https://doi.org/10.3390/v6114731