New Aspects of the Pathogenesis of Canine Distemper Leukoencephalitis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathogenesis of Canine Distemper
Neuropathogenesis of Distemper
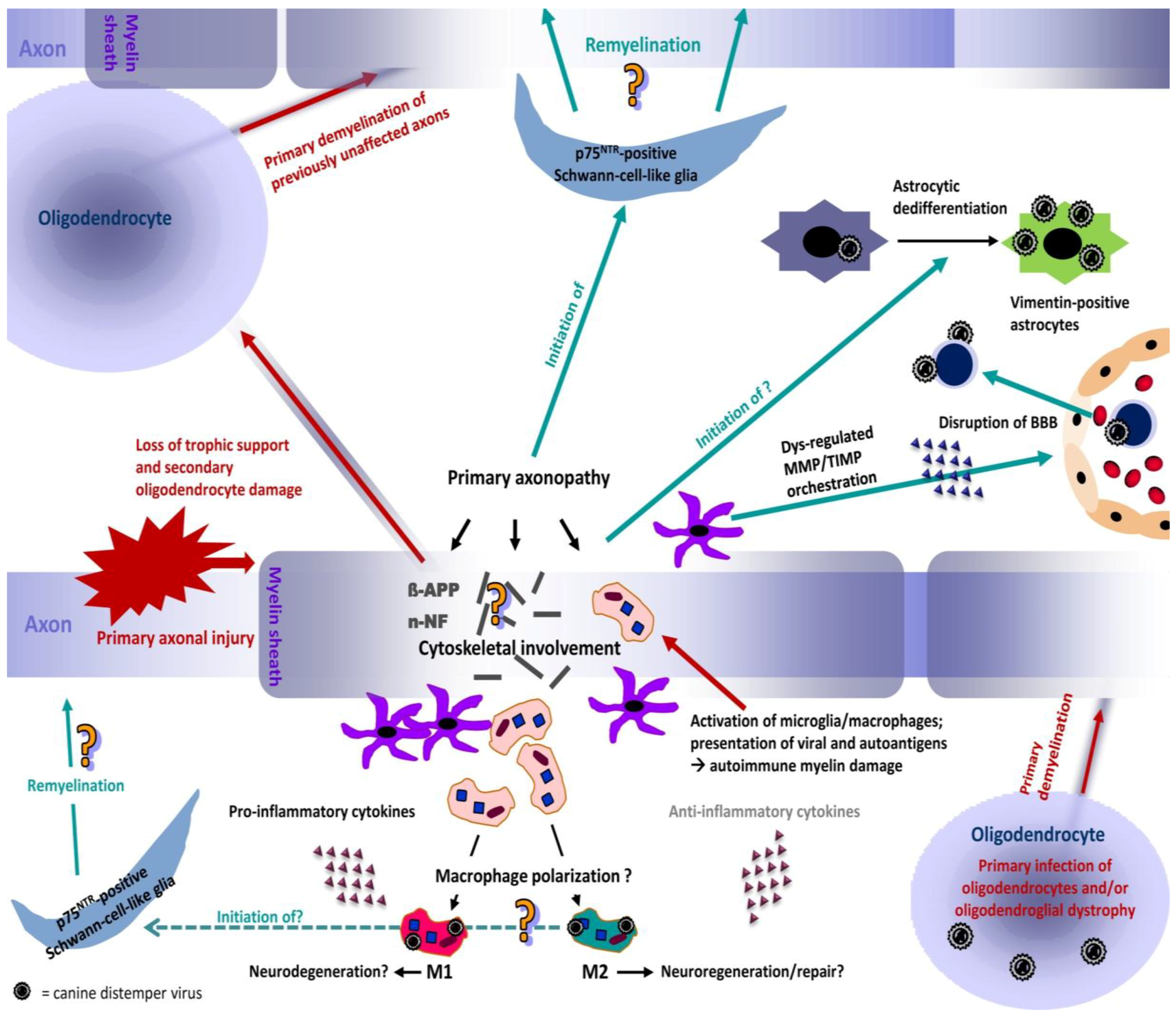
3. Canine Distemper Leukoencephalitis — Novel Aspects of its Pathogenesis
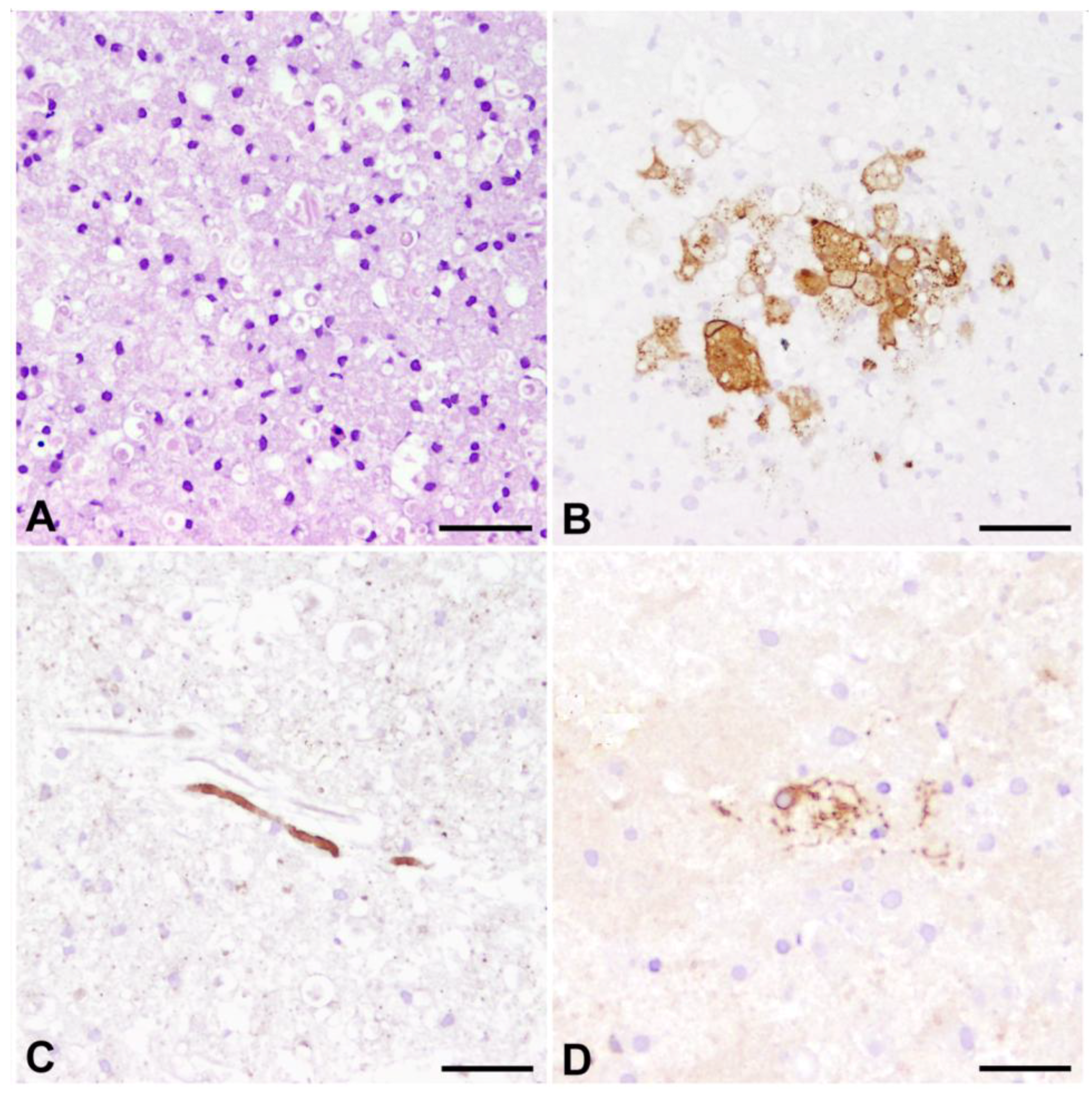
3.1. Pathogenesis and Morphology of Distemper Leukoencephalitis


3.2. Dominance of Pro-Inflammatory Cytokines in CDV-DL — A Hint for Macrophage Polarization?
3.3. The Role of Matrix Metalloproteinases and Their Inhibitors in CDV-DL
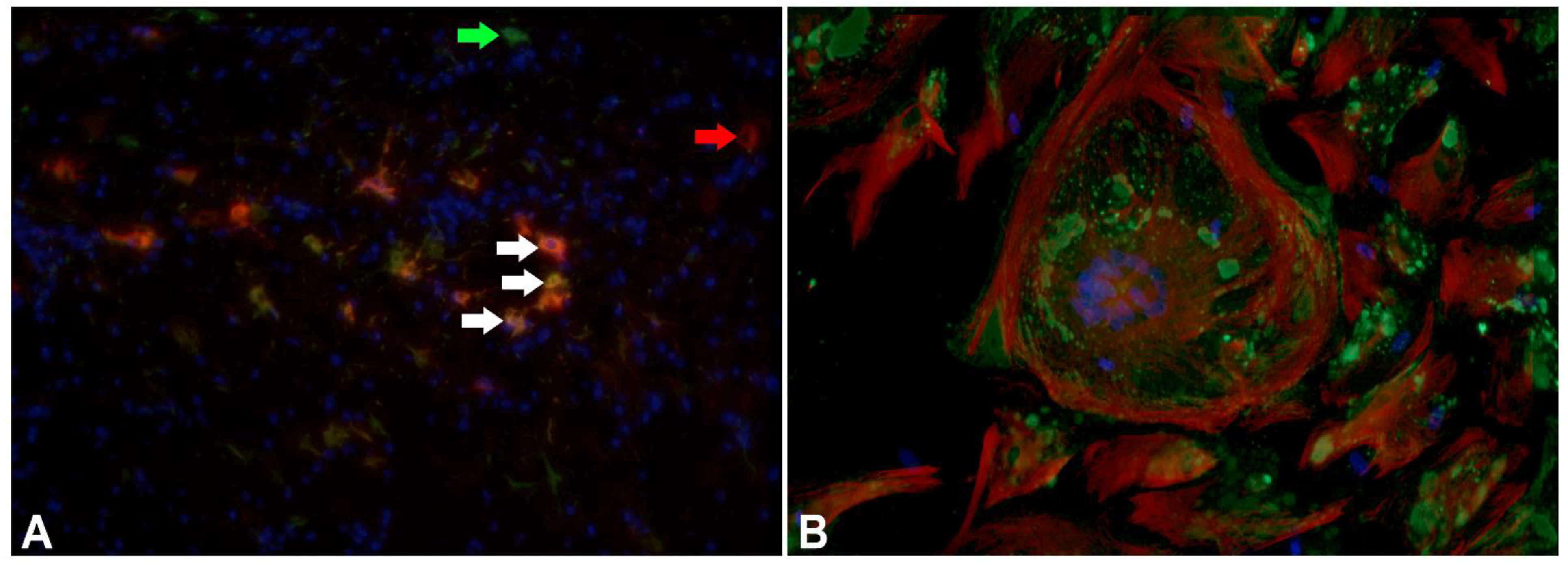
3.4. The Role of Astrocytic De-Differentiation in Chronic Lesions — Another Underestimated Mode of Virus Persistence and Spread?

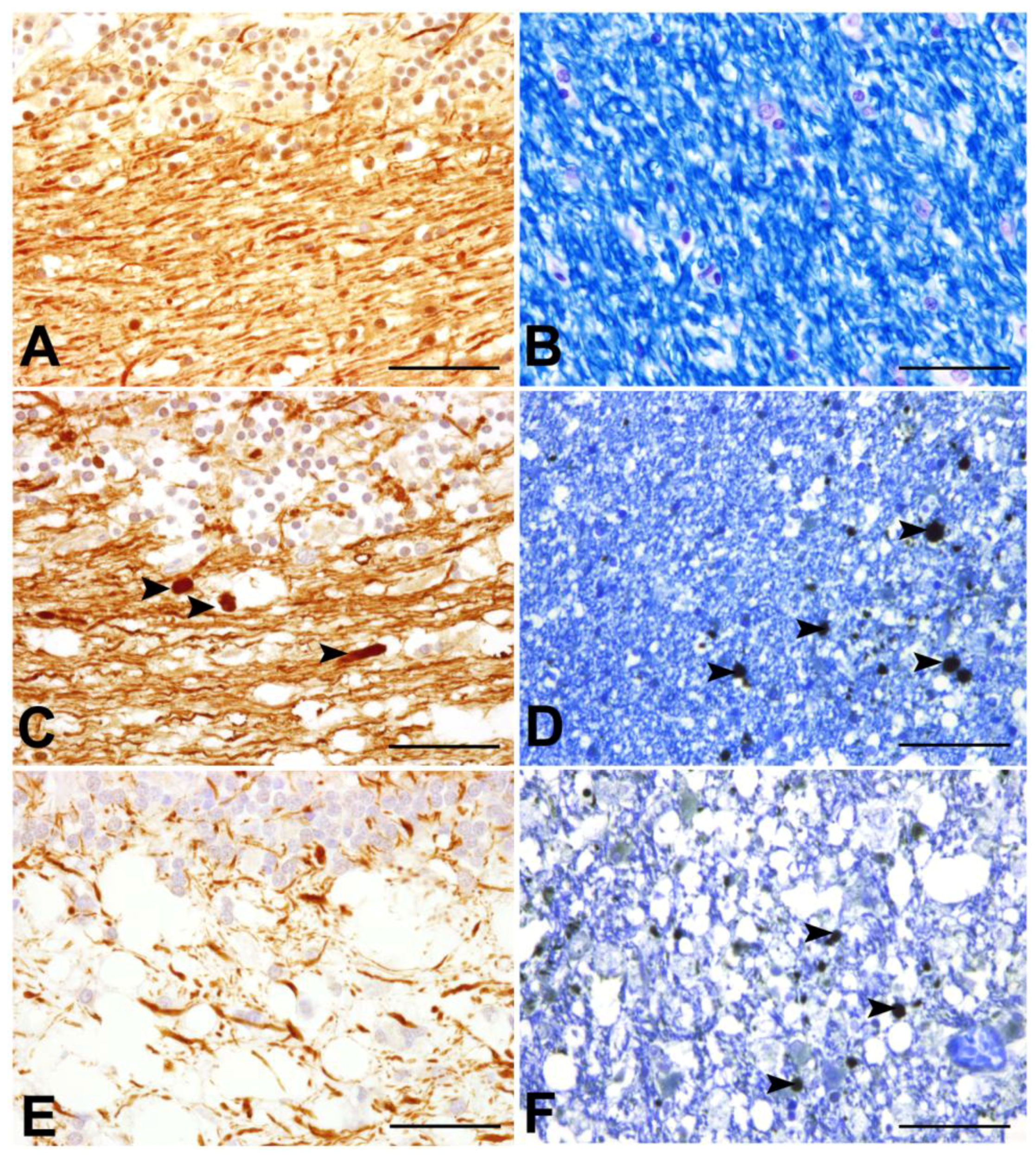
3.5. Early Axonal Damage as a Pivotal Triggering Mechanism — The End of an Old Dogma?

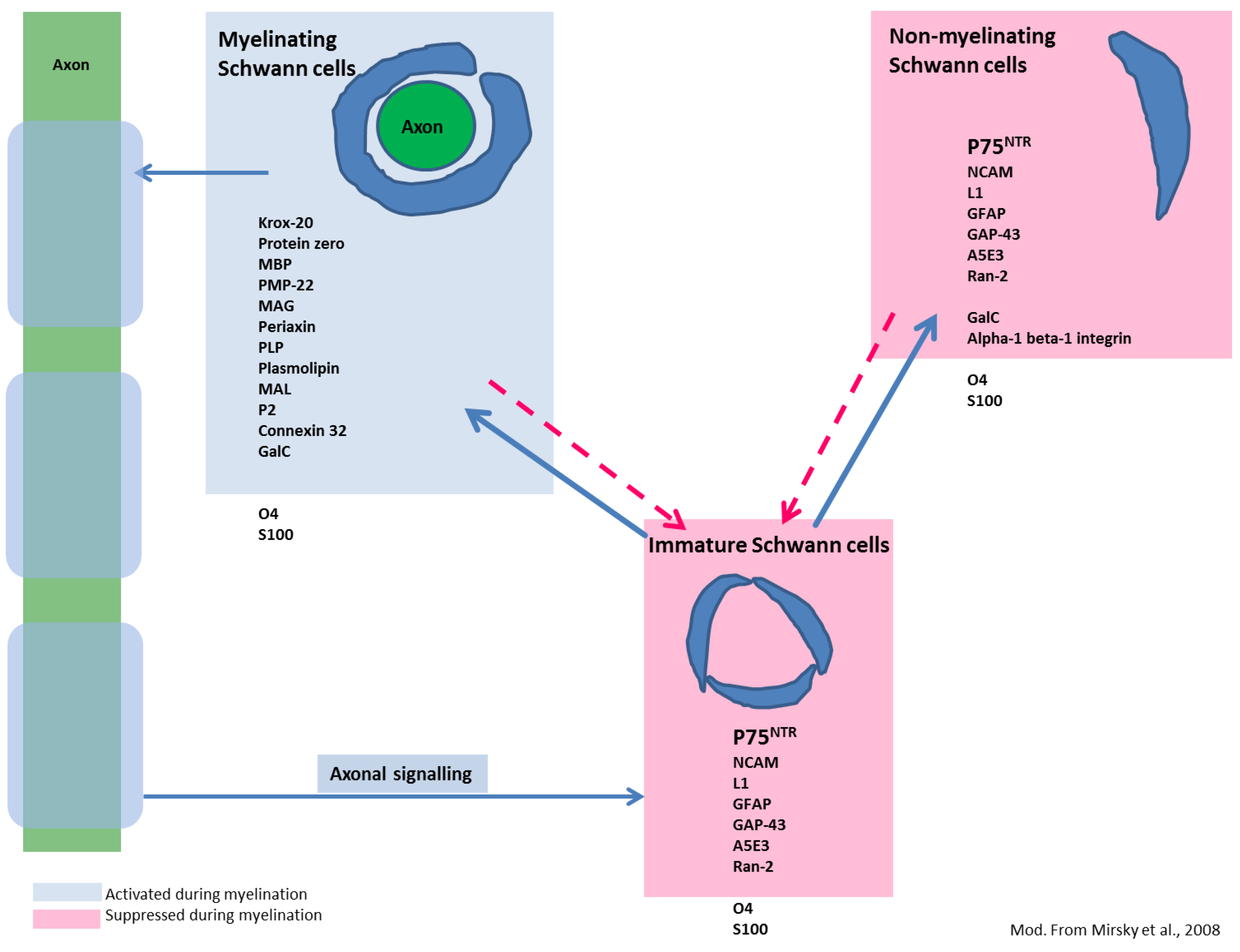
3.6. Aldynoglial p75NTR-Positive Glial Cells Emerge in Response to CDV Mediated CNS Damage — A First Step in Schwann Cell-Mediated Remyelination?

4. Organotypic Slice Cultures of the Canine CNS — A Promising in Vitro Tool for CDV-DL Research?

5. Future Perspectives and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Deem, S.L.; Spelman, L.H.; Yates, R.A.; Montali, R.J. Canine Distemper in Terrestrial Carnivores: A Review. J. Zoo Wildl. Med. 2000, 31, 441–451. [Google Scholar]
- Davies, M. Risk of Re-Emergence of Canine Distemper. Vet. Rec. 2014, 174, 178. [Google Scholar] [CrossRef]
- Beineke, A.; Puff, C.; Seehusen, F.; Baumgärtner, W. Pathogenesis and Immunopathology of Systemic and Nervous Canine Distemper. Vet. Immunol. Immunopathol. 2009, 127, 1–18. [Google Scholar] [CrossRef]
- Greene, C.E. Infectious Diseases of the Dog and Cat, 4th ed.; Elsevier/Saunders: St. Louis, MO, USA, 2012; p. 1354. [Google Scholar]
- Maeda, H.; Ozaki, K.; Takagi, Y.; Sawashima, K.; Narama, I. Distemper Skin Lesions in a Dog. Zentralbl. Veterinarmed. A 1994, 41, 247–250. [Google Scholar] [CrossRef]
- Gröne, A.; Engelhardt, P.; Zurbriggen, A. Canine Distemper Virus Infection: Proliferation of Canine Footpad Keratinocytes. Vet. Pathol. 2003, 40, 574–578. [Google Scholar] [CrossRef]
- Engelhardt, P.; Wyder, M.; Zurbriggen, A.; Gröne, A. Canine Distemper Virus Associated Proliferation of Canine Footpad Keratinocytes in vitro. Vet. Microbiol. 2005, 107, 1–12. [Google Scholar] [CrossRef]
- Tsunoda, I.; Fujinami, R.S. Neuropathogenesis of Theiler’s Murine Encephalomyelitis Virus Infection, an Animal Model for Multiple Sclerosis. J. Neuroimmune Pharmacol. 2010, 5, 355–369. [Google Scholar] [CrossRef]
- Ulrich, R.; Puff, C.; Wewetzer, K.; Kalkuhl, A.; Deschl, U.; Baumgärtner, W. Transcriptional Changes in Canine Distemper Virus-Induced Demyelinating Leukoencephalitis Favor a Biphasic Mode of Demyelination. PLoS One 2014, 9, e95917. [Google Scholar]
- Schirmer, L.; Antel, J.P.; Brück, W.; Stadelmann, C. Axonal Loss and Neurofilament Phosphorylation Changes Accompany Lesion Development and Clinical Progression in Multiple Sclerosis. Brain Pathol. 2011, 21, 428–440. [Google Scholar] [CrossRef]
- Appel, M.J. Pathogenesis of Canine Distemper. Am. J. Vet. Res. 1969, 30, 1167–1182. [Google Scholar]
- Ludlow, M.; Rennick, L.J.; Nambulli, S.; de Swart, R.L.; Paul Duprex, W. Using the Ferret Model to Study Morbillivirus Entry, Spread, Transmission and Cross-Species Infection. Curr. Opin. Virol. 2013, 4C, 15–23. [Google Scholar]
- Rudd, P.A.; Cattaneo, R.; von Messling, V. Canine Distemper Virus Uses Both the Anterograde and the Hematogenous Pathway for Neuroinvasion. J. Virol. 2006, 80, 9361–9370. [Google Scholar] [CrossRef]
- von Messling, V.; Springfeld, C.; Devaux, P.; Cattaneo, R. A Ferret Model of Canine Distemper Virus Virulence and Immunosuppression. J. Virol. 2003, 77, 12579–12591. [Google Scholar] [CrossRef]
- von Messling, V.; Milosevic, D.; Cattaneo, R. Tropism Illuminated: Lymphocyte-Based Pathways Blazed by Lethal Morbillivirus through the Host Immune System. Proc. Natl. Acad. Sci. USA 2004, 101, 14216–14221. [Google Scholar] [CrossRef]
- Appel, M.J. Distemper Pathogenesis in Dogs. J. Am. Vet. Med. Assoc. 1970, 156, 1681–1684. [Google Scholar]
- Krakowka, S.; Cockerell, G.; Koestner, A. Effects of Canine Distemper Virus Infection on Lymphoid Function in vitro and in vivo. Infect. Immun. 1975, 11, 1069–1078. [Google Scholar]
- Miele, J.A.; Krakowka, S. Antibody Responses to Virion Polypeptides in Gnotobiotic Dogs Infected with Canine Distemper Virus. Infect. Immun. 1983, 41, 869–871. [Google Scholar]
- Vandevelde, M.; Zurbriggen, A. Demyelination in Canine Distemper Virus Infection: A Review. Acta Neuropathol. 2005, 109, 56–68. [Google Scholar] [CrossRef]
- Baumgärtner, W.; Örvell, C.; Reinacher, M. Naturally Occurring Canine Distemper Virus Encephalitis: Distribution and Expression of Viral Polypeptides in Nervous Tissues. Acta Neuropathol. 1989, 78, 504–512. [Google Scholar] [CrossRef]
- Summers, B.A.; Greisen, H.A.; Appel, M.J. Canine Distemper Encephalomyelitis: Variation with Virus Strain. J. Comp. Pathol. 1984, 94, 65–75. [Google Scholar] [CrossRef]
- Mutinelli, F.; Vandevelde, M.; Griot, C.; Richard, A. Astrocytic Infection in Canine Distemper Virus-Induced Demyelination. Acta Neuropathol. 1989, 77, 333–335. [Google Scholar] [CrossRef]
- Frisk, A.L.; Konig, M.; Moritz, A.; Baumgärtner, W. Detection of Canine Distemper Virus Nucleoprotein Rna by Reverse Transcription-PCR Using Serum, Whole Blood, and Cerebrospinal Fluid from Dogs with Distemper. J. Clin. Microbiol. 1999, 37, 3634–3643. [Google Scholar]
- Krakowka, S.; Cork, L.C.; Winkelstein, J.A.; Axthelm, M.K. Establishment of Central Nervous System Infection by Canine Distemper Virus: Breach of the Blood-Brain Barrier and Facilitation by Antiviral Antibody. Vet. Immunol. Immunopathol. 1987, 17, 471–482. [Google Scholar] [CrossRef]
- Summers, B.A.; Greisen, H.A.; Appel, M.J. Early Events in Canine Distemper Demyelinating Encephalomyelitis. Acta Neuropathol. 1979, 46, 1–10. [Google Scholar] [CrossRef]
- Axthelm, M.K.; Krakowka, S. Canine Distemper Virus: The Early Blood-Brain Barrier Lesion. Acta Neuropathol. 1987, 75, 27–33. [Google Scholar]
- Higgins, R.J.; Krakowka, S.G.; Metzler, A.E.; Koestner, A. Primary Demyelination in Experimental Canine Distemper Virus Induced Encephalomyelitis in Gnotobiotic Dogs. Sequential Immunologic and Morphologic Findings. Acta Neuropathol. 1982, 58, 1–8. [Google Scholar] [CrossRef]
- Pratakpiriya, W.; Seki, F.; Otsuki, N.; Sakai, K.; Fukuhara, H.; Katamoto, H.; Hirai, T.; Maenaka, K.; Techangamsuwan, S.; Lan, N.T.; et al. Nectin4 Is an Epithelial Cell Receptor for Canine Distemper Virus and Involved in Neurovirulence. J. Virol. 2012, 86, 10207–10210. [Google Scholar] [CrossRef]
- Noyce, R.S.; Delpeut, S.; Richardson, C.D. Dog Nectin-4 Is an Epithelial Cell Receptor for Canine Distemper Virus That Facilitates Virus Entry and Syncytia Formation. Virology 2013, 436, 210–220. [Google Scholar] [CrossRef]
- Carvalho, O.V.; Botelho, C.V.; Ferreira, C.G.; Scherer, P.O.; Soares-Martins, J.A.; Almeida, M.R.; Silva Junior, A. Immunopathogenic and Neurological Mechanisms of Canine Distemper Virus. Adv. Virol. 2012, 2012, 163860. [Google Scholar]
- Nesseler, A.; Baumgärtner, W.; Zurbriggen, A.; Örvell, C. Restricted Virus Protein Translation in Canine Distemper Virus Inclusion Body Polioencephalitis. Vet. Microbiol. 1999, 69, 23–28. [Google Scholar] [CrossRef]
- Bestetti, G.; Fatzer, R.; Frankhauser, R. Encephalitis Following Vaccination against Distemper and Infectious Hepatitis in the Dog. An Optical and Ultrastructural Study. Acta Neuropathol. 1978, 43, 69–75. [Google Scholar] [CrossRef]
- Hartley, W.J. A Post-Vaccinal Inclusion Body Encephalitis in Dogs. Vet. Pathol. 1974, 11, 301–312. [Google Scholar] [CrossRef]
- Huynh, W.; Cordato, D.J.; Kehdi, E.; Masters, L.T.; Dedousis, C. Post-Vaccination Encephalomyelitis: Literature Review and Illustrative Case. J. Clin. Neurosci. 2008, 15, 1315–1322. [Google Scholar] [CrossRef]
- Lincoln, S.D.; Gorham, J.R.; Davis, W.C.; Ott, R.L. Studies of Old Dog Encephalitis. II. Electron Microscopic and Immunohistologic Findings. Vet. Pathol. 1973, 10, 124–129. [Google Scholar] [CrossRef]
- Headley, S.A.; Amude, A.M.; Alfieri, A.F.; Bracarense, A.P.; Alfieri, A.A.; Summers, B.A. Molecular Detection of Canine Distemper Virus and the Immunohistochemical Characterization of the Neurologic Lesions in Naturally Occurring Old Dog Encephalitis. J. Vet. Diagn. Invest. 2009, 21, 588–597. [Google Scholar] [CrossRef]
- Axthelm, M.K.; Krakowka, S. Experimental Old Dog Encephalitis (ODE) in a Gnotobiotic Dog. Vet. Pathol. 1998, 35, 527–534. [Google Scholar] [CrossRef]
- Nesseler, A.; Baumgärtner, W.; Gaedke, K.; Zurbriggen, A. Abundant Expression of Viral Nucleoprotein mRNA and Restricted Translation of the Corresponding Viral Protein in Inclusion Body Polioencephalitis of Canine Distemper. J. Comp. Pathol. 1997, 116, 291–301. [Google Scholar] [CrossRef]
- Vandevelde, M.; Higgins, R.J.; Kristensen, B.; Kristensen, F.; Steck, A.J.; Kihm, U. Demyelination in Experimental Canine Distemper Virus Infection: Immunological, Pathologic, and Immunohistological Studies. Acta Neuropathol. 1982, 56, 285–293. [Google Scholar] [CrossRef]
- Seehusen, F.; Baumgärtner, W. Axonal Pathology and Loss Precede Demyelination and Accompany Chronic Lesions in a Spontaneously Occurring Animal Model of Multiple Sclerosis. Brain Pathol. 2010, 20, 551–559. [Google Scholar] [CrossRef]
- Imbschweiler, I.; Seehusen, F.; Peck, C.T.; Omar, M.; Baumgärtner, W.; Wewetzer, K. Increased P75 Neurotrophin Receptor Expression in the Canine Distemper Virus Model of Multiple Sclerosis Identifies Aldynoglial Schwann Cells That Emerge in Response to Axonal Damage. Glia 2012, 60, 358–371. [Google Scholar] [CrossRef]
- Vandevelde, M.; Zurbriggen, A.; Higgins, R.J.; Palmer, D. Spread and Distribution of Viral Antigen in Nervous Canine Distemper. Acta Neuropathol. 1985, 67, 211–218. [Google Scholar] [CrossRef]
- Zurbriggen, A.; Yamawaki, M.; Vandevelde, M. Restricted Canine Distemper Virus Infection of Oligodendrocytes. Lab. Invest. 1993, 68, 277–284. [Google Scholar]
- Alldinger, S.; Baumgärtner, W.; Örvell, C. Restricted Expression of Viral Surface Proteins in Canine Distemper Encephalitis. Acta Neuropathol. 1993, 85, 635–645. [Google Scholar] [CrossRef]
- Zurbriggen, A.; Schmid, I.; Graber, H.U.; Vandevelde, M. Oligodendroglial Pathology in Canine Distemper. Acta Neuropathol. 1998, 95, 71–77. [Google Scholar] [CrossRef]
- Griot, C.; Vandevelde, M.; Schobesberger, M.; Zurbriggen, A. Canine Distemper, a Re-Emerging Morbillivirus with Complex Neuropathogenic Mechanisms. Anim. Health Res. Rev. 2003, 4, 1–10. [Google Scholar] [CrossRef]
- Schobesberger, M.; Zurbriggen, A.; Summerfield, A.; Vandevelde, M.; Griot, C. Oligodendroglial Degeneration in Distemper: Apoptosis or Necrosis? Acta Neuropathol. 1999, 97, 279–287. [Google Scholar] [CrossRef]
- Bollo, E.; Zurbriggen, A.; Vandevelde, M.; Fankhauser, R. Canine Distemper Virus Clearance in Chronic Inflammatory Demyelination. Acta Neuropathol. 1986, 72, 69–73. [Google Scholar] [CrossRef]
- Alldinger, S.; Wünschmann, A.; Baumgärtner, W.; Voss, C.; Kremmer, E. Up-Regulation of Major Histocompatibility Complex Class II Antigen Expression in the Central Nervous System of Dogs with Spontaneous Canine Distemper Virus Encephalitis. Acta Neuropathol. 1996, 92, 273–280. [Google Scholar] [CrossRef]
- Wünschmann, A.; Alldinger, S.; Kremmer, E.; Baumgärtner, W. Identification of CD4+ and CD8+ T Cell Subsets and B Cells in the Brain of Dogs with Spontaneous Acute, Subacute-, and Chronic-Demyelinating Distemper Encephalitis. Vet. Immunol. Immunopathol. 1999, 67, 101–116. [Google Scholar]
- Spitzbarth, I.; Baumgärtner, W.; Beineke, A. The Role of Pro- and Anti-Inflammatory Cytokines in the Pathogenesis of Spontaneous Canine CNS Diseases. Vet. Immunol. Immunopathol. 2012, 147, 6–24. [Google Scholar] [CrossRef]
- Qeska, V.; Barthel, Y.; Iseringhausen, M.; Tipold, A.; Stein, V.M.; Khan, M.A.; Baumgärtner, W.; Beineke, A. Dynamic Changes of Foxp3(+) Regulatory T Cells in Spleen and Brain of Canine Distemper Virus-Infected Dogs. Vet. Immunol. Immunopathol. 2013, 156, 215–222. [Google Scholar] [CrossRef]
- Gröne, A.; Fonfara, S.; Baumgärtner, W. Cell Type-Dependent Cytokine Expression after Canine Distemper Virus Infection. Viral Immunol. 2002, 15, 493–505. [Google Scholar]
- Spitzbarth, I.; Bock, P.; Haist, V.; Stein, V.M.; Tipold, A.; Wewetzer, K.; Baumgärtner, W.; Beineke, A. Prominent Microglial Activation in the Early Proinflammatory Immune Response in Naturally Occurring Canine Spinal Cord Injury. J. Neuropathol. Exp. Neurol. 2011, 70, 703–714. [Google Scholar] [CrossRef]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of Two Distinct Macrophage Subsets with Divergent Effects Causing Either Neurotoxicity or Regeneration in the Injured Mouse Spinal Cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar]
- Ahn, M.; Yang, W.; Kim, H.; Jin, J.K.; Moon, C.; Shin, T. Immunohistochemical Study of Arginase-1 in the Spinal Cords of Lewis Rats with Experimental Autoimmune Encephalomyelitis. Brain Res. 2012, 1453, 77–86. [Google Scholar] [CrossRef]
- David, S.; Kroner, A. Repertoire of Microglial and Macrophage Responses after Spinal Cord Injury. Nat. Rev. Neurosci. 2011, 12, 388–399. [Google Scholar] [CrossRef]
- Mikita, J.; Dubourdieu-Cassagno, N.; Deloire, M.S.; Vekris, A.; Biran, M.; Raffard, G.; Brochet, B.; Canron, M.H.; Franconi, J.M.; Boiziau, C.; et al. Altered M1/M2 Activation Patterns of Monocytes in Severe Relapsing Experimental Rat Model of Multiple Sclerosis. Amelioration of Clinical Status by M2 Activated Monocyte Administration. Mult. Scler. 2011, 17, 2–15. [Google Scholar]
- Gröne, A.; Alldinger, S.; Baumgärtner, W. Interleukin-1beta, -6, -12 and Tumor Necrosis Factor-Alpha Expression in Brains of Dogs with Canine Distemper Virus Infection. J. Neuroimmunol. 2000, 110, 20–30. [Google Scholar]
- Beineke, A.; Markus, S.; Borlak, J.; Thum, T.; Baumgärtner, W. Increase of Pro-Inflammatory Cytokine Expression in Non-Demyelinating Early Cerebral Lesions in Nervous Canine Distemper. Viral Immunol. 2008, 21, 401–410. [Google Scholar]
- Gröters, S.; Alldinger, S.; Baumgärtner, W. Up-Regulation of mRNA for Matrix Metalloproteinases-9 and -14 in Advanced Lesions of Demyelinating Canine Distemper Leukoencephalitis. Acta Neuropathol. 2005, 110, 369–382. [Google Scholar] [CrossRef]
- Puff, C.; Krudewig, C.; Imbschweiler, I.; Baumgärtner, W.; Alldinger, S. Influence of Persistent Canine Distemper Virus Infection on Expression of Reck, Matrix-Metalloproteinases and Their Inhibitors in a Canine Macrophage/Monocytic Tumour Cell Line (DH82). Vet. J. 2009, 182, 100–107. [Google Scholar] [CrossRef]
- Stein, V.M.; Puff, C.; Genini, S.; Contioso, V.B.; Baumgärtner, W.; Tipold, A. Variations on Brain Microglial Gene Expression of MMPs, RECK, and TIMPs in Inflammatory and Non-Inflammatory Diseases in Dogs. Vet. Immunol. Immunopathol. 2011, 144, 17–26. [Google Scholar] [CrossRef]
- Krakowka, S.; Axthelm, M.K.; Gorham, J.R. Effects of Induced Thrombocytopenia on Viral Invasion of the Central Nervous System in Canine Distemper Virus Infection. J. Comp. Pathol. 1987, 97, 441–450. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Matrix Metalloproteinases in Neuroinflammation. Glia 2002, 39, 279–291. [Google Scholar] [CrossRef]
- Matrisian, L.M. Metalloproteinases and Their Inhibitors in Matrix Remodeling. Trends Genet. 1990, 6, 121–125. [Google Scholar] [CrossRef]
- Woessner, J.F., Jr. Matrix Metalloproteinases and Their Inhibitors in Connective Tissue Remodeling. FASEB J. 1991, 5, 2145–2154. [Google Scholar]
- Stamenkovic, I. Extracellular Matrix Remodelling: The Role of Matrix Metalloproteinases. J. Pathol. 2003, 200, 448–464. [Google Scholar] [CrossRef]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue Inhibitors of Metalloproteinases: Evolution, Structure and Function. Biochim. Biophys. Acta 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Oh, J.; Takahashi, R.; Kondo, S.; Mizoguchi, A.; Adachi, E.; Sasahara, R.M.; Nishimura, S.; Imamura, Y.; Kitayama, H.; Alexander, D.B.; et al. The Membrane-Anchored MMP Inhibitor RECK Is a Key Regulator of Extracellular Matrix Integrity and Angiogenesis. Cell 2001, 107, 789–800. [Google Scholar] [CrossRef]
- Bregano, L.C.; Agostinho, S.D.; Roncatti, F.L.; Pires, M.C.; Riva, H.G.; Luvizotto, M.C.; Cardoso, T.C. Immunohistochemical Detection of Metalloproteinase-9 (MMP-9), Anti-Oxidant Like 1 Protein (AOP-1) and Synaptosomal-Associated Protein (SNAP-25) in the Cerebella of Dogs Naturally Infected with Spontaneous Canine Distemper. Folia Histochem. Cytobiol. 2011, 49, 41–48. [Google Scholar] [CrossRef]
- Miao, Q.; Baumgärtner, W.; Failing, K.; Alldinger, S. Phase-Dependent Expression of Matrix Metalloproteinases and Their Inhibitors in Demyelinating Canine Distemper Encephalitis. Acta Neuropathol. 2003, 106, 486–494. [Google Scholar] [CrossRef]
- Machado, G.F.; Melo, G.D.; Souza, M.S.; Machado, A.A.; Migliolo, D.S.; Moraes, O.C.; Nunes, C.M.; Ribeiro, E.S. Zymographic Patterns of MMP-2 and MMP-9 in the CSF and Cerebellum of Dogs with Subacute Distemper Leukoencephalitis. Vet. Immunol. Immunopathol. 2013, 154, 68–74. [Google Scholar] [CrossRef]
- Schwartz, M.; Puff, C.; Stein, V.M.; Baumgärtner, W.; Tipold, A. Marked MMP-2 Transcriptional up-Regulation in Mononuclear Leukocytes Invading the Subarachnoidal Space in Aseptic Suppurative Steroid-Responsive Meningitis-Arteritis in Dogs. Vet. Immunol. Immunopathol. 2010, 133, 198–206. [Google Scholar] [CrossRef]
- Alldinger, S.; Fonfara, S.; Kremmer, E.; Baumgärtner, W. Up-Regulation of the Hyaluronate Receptor CD44 in Canine Distemper Demyelinated Plaques. Acta Neuropathol. 2000, 99, 138–146. [Google Scholar] [CrossRef]
- Alldinger, S.; Gröters, S.; Miao, Q.; Fonfara, S.; Kremmer, E.; Baumgärtner, W. Roles of an Extracellular Matrix (ECM) Receptor and ECM Processing Enzymes in Demyelinating Canine Distemper Encephalitis. Dtsch. Tierarztl. Wochenschr. 2006, 113, 151–152, 154–156. [Google Scholar]
- Wyss-Fluehmann, G.; Zurbriggen, A.; Vandevelde, M.; Plattet, P. Canine Distemper Virus Persistence in Demyelinating Encephalitis by Swift Intracellular Cell-to-Cell Spread in Astrocytes Is Controlled by the Viral Attachment Protein. Acta Neuropathol. 2010, 119, 617–630. [Google Scholar] [CrossRef]
- Summers, B.A.; Appel, M.J. Aspects of Canine Distemper Virus and Measles Virus Encephalomyelitis. Neuropathol. Appl. Neurobiol. 1994, 20, 525–534. [Google Scholar] [CrossRef]
- Summers, B.A.; Greisen, H.A.; Appel, M.J. Canine Distemper and Experimental Allergic Encephalomyelitis in the Dog: Comparative Patterns of Demyelination. J. Comp. Pathol. 1984, 94, 575–589. [Google Scholar] [CrossRef]
- Vandevelde, M.; Zurbriggen, A.; Dumas, M.; Palmer, D. Canine Distemper Virus Does Not Infect Oligodendrocytes in vitro. J. Neurol. Sci. 1985, 69, 133–137. [Google Scholar] [CrossRef]
- Schnitzer, J.; Franke, W.W.; Schachner, M. Immunocytochemical Demonstration of Vimentin in Astrocytes and Ependymal Cells of Developing and Adult Mouse Nervous System. J. Cell Biol. 1981, 90, 435–447. [Google Scholar] [CrossRef]
- Pixley, S.K.; de Vellis, J. Transition between Immature Radial Glia and Mature Astrocytes Studied with a Monoclonal Antibody to Vimentin. Brain Res. 1984, 317, 201–209. [Google Scholar] [CrossRef]
- Saraga-Babic, M.; Stefanovic, V.; Saraga, M.; Wartiovaara, J.; Lehtonen, E. Expression of Intermediate Filaments and Desmosomal Proteins During Differentiation of the Human Spinal Cord. Acta Histochem. 2002, 104, 157–166. [Google Scholar] [CrossRef]
- Eng, L.F.; Ghirnikar, R.S. GFAP and Astrogliosis. Brain Pathol. 1994, 4, 229–237. [Google Scholar]
- Frisen, J.; Haegerstrand, A.; Risling, M.; Fried, K.; Johansson, C.B.; Hammarberg, H.; Elde, R.; Hokfelt, T.; Cullheim, S. Spinal Axons in Central Nervous System Scar Tissue Are Closely Related to Laminin-Immunoreactive Astrocytes. Neuroscience 1995, 65, 293–304. [Google Scholar] [CrossRef]
- Ekmark-Lewen, S.; Lewen, A.; Israelsson, C.; Li, G.L.; Farooque, M.; Olsson, Y.; Ebendal, T.; Hillered, L. Vimentin and GFAP Responses in Astrocytes after Contusion Trauma to the Murine Brain. Restor. Neurol. Neurosci. 2010, 28, 311–321. [Google Scholar]
- Li, L.; Lundkvist, A.; Andersson, D.; Wilhelmsson, U.; Nagai, N.; Pardo, A.C.; Nodin, C.; Stahlberg, A.; Aprico, K.; Larsson, K.; et al. Protective Role of Reactive Astrocytes in Brain Ischemia. J. Cereb. Blood Flow MeTable 2008, 28, 468–481. [Google Scholar] [CrossRef]
- Pekny, M.; Nilsson, M. Astrocyte Activation and Reactive Gliosis. Glia 2005, 50, 427–434. [Google Scholar] [CrossRef]
- Pekny, M.; Johansson, C.B.; Eliasson, C.; Stakeberg, J.; Wallen, A.; Perlmann, T.; Lendahl, U.; Betsholtz, C.; Berthold, C.H.; Frisen, J. Abnormal Reaction to Central Nervous System Injury in Mice Lacking Glial Fibrillary Acidic Protein and Vimentin. J. Cell Biol. 1999, 145, 503–514. [Google Scholar] [CrossRef]
- Pekny, M.; Wilhelmsson, U.; Pekna, M. The Dual Role of Astrocyte Activation and Reactive Gliosis. Neurosci. Lett. 2014, 565, 30–38. [Google Scholar] [CrossRef]
- Emsley, J.G.; Arlotta, P.; Macklis, J.D. Star-Cross’d Neurons: Astroglial Effects on Neural Repair in the Adult Mammalian Cns. Trends Neurosci. 2004, 27, 238–240. [Google Scholar] [CrossRef]
- Galou, M.; Colucci-Guyon, E.; Ensergueix, D.; Ridet, J.L.; Gimenez y Ribotta, M.; Privat, A.; Babinet, C.; Dupouey, P. Disrupted Glial Fibrillary Acidic Protein Network in Astrocytes from Vimentin Knockout Mice. J. Cell Biol. 1996, 133, 853–863. [Google Scholar] [CrossRef]
- Fuchs, E.; Cleveland, D.W. A Structural Scaffolding of Intermediate Filaments in Health and Disease. Science 1998, 279, 514–519. [Google Scholar] [CrossRef]
- Holley, J.E.; Gveric, D.; Newcombe, J.; Cuzner, M.L.; Gutowski, N.J. Astrocyte Characterization in the Multiple Sclerosis Glial Scar. Neuropathol. Appl. Neurobiol. 2003, 29, 434–444. [Google Scholar] [CrossRef]
- Yamada, T.; Kawamata, T.; Walker, D.G.; McGeer, P.L. Vimentin Immunoreactivity in Normal and Pathological Human Brain Tissue. Acta Neuropathol. 1992, 84, 157–162. [Google Scholar] [CrossRef]
- Spitzbarth, I.; Schenk, H.C.; Tipold, A.; Beineke, A. Immunohistochemical Characterization of Inflammatory and Glial Responses in a Case of Necrotizing Leucoencephalitis in a French Bulldog. J. Comp. Pathol. 2010, 142, 235–241. [Google Scholar] [CrossRef]
- Seehusen, F.; Orlando, E.A.; Wewetzer, K.; Baumgärtner, W. Vimentin-Positive Astrocytes in Canine Distemper: A Target for Canine Distemper Virus Especially in Chronic Demyelinating Lesions? Acta Neuropathol. 2007, 114, 597–608. [Google Scholar] [CrossRef]
- Eliasson, C.; Sahlgren, C.; Berthold, C.H.; Stakeberg, J.; Celis, J.E.; Betsholtz, C.; Eriksson, J.E.; Pekny, M. Intermediate Filament Protein Partnership in Astrocytes. J. Biol. Chem. 1999, 274, 23996–24006. [Google Scholar] [CrossRef]
- Lin, J.; Cai, W. Effect of Vimentin on Reactive Gliosis: In vitro and in vivo Analysis. J. Neurotrauma 2004, 21, 1671–1682. [Google Scholar] [CrossRef]
- Lundgaard, I.; Osorio, M.J.; Kress, B.T.; Sanggaard, S.; Nedergaard, M. White Matter Astrocytes in Health and Disease. Neuroscience 2013. [Google Scholar] [CrossRef]
- Povlishock, J.T. Traumatically Induced Axonal Injury: Pathogenesis and Pathobiological Implications. Brain Pathol. 1992, 2, 1–12. [Google Scholar]
- Povlishock, J.T.; Erb, D.E.; Astruc, J. Axonal Response to Traumatic Brain Injury: Reactive Axonal Change, Deafferentation, and Neuroplasticity. J. Neurotrauma 1992, 9, S189–S200. [Google Scholar]
- Coleman, M. Axon Degeneration Mechanisms: Commonality and Diversity. Nat. Rev. Neurosci. 2005, 6, 889–898. [Google Scholar] [CrossRef]
- Tsunoda, I.; Kuang, L.Q.; Libbey, J.E.; Fujinami, R.S. Axonal Injury Heralds Virus-Induced Demyelination. Am. J. Pathol. 2003, 162, 1259–1269. [Google Scholar] [CrossRef]
- Julien, J.P.; Mushynski, W.E. Neurofilaments in Health and Disease. Prog. Nucleic Acid Res. Mol. Biol. 1998, 61, 1–23. [Google Scholar] [CrossRef]
- Mancardi, G.; Hart, B.; Roccatagliata, L.; Brok, H.; Giunti, D.; Bontrop, R.; Massacesi, L.; Capello, E.; Uccelli, A. Demyelination and Axonal Damage in a Non-Human Primate Model of Multiple Sclerosis. J. Neurol. Sci. 2001, 184, 41–49. [Google Scholar]
- Petzold, A. Neurofilament Phosphoforms: Surrogate Markers for Axonal Injury, Degeneration and Loss. J. Neurol. Sci. 2005, 233, 183–198. [Google Scholar] [CrossRef]
- Brück, W. Inflammatory Demyelination Is Not Central to the Pathogenesis of Multiple Sclerosis. J. Neurol. 2005, 252, 10–15. [Google Scholar] [CrossRef]
- Ferguson, B.; Matyszak, M.K.; Esiri, M.M.; Perry, V.H. Axonal Damage in Acute Multiple Sclerosis Lesions. Brain 1997, 120, 393–399. [Google Scholar] [CrossRef]
- Kornek, B.; Storch, M.K.; Weissert, R.; Wallstroem, E.; Stefferl, A.; Olsson, T.; Linington, C.; Schmidbauer, M.; Lassmann, H. Multiple Sclerosis and Chronic Autoimmune Encephalomyelitis: A Comparative Quantitative Study of Axonal Injury in Active, Inactive, and Remyelinated Lesions. Am. J. Pathol. 2000, 157, 267–276. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Lingfeld, G.; Bitsch, A.; Schuchardt, J.; Brück, W. Acute Axonal Damage in Multiple Sclerosis Is Most Extensive in Early Disease Stages and Decreases over Time. Brain 2002, 125, 2202–2212. [Google Scholar] [CrossRef]
- De Vos, K.J.; Grierson, A.J.; Ackerley, S.; Miller, C.C. Role of Axonal Transport in Neurodegenerative Diseases. Annu. Rev. Neurosci. 2008, 31, 151–173. [Google Scholar] [CrossRef]
- Oehmichen, M.; Meissner, C.; Schmidt, V.; Pedal, I.; Konig, H.G.; Saternus, K.S. Axonal Injury—A Diagnostic Tool in Forensic Neuropathology? A Review. Forensic. Sci. Int. 1998, 95, 67–83. [Google Scholar] [CrossRef]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mork, S.; Bo, L. Axonal Transection in the Lesions of Multiple Sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef]
- Kreutzer, M.; Seehusen, F.; Kreutzer, R.; Pringproa, K.; Kummerfeld, M.; Claus, P.; Deschl, U.; Kalkul, A.; Beineke, A.; Baumgärtner, W.; et al. Axonopathy Is Associated with Complex Axonal Transport Defects in a Model of Multiple Sclerosis. Brain Pathol. 2012, 22, 454–471. [Google Scholar] [CrossRef]
- Schirmer, L.; Merkler, D.; König, F.B.; Brück, W.; Stadelmann, C. Neuroaxonal Regeneration Is More Pronounced in Early Multiple Sclerosis Than in Traumatic Brain Injury Lesions. Brain Pathol. 2013, 23, 2–12. [Google Scholar] [CrossRef]
- Bock, P.; Spitzbarth, I.; Haist, V.; Stein, V.M.; Tipold, A.; Puff, C.; Beineke, A.; Baumgärtner, W. Spatio-Temporal Development of Axonopathy in Canine Intervertebral Disc Disease as a Translational Large Animal Model for Nonexperimental Spinal Cord Injury. Brain Pathol. 2013, 23, 82–99. [Google Scholar]
- Tsunoda, I.; Fujinami, R.S. Two Models for Multiple Sclerosis: Experimental Allergic Encephalomyelitis and Theiler’s Murine Encephalomyelitis Virus. J. Neuropathol. Exp. Neurol. 1996, 55, 673–686. [Google Scholar] [CrossRef]
- Ulrich, R.; Seeliger, F.; Kreutzer, M.; Germann, P.G.; Baumgärtner, W. Limited Remyelination in Theiler's Murine Encephalomyelitis Due to Insufficient Oligodendroglial Differentiation of Nerve/Glial Antigen 2 (Ng2)-Positive Putative Oligodendroglial Progenitor Cells. Neuropathol. Appl. Neurobiol. 2008, 34, 603–620. [Google Scholar]
- Wang, D.; Ayers, M.M.; Catmull, D.V.; Hazelwood, L.J.; Bernard, C.C.; Orian, J.M. Astrocyte-Associated Axonal Damage in Pre-Onset Stages of Experimental Autoimmune Encephalomyelitis. Glia 2005, 51, 235–240. [Google Scholar]
- Tsunoda, I.; Fujinami, R.S. Inside-out Versus Outside-in Models for Virus Induced Demyelination: Axonal Damage Triggering Demyelination. Springer Semin. Immunopathol. 2002, 24, 105–125. [Google Scholar]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From Biology to Therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar]
- Crawford, A.H.; Chambers, C.; Franklin, R.J. Remyelination: The True Regeneration of the Central Nervous System. J. Comp. Pathol. 2013, 149, 242–254. [Google Scholar]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Brück, W.; Lucchinetti, C.; Lassmann, H. Remyelination Is Extensive in a Subset of Multiple Sclerosis Patients. Brain 2006, 129, 3165–3172. [Google Scholar] [CrossRef]
- Carroll, W.M.; Jennings, A.R.; Ironside, L.J. Identification of the Adult Resting Progenitor Cell by Autoradiographic Tracking of Oligodendrocyte Precursors in Experimental Cns Demyelination. Brain 1998, 121, 293–302. [Google Scholar] [CrossRef]
- Levine, J.M.; Reynolds, R. Activation and Proliferation of Endogenous Oligodendrocyte Precursor Cells During Ethidium Bromide-Induced Demyelination. Exp. Neurol. 1999, 160, 333–347. [Google Scholar] [CrossRef]
- Powers, B.E.; Sellers, D.L.; Lovelett, E.A.; Cheung, W.; Aalami, S.P.; Zapertov, N.; Maris, D.O.; Horner, P.J. Remyelination Reporter Reveals Prolonged Refinement of Spontaneously Regenerated Myelin. Proc. Natl. Acad. Sci. USA 2013, 110, 4075–4080. [Google Scholar] [CrossRef]
- Chang, A.; Nishiyama, A.; Peterson, J.; Prineas, J.; Trapp, B.D. Ng2-Positive Oligodendrocyte Progenitor Cells in Adult Human Brain and Multiple Sclerosis Lesions. J. Neurosci. 2000, 20, 6404–6412. [Google Scholar]
- Zawadzka, M.; Rivers, L.E.; Fancy, S.P.; Zhao, C.; Tripathi, R.; Jamen, F.; Young, K.; Goncharevich, A.; Pohl, H.; Rizzi, M.; et al. CNS-Resident Glial Progenitor/Stem Cells Produce Schwann Cells as Well as Oligodendrocytes During Repair of CNS Demyelination. Cell Stem Cell 2010, 6, 578–590. [Google Scholar]
- Blakemore, W.F. The Case for a Central Nervous System (CNS) Origin for the Schwann Cells That Remyelinate CNS Axons Following Concurrent Loss of Oligodendrocytes and Astrocytes. Neuropathol. Appl. Neurobiol. 2005, 31, 1–10. [Google Scholar] [CrossRef]
- Ghatak, N.R.; Hirano, A.; Doron, Y.; Zimmerman, H.M. Remyelination in Multiple Sclerosis with Peripheral Type Myelin. Arch. Neurol. 1973, 29, 262–267. [Google Scholar] [CrossRef]
- Itoyama, Y.; Webster, H.D.; Richardson, E.P., Jr.; Trapp, B.D. Schwann Cell Remyelination of Demyelinated Axons in Spinal Cord Multiple Sclerosis Lesions. Ann. Neurol. 1983, 14, 339–346. [Google Scholar] [CrossRef]
- Itoyama, Y.; Ohnishi, A.; Tateishi, J.; Kuroiwa, Y.; Webster, H.D. Spinal Cord Multiple Sclerosis Lesions in Japanese Patients: Schwann Cell Remyelination Occurs in Areas That Lack Glial Fibrillary Acidic Protein (GFAP). Acta Neuropathol. 1985, 65, 217–223. [Google Scholar] [CrossRef]
- Dal Canto, M.C.; Lipton, H.L. Schwann Cell Remyelination and Recurrent Demyelination in the Central Nervous System of Mice Infected with Attenuated Theiler's Virus. Am. J. Pathol. 1980, 98, 101–122. [Google Scholar]
- Guest, J.D.; Hiester, E.D.; Bunge, R.P. Demyelination and Schwann Cell Responses Adjacent to Injury Epicenter Cavities Following Chronic Human Spinal Cord Injury. Exp. Neurol. 2005, 192, 384–393. [Google Scholar] [CrossRef]
- Gilson, J.M.; Blakemore, W.F. Schwann Cell Remyelination Is Not Replaced by Oligodendrocyte Remyelination Following Ethidium Bromide Induced Demyelination. Neuroreport 2002, 13, 1205–1208. [Google Scholar] [CrossRef]
- Shields, S.A.; Blakemore, W.F.; Franklin, R.J. Schwann Cell Remyelination Is Restricted to Astrocyte-Deficient Areas after Transplantation into Demyelinated Adult Rat Brain. J. Neurosci. Res. 2000, 60, 571–578. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. Control of Schwann Cell Myelination. F1000 Biol. Rep. 2010, 2, 19. [Google Scholar]
- Jessen, K.R.; Mirsky, R. Signals That Determine Schwann Cell Identity. J. Anat. 2002, 200, 367–376. [Google Scholar] [CrossRef]
- Cragnolini, A.B.; Friedman, W.J. The Function of P75NTR in Glia. Trends Neurosci. 2008, 31, 99–104. [Google Scholar] [CrossRef]
- Mirsky, R.; Woodhoo, A.; Parkinson, D.B.; Arthur-Farraj, P.; Bhaskaran, A.; Jessen, K.R. Novel Signals Controlling Embryonic Schwann Cell Development, Myelination and Dedifferentiation. J. Peripher. Nerv. Syst. 2008, 13, 122–135. [Google Scholar] [CrossRef]
- Scherer, S.S.; Xu, Y.T.; Bannerman, P.G.; Sherman, D.L.; Brophy, P.J. Periaxin Expression in Myelinating Schwann Cells: Modulation by Axon-Glial Interactions and Polarized Localization During Development. Development 1995, 121, 4265–4273. [Google Scholar]
- Orlando, E.A.; Imbschweiler, I.; Gerhauser, I.; Baumgärtner, W.; Wewetzer, K. In Vitro Characterization and Preferential Infection by Canine Distemper Virus of Glial Precursors with Schwann Cell Characteristics from Adult Canine Brain. Neuropathol. Appl. Neurobiol. 2008, 34, 621–637. [Google Scholar] [CrossRef]
- Franklin, R.J.; Blakemore, W.F. Requirements for Schwann Cell Migration within CNS Environments: A Viewpoint. Int. J. Dev. Neurosci. 1993, 11, 641–649. [Google Scholar] [CrossRef]
- Jasmin, L.; Ohara, P.T. Remyelination within the CNS: Do Schwann Cells Pave the Way for Oligodendrocytes? Neuroscientist 2002, 8, 198–203. [Google Scholar] [CrossRef]
- Ulrich, R.; Imbschweiler, I.; Kalkuhl, A.; Lehmbecker, A.; Ziege, S.; Kegler, K.; Becker, K.; Deschl, U.; Wewetzer, K.; Baumgärtner, W. Transcriptional Profiling Predicts Overwhelming Homology of Schwann Cells, Olfactory Ensheathing Cells, and Schwann Cell-Like Glia. Glia 2014. [Google Scholar] [CrossRef]
- Krassioukov, A.V.; Ackery, A.; Schwartz, G.; Adamchik, Y.; Liu, Y.; Fehlings, M.G. An in vitro Model of Neurotrauma in Organotypic Spinal Cord Cultures from Adult Mice. Brain Res. Brain Res. Protoc. 2002, 10, 60–68. [Google Scholar] [CrossRef]
- Huuskonen, J.; Suuronen, T.; Miettinen, R.; van Groen, T.; Salminen, A. A Refined in vitro Model to Study Inflammatory Responses in Organotypic Membrane Culture of Postnatal Rat Hippocampal Slices. J. Neuroinflammation 2005, 2, 25. [Google Scholar] [CrossRef]
- Stavridis, S.I.; Dehghani, F.; Korf, H.W.; Hailer, N.P. Characterisation of Transverse Slice Culture Preparations of Postnatal Rat Spinal Cord: Preservation of Defined Neuronal Populations. Histochem. Cell Biol. 2005, 123, 377–392. [Google Scholar]
- Casha, S.; Yu, W.R.; Fehlings, M.G. Fas Deficiency Reduces Apoptosis, Spares Axons and Improves Function after Spinal Cord Injury. Exp. Neurol. 2005, 196, 390–400. [Google Scholar] [CrossRef]
- Pan, J.Z.; Ni, L.; Sodhi, A.; Aguanno, A.; Young, W.; Hart, R.P. Cytokine Activity Contributes to Induction of Inflammatory Cytokine mRNAs in Spinal Cord Following Contusion. J. Neurosci. Res. 2002, 68, 315–322. [Google Scholar] [CrossRef]
- Brandes, G.; Khayami, M.; Peck, C.T.; Baumgärtner, W.; Bugday, H.; Wewetzer, K. Cell Surface Expression of 27c7 by Neonatal Rat Olfactory Ensheathing Cells in situ and in vitro Is Independent of Axonal Contact. Histochem. Cell Biol. 2011, 135, 397–408. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lempp, C.; Spitzbarth, I.; Puff, C.; Cana, A.; Kegler, K.; Techangamsuwan, S.; Baumgärtner, W.; Seehusen, F. New Aspects of the Pathogenesis of Canine Distemper Leukoencephalitis. Viruses 2014, 6, 2571-2601. https://doi.org/10.3390/v6072571
Lempp C, Spitzbarth I, Puff C, Cana A, Kegler K, Techangamsuwan S, Baumgärtner W, Seehusen F. New Aspects of the Pathogenesis of Canine Distemper Leukoencephalitis. Viruses. 2014; 6(7):2571-2601. https://doi.org/10.3390/v6072571
Chicago/Turabian StyleLempp, Charlotte, Ingo Spitzbarth, Christina Puff, Armend Cana, Kristel Kegler, Somporn Techangamsuwan, Wolfgang Baumgärtner, and Frauke Seehusen. 2014. "New Aspects of the Pathogenesis of Canine Distemper Leukoencephalitis" Viruses 6, no. 7: 2571-2601. https://doi.org/10.3390/v6072571