RNA Virus Reverse Genetics and Vaccine Design

Abstract
:1. Introduction
2. RNA Virus Biology and Reverse Genetic Infectious Clone Design


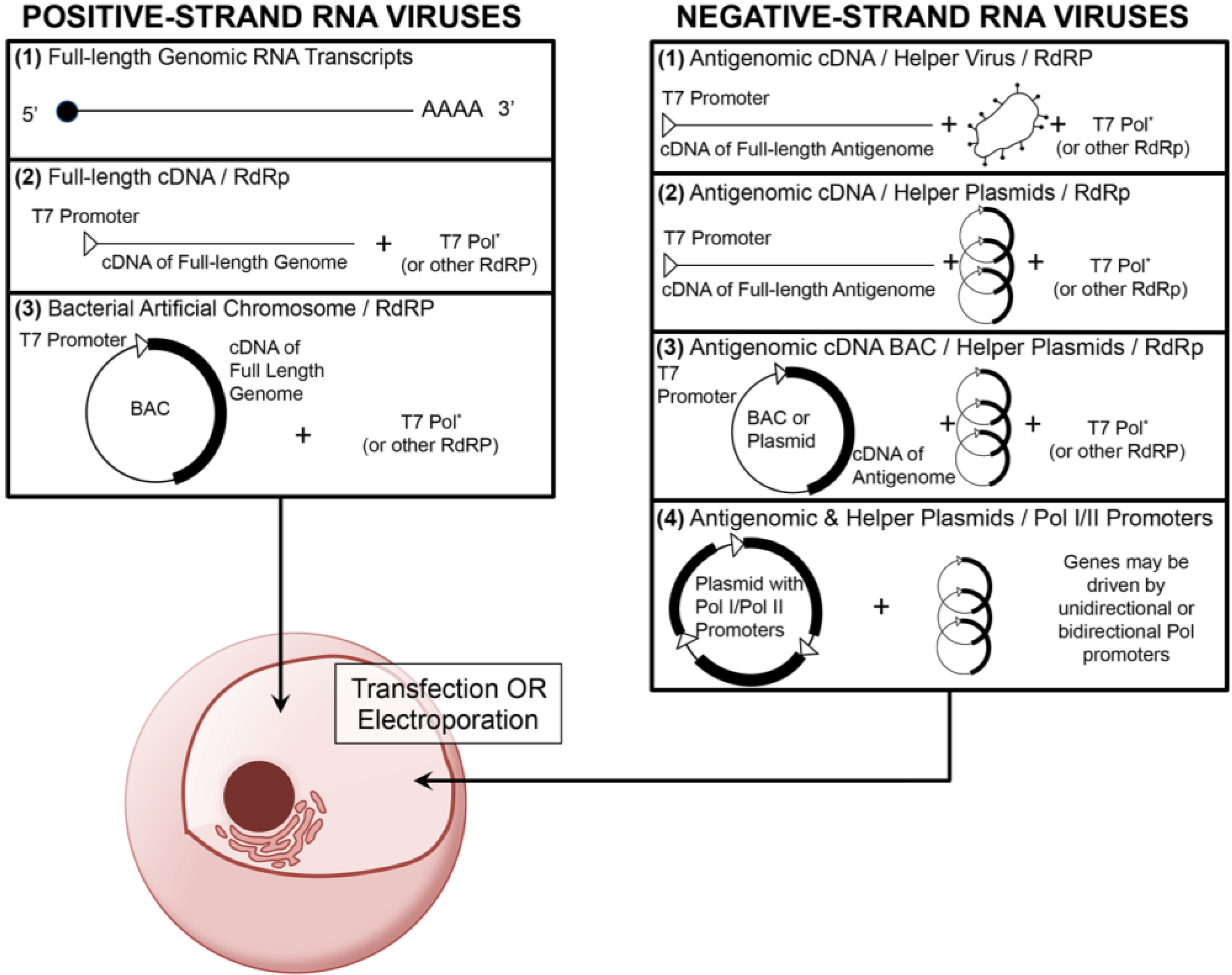{kind=link}
| Virus | Family | Genome | Genome Size | Year | Reference |
|---|---|---|---|---|---|
| Transmissible gastroenteritis virus (TGEV) | Coronaviridae | +ssRNA | 28 kb | 2000 | [4] |
| Japanese encephalitis virus (JEV) | Flaviviridae | +ssRNA | 11 kb | 2003 | [14] |
| Porcine reproductive and respiratory syndrome virus (PRRSV) | Arteriviridae | +ssRNA | 15 kb | 2006 | [15] |
| Human coronavirus OC43 | Coronaviridae | +ssRNA | 31 kb | 2006 | [16] |
| Severe acute respiratory syndrome coronavirus (SARS-CoV) | Coronaviridae | +ssRNA | 30 kb | 2007 | [17] |
| Dengue virus type 1 | Flaviviridae | +ssRNA | 11 kb | 2007 | [18] |
| Bovine viral diarrheal virus (BVDV) | Flaviviridae | +ssRNA | 12 kb | 2008 | [19] |
| Border disease virus (BDV) | Flaviviridae | +ssRNA | 12 kb | 2010 | [20] |
| Classical swine fever virus (CSFV) | Flaviviridae | +ssRNA | 12 kb | 2010 | [20] |
| Respiratory syncytial virus (RSV) | Paramyxoviridae | -ssRNA | 15 kb | 2012 | [21] |
| Feline infectious peritonitis virus (FIPV) | Coronaviridae | +ssRNA | 29 kb | 2012 | [22] |
| Middle East respiratory syndrome coronavirus (MERS-CoV) | Coronaviridae | +ssRNA | 30 kb | 2013 | [3] |
| Dengue virus type 2 | Flaviviridae | +ssRNA | 11 kb | 2014 | [23] |
3. Positive-Strand RNA Virus Biology, Reverse Genetics, and Vaccine Design
3.3. Flaviviruses: Yellow Fever, Dengue, and West Nile Viruses
4. Negative-Strand RNA Virus Biology, Reverse Genetics, and Vaccine Design
4.1. Paramyxoviruses: Measles Virus and Respiratory Syncytial Viruses
4.2. Orthomyxoviruses: Influenza virus
5. Synthetic Biology and the Future of RNA Virus Vaccine Design
5.1. Limitations and Challenges to the Application of Reverse Genetics to Vaccine Design
5.2. Current and Future Directions to Vaccine Design
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Roush, S.W.; Murphy, T.V.; Vaccine-Preventable Disease Table Working Group. Historical comparisons of morbidity and mortality for vaccine-preventable diseases in the united states. JAMA 2007, 298, 2155–2163. [Google Scholar] [CrossRef]
- Racaniello, V.R.; Baltimore, D. Cloned poliovirus complementary DNA is infectious in mammalian cells. Science 1981, 214, 916–919. [Google Scholar]
- Almazan, F.; DeDiego, M.L.; Sola, I.; Zuniga, S.; Nieto-Torres, J.L.; Marquez-Jurado, S.; Andres, G.; Enjuanes, L. Engineering a replication-competent, propagation-defective middle east respiratory syndrome coronavirus as a vaccine candidate. mBio 2013, 4, e00650–e00613. [Google Scholar]
- Almazan, F.; Gonzalez, J.M.; Penzes, Z.; Izeta, A.; Calvo, E.; Plana-Duran, J.; Enjuanes, L. Engineering the largest rna virus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 2000, 97, 5516–5521. [Google Scholar]
- Scobey, T.; Yount, B.L.; Sims, A.C.; Donaldson, E.F.; Agnihothram, S.S.; Menachery, V.D.; Graham, R.L.; Swanstrom, J.; Bove, P.F.; Kim, J.D.; et al. Reverse genetics with a full-length infectious cdna of the middle east respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2013, 110, 16157–16162. [Google Scholar] [CrossRef]
- Shi, P.Y.; Tilgner, M.; Lo, M.K.; Kent, K.A.; Bernard, K.A. Infectious cdna clone of the epidemic west nile virus from new york city. J. Virol. 2002, 76, 5847–5856. [Google Scholar] [CrossRef]
- Thiel, V.; Herold, J.; Schelle, B.; Siddell, S.G. Infectious rna transcribed in vitro from a cdna copy of the human coronavirus genome cloned in vaccinia virus. J. Gen. Virol. 2001, 82, 1273–1281. [Google Scholar]
- Yount, B.; Curtis, K.M.; Baric, R.S. Strategy for systematic assembly of large rna and DNA genomes: Transmissible gastroenteritis virus model. J. Virol. 2000, 74, 10600–10611. [Google Scholar] [CrossRef]
- Yount, B.; Curtis, K.M.; Fritz, E.A.; Hensley, L.E.; Jahrling, P.B.; Prentice, E.; Denison, M.R.; Geisbert, T.W.; Baric, R.S. Reverse genetics with a full-length infectious cdna of severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2003, 100, 12995–13000. [Google Scholar] [CrossRef]
- Yount, B.; Denison, M.R.; Weiss, S.R.; Baric, R.S. Systematic assembly of a full-length infectious cdna of mouse hepatitis virus strain a59. J. Virol. 2002, 76, 11065–11078. [Google Scholar] [CrossRef]
- Hoffmann, E.; Neumann, G.; Hobom, G.; Webster, R.G.; Kawaoka, Y. "Ambisense" approach for the generation of influenza a virus: Vrna and mrna synthesis from one template. Virology 2000, 267, 310–317. [Google Scholar] [CrossRef]
- Hoffmann, E.; Webster, R.G. Unidirectional rna polymerase i-polymerase ii transcription system for the generation of influenza a virus from eight plasmids. J. Gen. Virol. 2000, 81, 2843–2847. [Google Scholar]
- Flatz, L.; Bergthaler, A.; de la Torre, J.C.; Pinschewer, D.D. Recovery of an arenavirus entirely from rna polymerase i/ii-driven cdna. Proc. Natl. Acad. Sci. USA 2006, 103, 4663–4668. [Google Scholar] [CrossRef]
- Yun, S.I.; Kim, S.Y.; Rice, C.M.; Lee, Y.M. Development and application of a reverse genetics system for japanese encephalitis virus. J. Virol. 2003, 77, 6450–6465. [Google Scholar] [CrossRef]
- Choi, Y.J.; Yun, S.I.; Kang, S.Y.; Lee, Y.M. Identification of 5' and 3' cis-acting elements of the porcine reproductive and respiratory syndrome virus: Acquisition of novel 5' au-rich sequences restored replication of a 5'-proximal 7-nucleotide deletion mutant. J. Virol. 2006, 80, 723–736. [Google Scholar] [CrossRef]
- St-Jean, J.R.; Desforges, M.; Almazan, F.; Jacomy, H.; Enjuanes, L.; Talbot, P.J. Recovery of a neurovirulent human coronavirus oc43 from an infectious cdna clone. J. Virol. 2006, 80, 3670–3674. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Alvarez, E.; Almazan, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A severe acute respiratory syndrome coronavirus that lacks the e gene is attenuated in vitro and in vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef]
- Suzuki, R.; de Borba, L.; Duarte dos Santos, C.N.; Mason, P.W. Construction of an infectious cdna clone for a brazilian prototype strain of dengue virus type 1: Characterization of a temperature-sensitive mutation in ns1. Virology 2007, 362, 374–383. [Google Scholar] [CrossRef]
- Fan, Z.C.; Bird, R.C. An improved reverse genetics system for generation of bovine viral diarrhea virus as a bac cdna. J. Virol. Methods. 2008, 149, 309–315. [Google Scholar]
- Rasmussen, T.B.; Reimann, I.; Uttenthal, A.; Leifer, I.; Depner, K.; Schirrmeier, H.; Beer, M. Generation of recombinant pestiviruses using a full-genome amplification strategy. Vet. Microbiol. 2010, 142, 13–17. [Google Scholar] [CrossRef]
- Hotard, A.L.; Shaikh, F.Y.; Lee, S.; Yan, D.; Teng, M.N.; Plemper, R.K.; Crowe, J.E., Jr.; Moore, M.L. A stabilized respiratory syncytial virus reverse genetics system amenable to recombination-mediated mutagenesis. Virology 2012, 434, 129–136. [Google Scholar]
- Balint, A.; Farsang, A.; Zadori, Z.; Hornyak, A.; Dencso, L.; Almazan, F.; Enjuanes, L.; Belak, S. Molecular characterization of feline infectious peritonitis virus strain df-2 and studies of the role of orf3abc in viral cell tropism. J. Virol. 2012, 86, 6258–6267. [Google Scholar] [CrossRef]
- Usme-Ciro, J.A.; Lopera, J.A.; Enjuanes, L.; Almazan, F.; Gallego-Gomez, J.C. Development of a novel DNA-launched dengue virus type 2 infectious clone assembled in a bacterial artificial chromosome. Virus Res. 2014, 180, 12–22. [Google Scholar] [CrossRef]
- Kandolf, R.; Hofschneider, P.H. Molecular cloning of the genome of a cardiotropic coxsackie b3 virus: Full-length reverse-transcribed recombinant cdna generates infectious virus in mammalian cells. Proc. Natl. Acad. Sci. USA 1985, 82, 4818–4822. [Google Scholar] [CrossRef]
- Mizutani, S.; Colonno, R.J. In vitro synthesis of an infectious rna from cdna clones of human rhinovirus type 14. J. Virol. 1985, 56, 628–632. [Google Scholar]
- Cohen, J.I.; Ticehurst, J.R.; Feinstone, S.M.; Rosenblum, B.; Purcell, R.H. Hepatitis a virus cdna and its rna transcripts are infectious in cell culture. J. Virol. 1987, 61, 3035–3039. [Google Scholar]
- Roos, R.P.; Stein, S.; Routbort, M.; Senkowski, A.; Bodwell, T.; Wollmann, R. Theiler's murine encephalomyelitis virus neutralization escape mutants have a change in disease phenotype. J. Virol. 1989, 63, 4469–4473. [Google Scholar]
- Zibert, A.; Maass, G.; Strebel, K.; Falk, M.M.; Beck, E. Infectious foot-and-mouth disease virus derived from a cloned full-length cdna. J. Virol. 1990, 64, 2467–2473. [Google Scholar]
- Inoue, T.; Yamaguchi, S.; Saeki, T.; Sekiguchi, K. Production of infectious swine vesicular disease virus from cloned cdna in mammalian cells. J. Gen. Virol. 1990, 71, 1835–1838. [Google Scholar] [CrossRef]
- Blackburn, R.V.; Racaniello, V.R.; Righthand, V.F. Construction of an infectious cdna clone of echovirus 6. Virus Res. 1992, 22, 71–78. [Google Scholar] [CrossRef]
- Zimmermann, H.; Eggers, H.J.; Zimmermann, A.; Kraus, W.; Nelsen-Salz, B. Complete nucleotide sequence and biological properties of an infectious clone of prototype echovirus 9. Virus Res. 1995, 39, 311–319. [Google Scholar] [CrossRef]
- Palmenberg, A.C.; Spiro, D.; Kuzmickas, R.; Wang, S.; Djikeng, A.; Rathe, J.A.; Fraser-Liggett, C.M.; Liggett, S.B. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science 2009, 324, 55–59. [Google Scholar] [CrossRef]
- Waman, V.P.; Kolekar, P.S.; Kale, M.M.; Kulkarni-Kale, U. Population structure and evolution of rhinoviruses. PLoS One 2014, 9, e88981. [Google Scholar]
- Glanville, N.; McLean, G.R.; Guy, B.; Lecouturier, V.; Berry, C.; Girerd, Y.; Gregoire, C.; Walton, R.P.; Pearson, R.M.; Kebadze, T.; et al. Cross-serotype immunity induced by immunization with a conserved rhinovirus capsid protein. PLoS Pathog. 2013, 9, e1003669. [Google Scholar] [CrossRef]
- Rossmann, M.G.; Arnold, E.; Erickson, J.W.; Frankenberger, E.A.; Griffith, J.P.; Hecht, H.J.; Johnson, J.E.; Kamer, G.; Luo, M.; Mosser, A.G.; et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 1985, 317, 145–153. [Google Scholar] [CrossRef]
- Edlmayr, J.; Niespodziana, K.; Popow-Kraupp, T.; Krzyzanek, V.; Focke-Tejkl, M.; Blaas, D.; Grote, M.; Valenta, R. Antibodies induced with recombinant vp1 from human rhinovirus exhibit cross-neutralisation. Euro. Respir. J. 2011, 37, 44–52. [Google Scholar] [CrossRef]
- Bartlett, N.W.; Walton, R.P.; Edwards, M.R.; Aniscenko, J.; Caramori, G.; Zhu, J.; Glanville, N.; Choy, K.J.; Jourdan, P.; Burnet, J.; et al. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat. Med. 2008, 14, 199–204. [Google Scholar] [CrossRef]
- Dobrikova, E.Y.; Goetz, C.; Walters, R.W.; Lawson, S.K.; Peggins, J.O.; Muszynski, K.; Ruppel, S.; Poole, K.; Giardina, S.L.; Vela, E.M.; et al. Attenuation of neurovirulence, biodistribution, and shedding of a poliovirus:Rhinovirus chimera after intrathalamic inoculation in macaca fascicularis. J. Virol. 2012, 86, 2750–2759. [Google Scholar] [CrossRef]
- Jahan, N.; Wimmer, E.; Mueller, S. Polypyrimidine tract binding protein-1 (ptb1) is a determinant of the tissue and host tropism of a human rhinovirus/poliovirus chimera pv1(ripo). PLoS One 2013, 8, e60791. [Google Scholar] [CrossRef]
- Perlman, S.; Netland, J. Coronaviruses post-sars: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in saudi arabia. New Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Donaldson, E.F.; Yount, B.; Sims, A.C.; Burkett, S.; Pickles, R.J.; Baric, R.S. Systematic assembly of a full-length infectious clone of human coronavirus nl63. J. Virol. 2008, 82, 11948–11957. [Google Scholar] [CrossRef]
- Becker, M.M.; Graham, R.L.; Donaldson, E.F.; Rockx, B.; Sims, A.C.; Sheahan, T.; Pickles, R.J.; Corti, D.; Johnston, R.E.; Baric, R.S.; et al. Synthetic recombinant bat sars-like coronavirus is infectious in cultured cells and in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 19944–19949. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Lu, X.; Sperry, S.M.; Choi, L.; Denison, M.R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007, 81, 12135–12144. [Google Scholar] [CrossRef]
- Smith, E.C.; Blanc, H.; Vignuzzi, M.; Denison, M.R. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: Evidence for proofreading and potential therapeutics. PLoS Pathog. 2013, 9, e1003565. [Google Scholar] [CrossRef]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An rna proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011, 8, 270–279. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Becker, M.M.; Halpin, R.A.; Li, K.; Venter, E.; Lu, X.; Scherbakova, S.; Graham, R.L.; Baric, R.S.; Stockwell, T.B.; et al. Infidelity of sars-cov nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010, 6, e1000896. [Google Scholar] [CrossRef]
- Graham, R.L.; Becker, M.M.; Eckerle, L.D.; Bolles, M.; Denison, M.R.; Baric, R.S. A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nat. Med. 2012, 18, 1820–1826. [Google Scholar] [CrossRef]
- Hotez, P.J.; Bottazzi, M.E.; Tseng, C.T.; Zhan, B.; Lustigman, S.; Du, L.; Jiang, S. Calling for rapid development of a safe and effective mers vaccine. Microbes Infect. 2014. [Google Scholar] [CrossRef]
- Ying, T.; Du, L.; Ju, T.W.; Prabakaran, P.; Lau, C.C.; Lu, L.; Liu, Q.; Wang, L.; Feng, Y.; Wang, Y.; et al. Exceptionally potent neutralization of mers-cov by human monoclonal antibodies. J. Virol. 2014.
- Tang, X.C.; Agnihothram, S.S.; Jiao, Y.; Stanhope, J.; Graham, R.L.; Peterson, E.C.; Avnir, Y.; Tallarico, A.S.; Sheehan, J.; Zhu, Q.; et al. Identification of human neutralizing antibodies against mers-cov and their role in virus adaptive evolution. Proc. Natl. Acad. Sci. USA 2014, 111, E2018–E2026. [Google Scholar] [CrossRef]
- Ma, C.; Li, Y.; Wang, L.; Zhao, G.; Tao, X.; Tseng, C.T.; Zhou, Y.; Du, L.; Jiang, S. Intranasal vaccination with recombinant receptor-binding domain of mers-cov spike protein induces much stronger local mucosal immune responses than subcutaneous immunization: Implication for designing novel mucosal mers vaccines. Vaccine 2014, 32, 2100–2108. [Google Scholar] [CrossRef]
- Ishikawa, T.; Yamanaka, A.; Konishi, E. A review of successful flavivirus vaccines and the problems with those flaviviruses for which vaccines are not yet available. Vaccine 2014, 32, 1326–1337. [Google Scholar] [CrossRef]
- Reed, W.; Carroll, J. The prevention of yellow fever. Public Health Pap. Rep. 1901, 27, 113–129. [Google Scholar]
- Reed, W.; Carroll, J.; Agramonte, A. The etiology of yellow fever: An additional note. 1901. Mil. Med. 2001, 166, 44–53. [Google Scholar]
- Reed, W. Recent researches concerning the etiology, propagation, and prevention of yellow fever, by the united states army commission. J. Hyg. 1902, 2, 101–119. [Google Scholar] [CrossRef]
- Rice, C.M.; Grakoui, A.; Galler, R.; Chambers, T.J. Transcription of infectious yellow fever rna from full-length cdna templates produced by in vitro ligation. The New biologist 1989, 1, 285–296. [Google Scholar]
- Bredenbeek, P.J.; Kooi, E.A.; Lindenbach, B.; Huijkman, N.; Rice, C.M.; Spaan, W.J. A stable full-length yellow fever virus cdna clone and the role of conserved rna elements in flavivirus replication. J. Gen. Virol. 2003, 84, 1261–1268. [Google Scholar] [CrossRef]
- Kapoor, M.; Zhang, L.; Mohan, P.M.; Padmanabhan, R. Synthesis and characterization of an infectious dengue virus type-2 rna genome (new guinea c strain). Gene 1995, 162, 175–180. [Google Scholar] [CrossRef]
- Kinney, R.M.; Butrapet, S.; Chang, G.J.; Tsuchiya, K.R.; Roehrig, J.T.; Bhamarapravati, N.; Gubler, D.J. Construction of infectious cdna clones for dengue 2 virus: Strain 16681 and its attenuated vaccine derivative, strain pdk-53. Virology 1997, 230, 300–308. [Google Scholar] [CrossRef]
- Polo, S.; Ketner, G.; Levis, R.; Falgout, B. Infectious rna transcripts from full-length dengue virus type 2 cdna clones made in yeast. J. Virol. 1997, 71, 5366–5374. [Google Scholar]
- Puri, B.; Polo, S.; Hayes, C.G.; Falgout, B. Construction of a full length infectious clone for dengue-1 virus western pacific,74 strain. Virus Genes 2000, 20, 57–63. [Google Scholar] [CrossRef]
- Blaney, J.E., Jr.; Hanson, C.T.; Firestone, C.Y.; Hanley, K.A.; Murphy, B.R.; Whitehead, S.S. Genetically modified, live attenuated dengue virus type 3 vaccine candidates. Am. J. Trop. Med. Hyg. 2004, 71, 811–821. [Google Scholar]
- Blaney, J.E., Jr.; Hanson, C.T.; Hanley, K.A.; Murphy, B.R.; Whitehead, S.S. Vaccine candidates derived from a novel infectious cdna clone of an american genotype dengue virus type 2. BMC Infect. Dis. 2004, 4, 39. [Google Scholar] [CrossRef]
- Chen, W.; Kawano, H.; Men, R.; Clark, D.; Lai, C.J. Construction of intertypic chimeric dengue viruses exhibiting type 3 antigenicity and neurovirulence for mice. J. Virol. 1995, 69, 5186–5190. [Google Scholar]
- Lai, C.J.; Zhao, B.T.; Hori, H.; Bray, M. Infectious rna transcribed from stably cloned full-length cdna of dengue type 4 virus. Proc. Natl. Acad. Sci. USA 1991, 88, 5139–5143. [Google Scholar] [CrossRef]
- Sumiyoshi, H.; Hoke, C.H.; Trent, D.W. Infectious japanese encephalitis virus rna can be synthesized from in vitro-ligated cdna templates. J. Virol. 1992, 66, 5425–5431. [Google Scholar]
- Khromykh, A.A.; Westaway, E.G. Completion of kunjin virus rna sequence and recovery of an infectious rna transcribed from stably cloned full-length cdna. J. Virol. 1994, 68, 4580–4588. [Google Scholar]
- Gritsun, T.S.; Gould, E.A. Infectious transcripts of tick-borne encephalitis virus, generated in days by rt-pcr. Virology 1995, 214, 611–618. [Google Scholar] [CrossRef]
- Gritsun, T.S.; Gould, E.A. Development and analysis of a tick-borne encephalitis virus infectious clone using a novel and rapid strategy. J. Virol. Methods. 1998, 76, 109–120. [Google Scholar] [CrossRef]
- Mandl, C.W.; Ecker, M.; Holzmann, H.; Kunz, C.; Heinz, F.X. Infectious cdna clones of tick-borne encephalitis virus european subtype prototypic strain neudoerfl and high virulence strain hypr. J. Gen. Virol. 1997, 78, 1049–1057. [Google Scholar]
- Hurrelbrink, R.J.; Nestorowicz, A.; McMinn, P.C. Characterization of infectious murray valley encephalitis virus derived from a stably cloned genome-length cdna. J. Gen. Virol. 1999, 80, 3115–3125. [Google Scholar]
- Campbell, M.S.; Pletnev, A.G. Infectious cdna clones of langat tick-borne flavivirus that differ from their parent in peripheral neurovirulence. Virology 2000, 269, 225–237. [Google Scholar] [CrossRef]
- Yamshchikov, V.F.; Wengler, G.; Perelygin, A.A.; Brinton, M.A.; Compans, R.W. An infectious clone of the west nile flavivirus. Virology 2001, 281, 294–304. [Google Scholar] [CrossRef]
- Yoshii, K.; Igarashi, M.; Ito, K.; Kariwa, H.; Holbrook, M.R.; Takashima, I. Construction of an infectious cdna clone for omsk hemorrhagic fever virus, and characterization of mutations in ns2a and ns5. Virus Res. 2011, 155, 61–68. [Google Scholar] [CrossRef]
- Theiler, M.; Smith, H.H. The use of yellow fever virus modified by in vitro cultivation for human immunization. J. Exp. Med. 1937, 65, 787–800. [Google Scholar] [CrossRef]
- Bonaldo, M.C.; Sequeira, P.C.; Galler, R. The yellow fever 17d virus as a platform for new live attenuated vaccines. Hum. Vaccin. Immunother. 2014, 10, 1256–1265. [Google Scholar]
- Lefeuvre, A.; Marianneau, P.; Deubel, V. Current assessment of yellow fever and yellow fever vaccine. Curr. Infect. Dis. Rep. 2004, 6, 96–104. [Google Scholar] [CrossRef]
- Poland, J.D.; Calisher, C.H.; Monath, T.P.; Downs, W.G.; Murphy, K. Persistence of neutralizing antibody 30–35 years after immunization with 17d yellow fever vaccine. Bull. World Health Organ. 1981, 59, 895–900. [Google Scholar]
- Webster, L.T. Japanese b encephalitis virus: Its differentiation from st. Louis encephalitis virus and relationship to louping-ill virus. Science 1937, 86, 402–403. [Google Scholar]
- Normile, D. Tropical medicine. Surprising new dengue virus throws a spanner in disease control efforts. Science 2013, 342, 415. [Google Scholar] [CrossRef]
- Halstead, S.B. Immune enhancement of viral infection. Prog. Allergy 1982, 31, 301–364. [Google Scholar]
- Guirakhoo, F.; Arroyo, J.; Pugachev, K.V.; Miller, C.; Zhang, Z.X.; Weltzin, R.; Georgakopoulos, K.; Catalan, J.; Ocran, S.; Soike, K.; et al. Construction, safety, and immunogenicity in nonhuman primates of a chimeric yellow fever-dengue virus tetravalent vaccine. J. Virol. 2001, 75, 7290–7304. [Google Scholar] [CrossRef]
- Caufour, P.S.; Motta, M.C.; Yamamura, A.M.; Vazquez, S.; Ferreira, II; Jabor, A.V.; Bonaldo, M.C.; Freire, M.S.; Galler, R. Construction, characterization and immunogenicity of recombinant yellow fever 17d-dengue type 2 viruses. Virus Res. 2001, 79, 1–14. [Google Scholar] [CrossRef]
- Guy, B.; Guirakhoo, F.; Barban, V.; Higgs, S.; Monath, T.P.; Lang, J. Preclinical and clinical development of yfv 17d-based chimeric vaccines against dengue, west nile and japanese encephalitis viruses. Vaccine 2010, 28, 632–649. [Google Scholar] [CrossRef]
- Arroyo, J.; Miller, C.; Catalan, J.; Myers, G.A.; Ratterree, M.S.; Trent, D.W.; Monath, T.P. Chimerivax-west nile virus live-attenuated vaccine: Preclinical evaluation of safety, immunogenicity, and efficacy. J. Virol. 2004, 78, 12497–12507. [Google Scholar] [CrossRef]
- Schnell, M.J.; Mebatsion, T.; Conzelmann, K.K. Infectious rabies viruses from cloned cdna. EMBO J. 1994, 13, 4195–4203. [Google Scholar]
- Conzelmann, K.K.; Schnell, M. Rescue of synthetic genomic rna analogs of rabies virus by plasmid-encoded proteins. J. Virol. 1994, 68, 713–719. [Google Scholar]
- Radecke, F.; Spielhofer, P.; Schneider, H.; Kaelin, K.; Huber, M.; Dotsch, C.; Christiansen, G.; Billeter, M.A. Rescue of measles viruses from cloned DNA. EMBO J. 1995, 14, 5773–5784. [Google Scholar]
- Clarke, D.K.; Sidhu, M.S.; Johnson, J.E.; Udem, S.A. Rescue of mumps virus from cdna. J. Virol. 2000, 74, 4831–4838. [Google Scholar]
- Marsh, G.A.; Virtue, E.R.; Smith, I.; Todd, S.; Arkinstall, R.; Frazer, L.; Monaghan, P.; Smith, G.A.; Broder, C.C.; Middleton, D.; et al. Recombinant hendra viruses expressing a reporter gene retain pathogenicity in ferrets. Virol. J. 2013, 10, 95. [Google Scholar] [CrossRef]
- Yoneda, M.; Guillaume, V.; Ikeda, F.; Sakuma, Y.; Sato, H.; Wild, T.F.; Kai, C. Establishment of a nipah virus rescue system. Proc. Natl. Acad. Sci. USA 2006, 103, 16508–16513. [Google Scholar] [CrossRef]
- Collins, P.L.; Murphy, B.R. New generation live vaccines against human respiratory syncytial virus designed by reverse genetics. Proc. Am. Thorac. Soc. 2005, 2, 166–173. [Google Scholar] [CrossRef]
- Katz, S.L. John F. Enders and measles virus vaccine—A reminiscence. Curr. Top. Microbiol. Immunol. 2009, 329, 3–11. [Google Scholar]
- Buynak, E.B.; Hilleman, M.R. Live attenuated mumps virus vaccine. 1. Vaccine development. Exp. Biol. Med. 1966, 123, 768–775. [Google Scholar] [CrossRef]
- Stokes, J., Jr.; Weibel, R.E.; Villarejos, V.M.; Arguedas, J.A.; Buynak, E.B.; Hilleman, M.R. Trivalent combined measles-mumps-rubella vaccine. Findings in clinical-laboratory studies. JAMA 1971, 218, 57–61. [Google Scholar] [CrossRef]
- Blount, R.E., Jr.; Morris, J.A.; Savage, R.E. Recovery of cytopathogenic agent from chimpanzees with coryza. Exp. Biol. Med. 1956, 92, 544–549. [Google Scholar] [CrossRef]
- Hall, C.B.; Weinberg, G.A.; Iwane, M.K.; Blumkin, A.K.; Edwards, K.M.; Staat, M.A.; Auinger, P.; Griffin, M.R.; Poehling, K.A.; Erdman, D.; et al. The burden of respiratory syncytial virus infection in young children. New Engl. J. Med. 2009, 360, 588–598. [Google Scholar]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O'Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar]
- Glezen, W.P.; Taber, L.H.; Frank, A.L.; Kasel, J.A. Risk of primary infection and reinfection with respiratory syncytial virus. Am. J. Dis. Child. 1986, 140, 543–546. [Google Scholar]
- Subramanian, K.N.; Weisman, L.E.; Rhodes, T.; Ariagno, R.; Sanchez, P.J.; Steichen, J.; Givner, L.B.; Jennings, T.L.; Top, F.H., Jr.; Carlin, D.; et al. Safety, tolerance and pharmacokinetics of a humanized monoclonal antibody to respiratory syncytial virus in premature infants and infants with bronchopulmonary dysplasia. Medi-493 study group. J. Pediatr. Infect. Dis. 1998, 17, 110–115. [Google Scholar] [CrossRef]
- Abarca, K.; Jung, E.; Fernandez, P.; Zhao, L.; Harris, B.; Connor, E.M.; Losonsky, G.A.; Motavizumab Study, G. Safety, tolerability, pharmacokinetics, and immunogenicity of motavizumab, a humanized, enhanced-potency monoclonal antibody for the prevention of respiratory syncytial virus infection in at-risk children. J. Pediatr. Infect. Dis. 2009, 28, 267–272. [Google Scholar]
- Kamal-Bahl, S.; Doshi, J.; Campbell, J. Economic analyses of respiratory syncytial virus immunoprophylaxis in high-risk infants: A systematic review. Arch. Pediatr. Adolesc. Med. 2002, 156, 1034–1041. [Google Scholar] [CrossRef]
- Kim, H.W.; Leikin, S.L.; Arrobio, J.; Brandt, C.D.; Chanock, R.M.; Parrott, R.H. Cell-mediated immunity to respiratory syncytial virus induced by inactivated vaccine or by infection. Pediatr. Res. 1976, 10, 75–78. [Google Scholar] [CrossRef]
- Collins, P.L.; Melero, J.A. Progress in understanding and controlling respiratory syncytial virus: Still crazy after all these years. Virus Res. 2011, 162, 80–99. [Google Scholar] [CrossRef]
- Karron, R.A.; Wright, P.F.; Belshe, R.B.; Thumar, B.; Casey, R.; Newman, F.; Polack, F.P.; Randolph, V.B.; Deatly, A.; Hackell, J.; et al. Identification of a recombinant live attenuated respiratory syncytial virus vaccine candidate that is highly attenuated in infants. J. Infect. Dis. 2005, 191, 1093–1104. [Google Scholar] [CrossRef]
- Schickli, J.H.; Kaur, J.; Tang, R.S. Nonclinical phenotypic and genotypic analyses of a phase 1 pediatric respiratory syncytial virus vaccine candidate medi-559 (ra2cp248/404/1030deltash) at permissive and non-permissive temperatures. Virus Res. 2012, 169, 38–47. [Google Scholar] [CrossRef]
- Luongo, C.; Winter, C.C.; Collins, P.L.; Buchholz, U.J. Increased genetic and phenotypic stability of a promising live-attenuated respiratory syncytial virus vaccine candidate by reverse genetics. J. Virol. 2012, 86, 10792–10804. [Google Scholar] [CrossRef]
- Graham, B.S. Biological challenges and technological opportunities for respiratory syncytial virus vaccine development. Immunol. Rev. 2011, 239, 149–166. [Google Scholar] [CrossRef]
- Engelhardt, O.G. Many ways to make an influenza virus—Review of influenza virus reverse genetics methods. Influenza Other Respir. Viruses 2013, 7, 249–256. [Google Scholar] [CrossRef]
- Enami, M.; Luytjes, W.; Krystal, M.; Palese, P. Introduction of site-specific mutations into the genome of influenza virus. Proc. Natl. Acad. Sci. USA 1990, 87, 3802–3805. [Google Scholar] [CrossRef]
- Luytjes, W.; Krystal, M.; Enami, M.; Parvin, J.D.; Palese, P. Amplification, expression, and packaging of foreign gene by influenza virus. Cell 1989, 59, 1107–1113. [Google Scholar] [CrossRef]
- Fodor, E.; Devenish, L.; Engelhardt, O.G.; Palese, P.; Brownlee, G.G.; Garcia-Sastre, A. Rescue of influenza a virus from recombinant DNA. J. Virol. 1999, 73, 9679–9682. [Google Scholar]
- Neumann, G.; Kawaoka, Y. Generation of influenza a virus from cloned cdnas--historical perspective and outlook for the new millenium. Rev. Med. Virol. 2002, 12, 13–30. [Google Scholar] [CrossRef]
- Koudstaal, W.; Hartgroves, L.; Havenga, M.; Legastelois, I.; Ophorst, C.; Sieuwerts, M.; Zuijdgeest, D.; Vogels, R.; Custers, J.; de Boer-Luijtze, E.; et al. Suitability of per.C6 cells to generate epidemic and pandemic influenza vaccine strains by reverse genetics. Vaccine 2009, 27, 2588–2593. [Google Scholar] [CrossRef]
- de Wit, E.; Spronken, M.I.; Vervaet, G.; Rimmelzwaan, G.F.; Osterhaus, A.D.; Fouchier, R.A. A reverse-genetics system for influenza a virus using t7 rna polymerase. J. Gen. Virol. 2007, 88, 1281–1287. [Google Scholar] [CrossRef]
- Crescenzo-Chaigne, B.; van der Werf, S. Rescue of influenza c virus from recombinant DNA. J. Virol. 2007, 81, 11282–11289. [Google Scholar] [CrossRef]
- Muraki, Y.; Murata, T.; Takashita, E.; Matsuzaki, Y.; Sugawara, K.; Hongo, S. A mutation on influenza c virus m1 protein affects virion morphology by altering the membrane affinity of the protein. J. Virol. 2007, 81, 8766–8773. [Google Scholar] [CrossRef]
- Massin, P.; Rodrigues, P.; Marasescu, M.; van der Werf, S.; Naffakh, N. Cloning of the chicken rna polymerase i promoter and use for reverse genetics of influenza a viruses in avian cells. J. Virol. 2005, 79, 13811–13816. [Google Scholar] [CrossRef]
- Murakami, S.; Horimoto, T.; Yamada, S.; Kakugawa, S.; Goto, H.; Kawaoka, Y. Establishment of canine rna polymerase i-driven reverse genetics for influenza a virus: Its application for h5n1 vaccine production. J. Virol. 2008, 82, 1605–1609. [Google Scholar] [CrossRef]
- Neumann, G.; Fujii, K.; Kino, Y.; Kawaoka, Y. An improved reverse genetics system for influenza a virus generation and its implications for vaccine production. Proc. Natl. Acad. Sci. USA 2005, 102, 16825–16829. [Google Scholar] [CrossRef]
- Vinnemeier, C.D.; Fischer-Herr, J.; Meyer, S.; Liebig, K.; Theess, W.; Burchard, G.D.; Cramer, J.P. Immunogenicity and safety of an inactivated 2012/2013 trivalent influenza vaccine produced in mammalian cell culture (optaflu): An open label, uncontrolled study. Hum. Vaccin. Immunother. 2013, 10, 441–448. [Google Scholar]
- Doroshenko, A.; Halperin, S.A. Trivalent mdck cell culture-derived influenza vaccine optaflu (novartis vaccines). Expert Rev. Vaccin. 2009, 8, 679–688. [Google Scholar] [CrossRef]
- Bright, R.A.; Carter, D.M.; Daniluk, S.; Toapanta, F.R.; Ahmad, A.; Gavrilov, V.; Massare, M.; Pushko, P.; Mytle, N.; Rowe, T.; et al. Influenza virus-like particles elicit broader immune responses than whole virion inactivated influenza virus or recombinant hemagglutinin. Vaccine 2007, 25, 3871–3878. [Google Scholar] [CrossRef]
- Pushko, P.; Tumpey, T.M.; Van Hoeven, N.; Belser, J.A.; Robinson, R.; Nathan, M.; Smith, G.; Wright, D.C.; Bright, R.A. Evaluation of influenza virus-like particles and novasome adjuvant as candidate vaccine for avian influenza. Vaccine 2007, 25, 4283–4290. [Google Scholar] [CrossRef]
- Pushko, P.; Tumpey, T.M.; Bu, F.; Knell, J.; Robinson, R.; Smith, G. Influenza virus-like particles comprised of the ha, na, and m1 proteins of h9n2 influenza virus induce protective immune responses in balb/c mice. Vaccine 2005, 23, 5751–5759. [Google Scholar] [CrossRef]
- Galarza, J.M.; Latham, T.; Cupo, A. Virus-like particle vaccine conferred complete protection against a lethal influenza virus challenge. Viral Immunol. 2005, 18, 365–372. [Google Scholar] [CrossRef]
- Quan, F.S.; Huang, C.; Compans, R.W.; Kang, S.M. Virus-like particle vaccine induces protective immunity against homologous and heterologous strains of influenza virus. J. Virol. 2007, 81, 3514–3524. [Google Scholar] [CrossRef]
- Treanor, J.J.; Schiff, G.M.; Hayden, F.G.; Brady, R.C.; Hay, C.M.; Meyer, A.L.; Holden-Wiltse, J.; Liang, H.; Gilbert, A.; Cox, M. Safety and immunogenicity of a baculovirus-expressed hemagglutinin influenza vaccine: A randomized controlled trial. JAMA 2007, 297, 1577–1582. [Google Scholar] [CrossRef]
- Traynor, K. First recombinant flu vaccine approved. AJHP 2013, 70, 382. [Google Scholar]
- Yang, L.P. Recombinant trivalent influenza vaccine (flublok(®)): A review of its use in the prevention of seasonal influenza in adults. Drugs 2013, 73, 1357–1366. [Google Scholar] [CrossRef]
- Dormitzer, P.R.; Suphaphiphat, P.; Gibson, D.G.; Wentworth, D.E.; Stockwell, T.B.; Algire, M.A.; Alperovich, N.; Barro, M.; Brown, D.M.; Craig, S.; et al. Synthetic generation of influenza vaccine viruses for rapid response to pandemics. Sci. Transl. Med. 2013, 5, 185ra168. [Google Scholar]
- Burns, C.C.; Shaw, J.; Campagnoli, R.; Jorba, J.; Vincent, A.; Quay, J.; Kew, O. Modulation of poliovirus replicative fitness in hela cells by deoptimization of synonymous codon usage in the capsid region. J. Virol. 2006, 80, 3259–3272. [Google Scholar] [CrossRef]
- Mueller, S.; Papamichail, D.; Coleman, J.R.; Skiena, S.; Wimmer, E. Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J. Virol. 2006, 80, 9687–9696. [Google Scholar] [CrossRef]
- Dormitzer, P.R.; Grandi, G.; Rappuoli, R. Structural vaccinology starts to deliver. Nat. Rev. Microbiol. 2012, 10, 807–813. [Google Scholar] [CrossRef]
- Correia, B.E.; Bates, J.T.; Loomis, R.J.; Baneyx, G.; Carrico, C.; Jardine, J.G.; Rupert, P.; Correnti, C.; Kalyuzhniy, O.; Vittal, V.; et al. Proof of principle for epitope-focused vaccine design. Nature 2014, 507, 201–206. [Google Scholar] [CrossRef]
- McLellan, J.S.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.; Yang, Y.; Zhang, B.; Chen, L.; Srivatsan, S.; Zheng, A.; et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013, 342, 592–598. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stobart, C.C.; Moore, M.L. RNA Virus Reverse Genetics and Vaccine Design. Viruses 2014, 6, 2531-2550. https://doi.org/10.3390/v6072531
Stobart CC, Moore ML. RNA Virus Reverse Genetics and Vaccine Design. Viruses. 2014; 6(7):2531-2550. https://doi.org/10.3390/v6072531
Chicago/Turabian StyleStobart, Christopher C., and Martin L. Moore. 2014. "RNA Virus Reverse Genetics and Vaccine Design" Viruses 6, no. 7: 2531-2550. https://doi.org/10.3390/v6072531
APA StyleStobart, C. C., & Moore, M. L. (2014). RNA Virus Reverse Genetics and Vaccine Design. Viruses, 6(7), 2531-2550. https://doi.org/10.3390/v6072531