Immunogenetics of Small Ruminant Lentiviral Infections

1
Centre for the Genetic Improvement of Livestock, Department of Animal and Poultry Science, University of Guelph, Guelph, ON N1G 2W1, Canada
2
Department of Pathobiology, Ontario Veterinary College, University of Guelph, Guelph, ON N1G 2W1, Canada
*
Author to whom correspondence should be addressed.
Viruses 2014, 6(8), 3311-3333; https://doi.org/10.3390/v6083311
Submission received: 23 May 2014
/
Revised: 18 August 2014
/
Accepted: 19 August 2014
/
Published: 22 August 2014
(This article belongs to the Special Issue Small Ruminant Lentiviruses)
{kind=link}
Abstract
:The small ruminant lentiviruses (SRLV) include the caprine arthritis encephalitis virus (CAEV) and the Maedi-Visna virus (MVV). Both of these viruses limit production and can be a major source of economic loss to producers. Little is known about how the immune system recognizes and responds to SRLVs, but due to similarities with the human immunodeficiency virus (HIV), HIV research can shed light on the possible immune mechanisms that control or lead to disease progression. This review will focus on the host immune response to HIV-1 and SRLV, and will discuss the possibility of breeding for enhanced SRLV disease resistance.
1. Introduction
The caprine arthritis encephalitis virus (CAEV) and the Maedi-Visna virus (MVV) are enveloped RNA viruses in the lentivirus genus of the Retroviridae family [1,2]. While small ruminant lentiviruses (SRLVs) were once considered to be species-specific, recent studies suggest that they can be transmitted between sheep and goats [3], and can recombine to form new CAEV-MVV strains [4]. These viruses primarily infect monocytes, macrophages, and dendritic cells [5], and like the human immunodeficiency virus (HIV), infection is lifelong and can persist for months or years in a latent or sub-clinical state [6]. When disease symptoms do emerge, goats predominantly show signs of arthritis or mastitis, while in sheep the disease tends to manifest as pneumonia or mastitis [7]. Encephalitis can also be a symptom in either lambs or kids but is less common [8].
SRLVs are predominantly vertically transmitted to offspring through the shedding of virus particles, and infected macrophages and epithelial cells in the colostrum and milk [9]. Horizontal transmission however, can occur through prolonged direct contact with bodily secretions, and sexual transmission of the virus may also be possible [10]. There is currently no effective treatment for SRLV infections and due to a high mutation rate, effective vaccine development has been and will continue to be challenging [11]. Therefore, the most effective means of controlling the virus is through herd management that prevents viral transmission.
The dynamics of the host immune response to SRLV infections remain unclear, but due to the similarities between SRLV and HIV, a great deal of our knowledge of the immune responses to HIV can be used to enhance our understanding of the host response to SRLV. This review will discuss the immune response to lentiviral infections, and the possibility of breeding for enhanced SRLV resistance will be addressed.
2. Lentiviral Characteristics
To fully understand the complex interaction between the host and virus, it is first necessary to understand the structural and genomic organization of the lentiviruses. Although an in-depth discussion of the structural and functional characteristics of lentiviruses is beyond the scope of this paper, a brief overview of SRLV and HIV-1 organization is given.
Genomic Organization
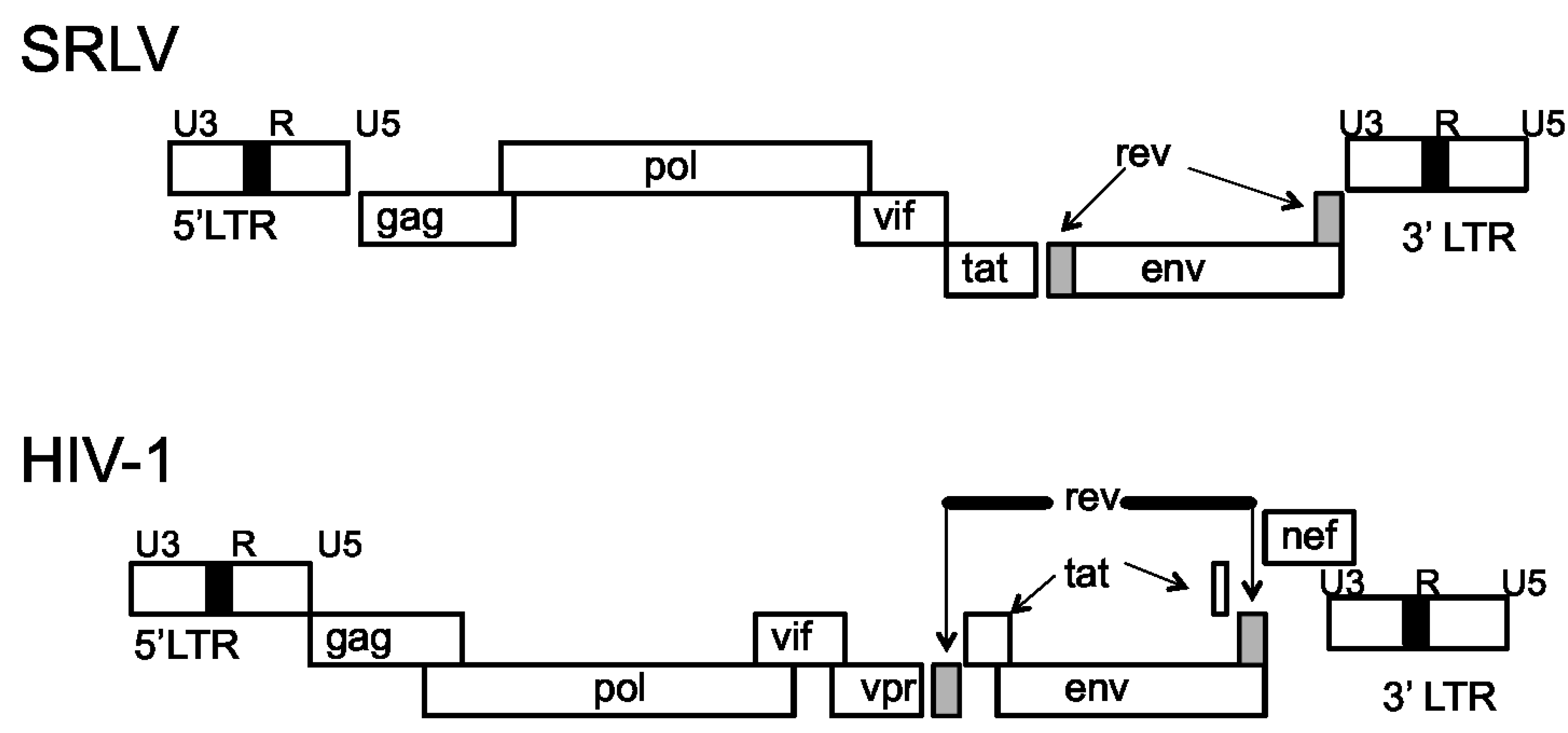
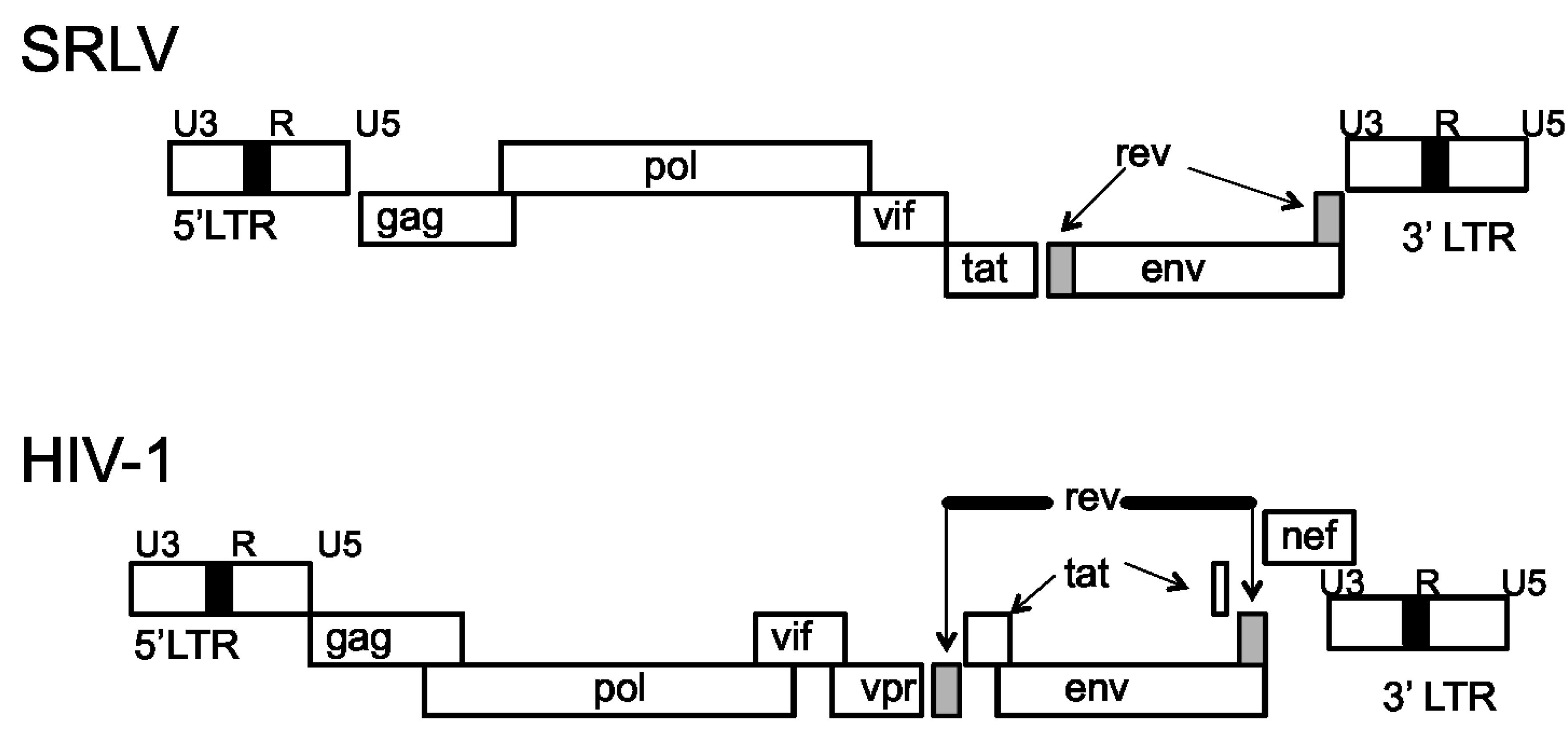
The SRLV genome is comprised of three structural genes, gag, pol and env, and three accessory genes, vif, tat, and rev (Figure 1) [12,13]. The gag gene encodes the capsid proteins, pol encodes the viral enzymes protease, reverse transcriptase, and integrase, and env encodes the envelope glycoproteins, gp135 (SU) and gp38 (TM) [14]. While the tat gene is dispensable for efficient viral replication [15], Vif is absolutely required for efficient in vivo virus replication and pathogenicity [16,17]. The flanking ends of the proviral DNA are regions of long terminal repeats (LTRs) that are divided into the U3, R, and U5 regions [18,19,20]. These regions provide the signals required for viral transcription and integration into the host genome [19]. Small ruminant lentiviruses differ from the primate lentiviruses in that their Tat proteins do not trans-activate the viral LTR promoters [21]. Rather, the SRLV Tat protein is functionally similar to the HIV type 1 (HIV-1) Vpr protein [22].
The HIV-1 genome is similar to the SRLV genome, but is more complex and contains additional genes [23]. In contrast to SRLV, HIV-1 contains nine genes encoding 15 proteins; these genes, include the structural gag, pol, and env genes that encode the capsid proteins, the viral enzymes, and the envelope glycoproteins, gp120 (SU) and gp41 (TM), respectively [24]. The HIV-1 non-structural genes consist of vif, tat, rev, nef, vpu, and vpr, and are associated with HIV-1 pathogenesis and immune evasion [25]. Like the SRLV genome, the flanking ends of the provirus contain LTRs consisting of U3, R, and U5 regions [26]. Additionally, a variety of promoter and enhancer elements have been identified in the LTR region of HIV-1. Some of these response elements include AP-1, NF-κB, and Sp1 [27], important host immune related transcriptional factors which all lead to transcriptional activation and virus replication.
Figure 1.
Genomic organization of small ruminant lentiviruses (SRLV) and HIV-1 [23].
Figure 1.
Genomic organization of small ruminant lentiviruses (SRLV) and HIV-1 [23].
5. Conclusions
The immune response to lentiviral infections is a complex and dynamic response, and to date very little is known about how the immune system responds to these infections. Extensive research on HIV-1 has greatly contributed to our knowledge of the dynamics of the host–virus interaction; however, HIV and SRLV, despite their similarities are two distinct viruses and extrapolating knowledge from HIV research must be approached with caution. It is evident from the research presented here that gaps exist in our knowledge of SRLV, and before exploring the possibility of breeding for resistance, extensive research needs to be done to better understand SRLV immune responses. These knowledge gaps primarily lie in the areas that focus on how SRLV modulate the host immune response and the dynamics of early infection. As previously discussed, for example, the Th1/Th2 paradigm appears to apply to SRLV infection; however, if in fact SRLV infection steers macrophage polarization towards a M2 phenotype favoring the Th2 immune response, then one would expect that the disease would progress rapidly and clinical disease would be apparent in a large proportion of animals. However, it often takes years before clinical SRLV infection becomes apparent. Therefore, future studies should firstly investigate how SRLV infection alters macrophage function as a whole. To start, cytokine production and intracellular signaling should be investigated. It would also be beneficial to investigate how endogenous stress hormone levels affect disease resistance and progression, and how host macrophages respond to SRLV during co-infection with other pathogens. Since macrophages play an important role as both innate effector cells and antigen presenting cells, an improved understanding of SRLV-infected macrophage function will improve our understanding of how SRLV is controlled during early infection, how the acquired immune response is induced, and how SRLVs modulate the host immune response.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Fieni, F.; Rowe, J.; van Hoosear, K.; Burucoa, C.; Oppenheim, S.; Anderson, G.; Murray, J.; BonDurant, R. Presence of caprine arthritis encephalitis virus (CAEV) proviral DNA in genital tract tissues for superovulated dairy goat does. Theriogeneology 2003, 59, 1515–1523. [Google Scholar] [CrossRef]
- Konshi, M.; Tsuduku, S.; Haritani, M.; Murakami, K.; Tsuboi, T.; Kobayashi, C.; Yoshikawa, K.; Kimura, K.; Sentsui, H. An epidemic of caprine arthritis encephalitis in Japan: Isolation of the virus. Virology 2004, 66, 911–917. [Google Scholar]
- Pisoni, G.; Quasso, A.; Moroni, P. Phylogentic analysis of small ruminant lentivirus subtype B1 in mixed flocks: Evidence for natural transmission from goats to sheep. Virology 2005, 399, 147–152. [Google Scholar] [CrossRef]
- Pisoni, G.; Bertoni, G.; Puricelli, M.; Maccalli, M.; Moroni, P. Demonstration of coinfection with and recombination by caprine arthritis encephalitis virus and maedi visna virus in naturally infected goats. J. Virol. 2007, 81, 4948–4955. [Google Scholar] [CrossRef]
- Ravazzolo, A.; Nenci, C.; Vogt, H.; Waldvogel, A.; Obexer-Ruff, G.; Peterhans, E.; Bertoni, G. Viral load, organ distribution, histopathological lesions, and cytokine mRNA expression in goats infected with a molecular clone of the caprine arthritis encephalitis virus. Virology 2006, 350, 116–127. [Google Scholar]
- Dufour, S.; Zahno, M.; Vogt, H.; Peterhans, E.; Bertoni, G. Production of monospecific antibody to immunodominant epitopes of the caprine arthritis encephalitis virus transmembrane glycoprotein and analysis of their activity in vitro. Intervirology 2001, 45, 177–182. [Google Scholar]
- Olech, M.; Rachid, A.; Croise, B.; Kuzmak, J.; Valas, S. Genetic and antigenic characterization of small ruminant lentiviruses circulating in Poland. Virus Res. 2012, 163, 528–563. [Google Scholar]
- Polledo, L.; Gonzalez, J.; Benavides, J.; Morales, S.; Martinez-Fernandez, B.; Delago, L.; Reina, R.; Glaria, I.; Perez, V.; Ferreras, M.; et al. Patterns of lesion and local host cellular immune response in natural cases of ovine maedi visna. J. Comp. Path. 2011, 147, 1–10. [Google Scholar]
- Bolea, R.; Monleon, E.; Carrasco, L.; Vargas, A.; de Andres, D.; Amorena, B.; Badiola, J.; Jujan, L. Maedi-Visna virus infection of ovine mammary epithelial cells. Vet. Res. 2006, 37, 133–144. [Google Scholar]
- Ali Al Ahmad, M.; Chebloune, Y.; Chatagnon, G.; Pellerin, J.; Fieni, F. Is caprine arthritis encephalitis virus (CAEV) transmitted vertically to early embryo development stages (morulae or blastocyst) via in vitro infected frozen semen. Theriogenology 2012, 77, 1673–1678. [Google Scholar] [CrossRef]
- Modolo, J.; Stachissini, A.; Padovani, C.; Araujo Junior, J.; Castro, R.; Ravazzolo, A.; Leite, B. PCR associated with agar gel immunodifusion assay improve caprine arthritis encephalaitis (CAEV) control. Small Rumin. Res. 2009, 81, 18–20. [Google Scholar] [CrossRef]
- Reina, R.; Grego, E.; Bertolotti, L.; de Meneghi, D.; Rosati, S. Genome analysis of small ruminant lentivirus genotype E: A caprine lentivirus with natural deletions of the dUTPase subunit, vpr-like accessory gene, and 70-base-pair repeat of the U3 regions. J. Virol. 2009, 83, 1152–1155. [Google Scholar] [CrossRef]
- L’Homme, Y.; Ouardani, M.; Levesque, V.; Bertoni, G.; Simard, C.; Pisoni, G. Molecular characterization and phylogenetic analysis of small ruminant lentiviruses isolatd from Canadian sheep and goats. Virol. J. 2011, 8, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Abelson, M.; Schoborg, R. Characterization of the caprine arthritis encephalitis virus (CAEV) rev N-terminal elements required for efficient interaction with the RRE. Virus Res. 2003, 92, 23–35. [Google Scholar] [CrossRef]
- Harmache, A.; Bouyac, M.; Audoly, G.; Hieblot, C.; Peveri, P.; Vigne, R.; Suzan, M. The vif gene is essential for efficient replication of caprine arthritis encephalitis virus in goat synovial membrane cells and affects the late steps of the virus replication cycle. J. Virol. 1995, 69, 3247–3257. [Google Scholar]
- Harmache, A.; Russo, P.; Guiguen, F.; Vtu, C.; Vignoni, M.; Bouyac, M.; Heiblot, C.; Pepin, M.; Vigne, R.; Suzan, M. Requirement of caprine arthritis encephalitis virus vif gene for in vitro replication. Virology 1996, 224, 246–255. [Google Scholar] [CrossRef]
- Kristbjornsdottir, H.; Andresdottir, V.; Svansson, V.; Throsteinsdottir, S.; Matthiasdottir, S.; Andresson, O. The vif gene of madei-visna is essential for infectivity in vivo and vitro. Virology 2004, 318, 350–359. [Google Scholar] [CrossRef]
- Angelopoulou, K.; Brellou, G.; Greenland, T.; Vlemmas, I. A novel eletion in the LTR region of a Greek small ruminant lentivirus may be associated with low pathogenicity. Virus Res. 2006, 118, 174–184. [Google Scholar]
- Angelopoulou, K.; Poutahidis, T.; Brellou, G.; Greenland, T.; Vlemmas, I. 2008. A deletion in the R region of long terminal repeats in small ruminant lentiviruses is associated with decreased pathology in the lung. Vet. J. 2008, 175, 346–355. [Google Scholar] [CrossRef]
- Leroux, C.; Mornex, J. Retroviral infections in sheep and the associated diseases. Small Rumin. Res. 2008, 76, 68–76. [Google Scholar] [CrossRef]
- Villet, S.; Faure, C.; Bouzar, B.; Morin, T.; Verdier, G.; Chebloune, Y.; Legras, C. Lack of trans-activation function for madei visna virus and caprine arthritis encephalitis virus tat proteins. Virology 2003, 307, 317–327. [Google Scholar] [CrossRef]
- Villet, S.; Bouzar, B.; Morin, T.; Verdier, G.; Legras, C.; Chebloune, Y. Madei-Visna and caprine arthritis encelphalitis vurs genomes encode a vpr-like but no tat protein. J. Virol. 2003, 77, 9632–9638. [Google Scholar] [CrossRef]
- Valas, S.; Rolland, M.; Perrin, C.; Perrin, G.; Mamoun, R. Characterization of a new 5’ splice site within the caprine arthritis encephalitis virus genome: evidence for a novel auxiliary protein. Retrovirology 2008, 5, 1–17. [Google Scholar] [CrossRef]
- Watts, J.; Dang, K.; Gorelick, R.; Leonard, C.; Bess, J.; Swanstrom, R.; Burch, C.; Weeks, K. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 2009, 460, 711–719. [Google Scholar] [CrossRef]
- Malim, M.; Emerman, M. HIV-1 accessory proteins—Ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef]
- Arellano, E.; Alcami, J.; Lopez, M.; Soriano, V.; Holguin, A. Drastic decrease of transcription activity due to hypermutated long terminal repeat (LTR) region in different HIV-1 subtypes and recombinants. Antivir. Res. 2010, 88, 152–159. [Google Scholar] [CrossRef]
- Katagiri, D.; Hayashi, H.; Victoriano, A.; Okamoto, T.; Onozaki, K. Estrogen stimulates transcription of human immunodeficiency virus type 1 (HIV-1). Int. Immunopharmacol. 2006, 6, 170–181. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-Like receptor signaling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar]
- Oh, D.; Baumann, K.; Hamouda, O.; Eckert, J.; Neumann, K.; Kucherer, C.; Bartmeyer, B.; Poggensee, G.; Oh, N.; Pruss, A.; et al. A frequent functional toll-like receptor 7 polymorphism is associated with accelerated HIV-1 disease progression. AIDS 2009, 23, 297–307. [Google Scholar] [CrossRef]
- Chang, J.; Lacas, A.; Lindsay, R.; Doyle, E.; Axten, K.; Pereyra, F.; Rosenberg, E.; Walker, B.; Allen, T.; Altfeld, M. Differential regulation of toll-like receptor pathways in acute and chronic HIV-1 infection. AIDS 2012, 26, 533–541. [Google Scholar] [CrossRef]
- Ranjbar, S.; Jasenosky, L.; Chow, N.; Goldfield, A. Regulation of mycobacterium tuberculosis-dependant HIV-1 transcription reveals a new role of NFAT5 in the toll-like receptor pathway. PLoS Pathog. 2012, 8, e1002620. [Google Scholar] [CrossRef]
- Goujon, C.; Malim, M. Characterization of the alpha interfearon-induced postentry block to HIV-1 infection in primary human macrophages and T cells. J. Virol. 2010, 84, 9254–9266. [Google Scholar] [CrossRef]
- Pham, Q.; Bouchard, A.; Grutter, M.; Berthoux, L. Generation of human TRIM5α mutants with high HIV-1 restriction activity. Gene Ther. 2010, 17, 859–871. [Google Scholar] [CrossRef]
- Campbell, E.; Perez, O.; Anderson, J.; Hope, T. Visualization of proteasome-independent intermediate during restriction of HIV-1 by rehesus TRIM5α. J. Cell Biol. 2008, 180, 549–561. [Google Scholar] [CrossRef]
- Lukic, Z.; Hausmann, S.; Sebastian, S.; Rucci, J.; Sastrji, J.; Robia, S.; Juban, J.; Campbell, E. TRIM5α associates with proteosomal subunits in cells while in complex with HIV-1 virons. Retrovirology 2011, 8, 1–15. [Google Scholar] [CrossRef]
- Casartelli, N.; Guivel-Benhassine, F.; Bouziat, R.; Brandler, S.; Schwartz, O.; Moris, A. The antiviral factor APOBEC3G improves CTL recognition of cultured HIV-infected T cells. J. Exp. Med. 2010, 207, 39–49. [Google Scholar] [CrossRef]
- Khan, M.; Golia-Gaur, R.; Kao, S.; Miyagi, E.; Walker, R.; Strebel, K. Encapsidation of APOBEC3G into HIV-1 virons involves lip raft assocaiation and does not correlate with APOBEC3G oligomerization. Retrovirology 2009, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Love, R.; Chelico, L. HIV-1 viral infectivity factor (vif) alters possessive single stranded DNA scanning of the retroviral restriction factor, APOBEC3G. J. Biol. Chem. 2013, 288, 6083–6094. [Google Scholar] [CrossRef]
- Britan-Rosich, E.; Nowarski, R.; Kotler, M. Multifaceted counter-APOBEC3G mechanisms employed by HIV-1 vif. J. Mol. Biol. 2011, 410, 1065–1076. [Google Scholar] [CrossRef]
- Wang, X.; Ao, Z.; Chen, L.; Kobinger, G.; Peng, J.; You, X. The cellular antiviral protein APOBEC3G interacts with HIV-1 reverse transcriptase and inhibits its function during viral replication. J. Virol. 2012, 86, 3777–3786. [Google Scholar] [CrossRef]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.; Gregory, D.; Johnson, M.; Bieniasz, P. Tetherin inhibits HIV-1 release by directly tethering virons to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef]
- Dube, M.; Roy, B.; Guiot-Guillain, P.; Binette, J.; Mercier, J.; Chiasson, A.; Cohen, E. Antagonism of thetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010, 6, e1000856. [Google Scholar] [CrossRef] [Green Version]
- Miyakawa, K.; Ryo, A.; Murakami, T.; Ohba, K.; Yamaoka, S.; Fukuda, M.; Guatelli, J.; Yammamoto, N. BCA2/Rabring7 promotes tetherin-dependant HIV-1 restriction. PLoS Pathog. 2009, 5, e1000700. [Google Scholar] [CrossRef]
- Kueck, T.; Neil, S. A cytoplasmic tail determinant in HIV-1 Vpu mediates targeting of tetherin for endosomal degradation and counteracts interfearon-induced restricton. PLoS Pathog. 2012, 8, e1002609. [Google Scholar] [CrossRef]
- Sze, A.; Olagnier, D.; Lin, R.; van Grevenyghe, J.; Hiscott, J. SAMHD1 host restriction factor: A link with innate immune sensing of retrovirus infection. J. Mol. Biol. 2013, 425, 4981–4494. [Google Scholar] [CrossRef]
- Baldauf, H.M.; Pan, X.; Erikson, E.; Schmidt, S.; Daddacha, W.; Burggraf, M.; Schenkova, K.; Abbiel, I.; Wabintz, G.; Gramberg, T.; et al. SAMHD1 restricts HIV-1 infection in resting CD4+ T cells. Nat. Med. 2012, 18, 1682–1689. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, G.; Lv, F.; Wang, X.; Ji, X.; Xu, Y.; Sun, J.; Wu, L.; Zheng, Y.; Gao, G. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proc. Natl. Acad. Sci. USA 2011, 108, 15834–15839. [Google Scholar]
- Alter, G.; Teigen, N.; Ahern, R.; Streeck, H.; Meier, A.; Rosenberg, E.; Atfeld, M. Evolution of innate and adaptive effector cell functions during acute HIV-1 infection. J. Infect. Dis. 2007, 195, 1452–1460. [Google Scholar] [CrossRef]
- Jia, M.; Li, D.; He, X.; Zhao, Y.; Peng, H.; Ma, P.; Hong, K.; Liang, H.; Shao, Y. Impaired natural killer cell induced antibody-dependant cell-mediated cytotoxicity is associated with human immunodeficiency virus-1 disease progression. Clin. Exp. Immunol. 2012, 171, 107–116. [Google Scholar]
- Naranbhai, V.; Altfeld, M.; Karim, S.; Ndung’u, T.; Karim, Q.; Carr, W. Changes in natural killer cell activation and function during primary HIV-1 infection. PLoS One 2013, 8, e53251. [Google Scholar]
- Thobakgale, C.; Fadda, L.; Lane, K.; Toth, I.; Pereyra, F.; Bazner, S.; Ndung’u, T.; Walker, B.; Rosenberg, E.; Alter, G.; et al. Frequent and strong antibody-mediated natural killer cell activation in response to HIV-1 env in individuals with chronic HIV-1 infection. J. Virol. 2012, 86, 6986–6993. [Google Scholar] [CrossRef]
- Fenogilo, D.; Poggi, A.; Catellani, S.; Battagila, F.; Ferrera, A.; Setti, M.; Murdaca, G.; Zocchi, M. Vδ1 T lymphocytes producing IFN-γ and IL-17 are expanded in HIV-1 infected patients and respond to candida albicans. Blood 2009, 113, 6611–6618. [Google Scholar] [CrossRef]
- Zheng, N.; McElrath, J.; Sow, P.; Mesher, A.; Hawes, S.; Stern, J.; Gottlieb, G.; De Rosa, S.; Kiviat, N. Association between peripheral γδ T-cell profile and disease progression in individuals infected with HIV-1 or HIV-2 in West Africa. J. Acquir. Immune Defic. Syndr. 2011, 57, 92–100. [Google Scholar] [CrossRef]
- Poonia, B.; Pauza, C. Gamma delta T cells form HIV+ donor can be extended in vitro by zoledronate/interlukin-2 to become cytotoxic effectors for antibody-dependant cellular cytotoxicity. Cytotherapy 2012, 14, 173–181. [Google Scholar] [CrossRef]
- Kindt, T.; Goldsby, R.; Osborne, B. Kuby Immunology, 6th ed.; W.H. Freeman and Company: New York, NY, USA, 2007; pp. 314–315. [Google Scholar]
- Munier, C.; Kelleher, A.; Kent, S.; De Rose, R. The role of T cell immunity in HIV-1 infection. Curr. Opin. Virol. 2013, 3, 1–9. [Google Scholar] [CrossRef]
- Buckheit, R.; Siliciano, R.; Lankson, J. Primary CD8+ T cells form elite suppressors effectively eliminate non-productively HIV-1 infected resting and activated CD4+ T cells. Retrovirology 2013, 10, 1–12. [Google Scholar] [CrossRef]
- Okoye, A.; Rohankhedkar, M.; Abana, C.; Pattenn, A.; Reyes, M.; Pexton, C.; Lum, R.; Sylwester, A.; Planer, S.; Legasse, A.; et al. Naive T cells are dispensable for memory CD4+ T cell homeostatis in progressive simian immunodeficiency virus infection. J. Exp. Med. 2012, 209, 641–651. [Google Scholar] [CrossRef]
- Ranasinghe, S.; Flanders, M.; Cutler, S.; Soghian, Z.; Ghebremichael, M.; Davis, I.; Lindquvist, M.; Pereyra, F.; Walker, B.; Heckerman, D.; et al. HIV-specific CD4 T cell responses to different viral proteins hve discordant assocaitions with viral load and clinical outcome. J. Virol. 2011, 86, 277–283. [Google Scholar]
- Goonetilleke, N.; Liu, M.; Salazar-Gonzalez, J.; Ferrari, G.; Giorgi, E.; Ganusov, V.; Keele, B.; Learn, G.; Turnbull, E.; Salazar, M.; et al. The first T cell response to transmitted/founder virus contributes to the control of acute virema in HIV-1 infection. J. Exp. Med. 2009, 206, 1253–1272. [Google Scholar] [CrossRef]
- Cohen, G.; Gandhi, R.; Davis, D.; Mandelboim, O.; Chen, B.; Strominger, J.; Baltimore, D. The selective downregulation of class 1 major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 1999, 10, 661–671. [Google Scholar] [CrossRef]
- Frahm, N.; Korber, B.; Adams, C.; Szinger, J.; Draenert, R.; Addo, M.; Feeney, M.; Yusim, K.; Sango, K.; Brown, N.; et al. Constant cytotoxic T lymphocyte targeting of immunodominant regions in human immunodeficiency virus across multiple ethnicities. J. Virol. 2004, 78, 2178–2200. [Google Scholar]
- Chevalier, M.; Julg, B.; Pyo, A.; Flanders, M.; Ranasinghe, S.; Soghoian, D.; Kwon, D.; Rychert, J.; Lian, J.; Muller, M.; et al. HIV-1-specific interleukin-21+ CD4+ T cell responses contribute to durable viral control through the modulation of HIV-specific CD8+ T cell function. J. Virol. 2011, 85, 733–741. [Google Scholar] [CrossRef]
- Wang, Y.; Li, B.; Carlson, J.; Streeck, H.; Gladden, A.; Goodman, R.; Schneidewind, A.; Power, K.; Toth, I.; Frahm, N.; et al. Protective HLA class I alleles that restrict acute phase CD8+ T cell responses are associated with viral escape mutation located in highly conserved region of human immunodeficiency virus type 1. J. Virol. 2009, 83, 1845–1855. [Google Scholar] [CrossRef]
- Ferrari, G.; Korber, B.; Goonetilleke, N.; Liu, M.; Turnbull, E.; Salazar-Gonzalez, J.; Hawkins, N.; Self, S.; Watson, S.; Betts, M.; et al. Relationship between functional profile of HIV-1 specific CD8 T cells and epitope variability with the selection of escape mutants in acute HIV-1 infection. PLoS Pathog. 2011, 7, e1001273. [Google Scholar] [CrossRef]
- Sanchez-Merino, V.; Farrow, M.; Brewster, F.; Somasundaran, M.; Luzuriaga, K. Identification and characterization of HIV-1 CD8+ T cell escape variants with impaired fitness. JID 2008, 197, 300–308. [Google Scholar] [CrossRef]
- Troyer, R.; McNevin, J.; Liu, Y.; Zhang, S.; Krizan, R.; Abraha, A.; Tebit, D.; Zhao, H.; Avila, S.; Lobritz, M.; et al. Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog. 2013, 5, e1000365. [Google Scholar]
- Ganusov, V.; Goonetilleke, N.; Liu, M.; Ferrari, G.; Shaw, G.; McMicheal, A.; Borrow, P.; Korber, B.; Perelson, A. Fitness costs and diversity of the cytotoxic T lymphocyte response determine the rate of CTL escape during acute and chronic phases of HIV infection. J. Virol. 2011, 85, 10518–10528. [Google Scholar] [CrossRef]
- Akahoshi, T.; Chikatat, T.; Tamura, Y.; Gatanaga, H.; Oka, S.; Takiguchi, M. Selection and accumulation of an HIV-1 escape mutant by three types of HIV-1 specific cytotoxic T lymphocytes recognizing wild-type and/or escape mutant epitopes. J. Virol. 2012, 86, 1971–1981. [Google Scholar] [CrossRef]
- Rueda, C.; Veilla, P.; Chougnet, C.; Montoya, C.; Rugeles, M. HIV-induced T-cell activation/exhaustion in rectal mucosa is controlled only partially by antiretroviral treatment. PLoS One 2012, 7, e30307. [Google Scholar]
- Sauce, D.; Larsen, M.; Fastenackels, S.; Pauchard, M.; Ait-Mohand, H.; Schneider, L.; Guihot, A.; Boufassa, F.; Zaunders, J.; Iguertsira, M.; et al. HIV disease progression despite suppression of viral replication is associated with exhaustion of lymphopoesis. Blood 2011, 117, 5142–5151. [Google Scholar] [CrossRef]
- Yamamoto, T.; Price, D.; Casazza; Ferrari, G.; Nason, M.; Chattopadhay, P.; Roederer, M.; Gostik, E.; Katsikis, P.; Douek, D.; Jaubrich, R.; et al. Surface expression patterns of negative regulator molecules identify determinants of virus specific CD8+ T cell exhaustion in HIV infection. Blood 2011, 117, 4805–4815. [Google Scholar] [CrossRef]
- Tomaras, G.; Yates, N.; Liu, P.; Qin, L.; Fouda, G.; Chavez, L.; Decamp, A.; Parks, R.; Ashley, V.; Lucas, J.; et al. Initial B-cell responses to transmitted human immunodeficiency virus type 1, viron-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J. Virol. 2008, 82, 12449–12463. [Google Scholar] [CrossRef]
- Lynch, R.; Tran, L.; Louder, M.; Schmidt, S.; Cohen, M.; DerSimonian, R.; Euler, Z.; Gray, E.; Karim, S.; Kirchherr, J.; et al. The development of CD4 binding site antibodies during HIV-1 infection. J. Virol. 2012, 86, 7588–7595. [Google Scholar] [CrossRef]
- Liu, P.; Overman, R.; yates, N.; Alam, M.; Vandergrift, N.; Chen, Y.; Graw, F.; Freel, S.; Kappes, J.; Ochsenbauer, C.; et al. Dynamic antibody specificities and viron concentrations in circulating immune complexes in acute to chronic HIV-1 infection. J. Virol. 2011, 85, 11196–11207. [Google Scholar] [CrossRef]
- Smalls-Mantey, A.; Doria-Rose, N.; Klein, R.; Patamawenu, A.; Migueles, S.; Ko, S.; Hallahan, C.; Wong, H.; Liu, B.; You, L.; et al. Antibody-Dependent cellular cytotoxicity against primary HIV-infected CD4+ T cells is directly associated with the magnitude of surface IgG binding. J. Virol. 2012, 86, 8672–8680. [Google Scholar] [CrossRef]
- Mikell, I.; Sather, D.; Kalams, S.; Altfeld, M.; Alter, G.; Stamatatos, L. Characteristics of the earliest cross-neutralizing antibody response to HIV-1. PLoS Pathog. 2011, 7, e1001251. [Google Scholar] [CrossRef]
- Euler, Z.; Gils, M.; Bunnik, E.; Phung, P.; Schweighardt, B.; Wrin, T.; Schuitemaker, H. Cross-Reactive neutralizing humoral immunity does ot protect for HIV type 1 disease progression. J. Infect. Dis. 2010, 201, 1045–1053. [Google Scholar] [CrossRef]
- Salado, M.; Rallon, N.; Rodes, B.; Lopez, M.; Soriano, V.; Benito, J. Long-Term non-progressors display a greater number of Th17 cells than HIV-infected typical progressors. Clin. Immunol. 2011, 139, 110–114. [Google Scholar] [CrossRef]
- Li, D.; Chen, J.; Jia, M.; Hong, K.; Ruan, Y.; Liang, H.; Zhang, X.; Zhao, H.; Peng, H.; Ma, P.; et al. Loss of balance between T helper type 17 and regulatory T cells in chronic human immunodeficiency virus infection. Clin. Exp. Immunol. 2011, 165, 363–371. [Google Scholar] [CrossRef]
- Kwon, D.; Angin, M.; Hongo, T.; Law, K.; Johnson, J.; Porichis, F.; Hart, M.; Pavlik, D.; Tighe, D.; Kavanagh, D.; et al. CD4+ CD25+ regulatory T cell impair HIV-1 specific CD4 T cell responses by upregulating interlukin-10 production in monocytes. J. Virol. 2012, 86, 6586–6594. [Google Scholar] [CrossRef]
- Card, C.; McLaren, P.; Wachihi, C.; Kimani, J.; Plummer, F.; Fowke, K. Decreased immune activation in resistance to HIV-1 infection is associated with an elevated frequency of CD4+CD25+FOXP3+ regulatory T cells. J. Infect. Dis. 2009, 199, 1318–1322. [Google Scholar] [CrossRef]
- Prendergast, A.; Prado, J.; Kang, U.; Chen, F.; Riddell, L.; Luzzi, G.; Goulder, P.; Klenerman, P. HIV-1 infection is characterized by profound depletion of CD161+ Th17 cells and gradual decline in regulator T cells. AIDS 2010, 24, 491–502. [Google Scholar] [CrossRef]
- Blacklaws, B. Small ruminant lentiviruses: Immunopathogenesis of visna-maedi and caprine arthritis encephalitis virus. Comp. Immunol. Microbiol. Infect. Dis. 2012, 35, 259–269. [Google Scholar]
- Jauregui, P.; Crespo, H.; Glaria, I.; Lujan, L.; Conteras, A.; Rosati, S.; de Andres, D.; Amorena, B.; Towers, G.; Reina, R. Ovine TRIM5α can restrict visna/maedi virus. J. Virol. 2012, 86, 9504–9509. [Google Scholar] [CrossRef]
- LaRue, R.; Jónsson, S.; Silverstein, K.; Lajoie, M.; Bertrand, D.; El-Mabrouk, N.; Hotzel, I.; Andresdottir, V.; Smith, T.; Harris, R. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol. Biol. 2008, 9, 104. [Google Scholar] [CrossRef]
- Arnaud, F.; Black, S.; Murphy, L.; Griffiths, D.; Neil, S.; Spencer, T.; Palmarini, M. Interplay between ovine bone marrow stromal cell antigen 2/tetherin and endogenous retroviruses. J. Virol. 2010, 84, 4415–4425. [Google Scholar] [CrossRef]
- Singh, I.; McConnell, I.; Dalziel, R.; Blacklaws, B. Serum containing ovine IgG2 antibody specific for maedi visna virus envelope glycoprotein mediates antibody dependent cellular cytotoxicity. Vet. Immunol. Immunopathol. 2006, 113, 357–366. [Google Scholar] [CrossRef]
- Kaba, J.; Winnicka, A.; Zaleska, M.; Nowicki, M.; Bagnicka, E. Influence of chronic caprine arthritis encephalitis virus infection on the population of peripheral blood leukocytes. Pol. J. Vet. Sci. 2011, 14, 585–590. [Google Scholar]
- Jolly, P.; Gangpoadhyay, A.; Chen, S.; Gopal Reddy, P.; Weiss, H.; Sapp, W. Changes in the leukocyte phenotype profile of goats infected with the caprine arthritis encephalitis virus. Vet. Immunol. Immunopathol. 1997, 56, 97–106. [Google Scholar] [CrossRef]
- Ponti, W.; Paape, M.; Bronzo, V.; Pisoni, G.; Pollera, C.; Moroni, P. Phenotypic alteration of blood and milk leukocytes in goats naturally infected with caprine arthritis-encephalitis virus (CAEV). Small Rumin. Res. 2008, 78, 176–180. [Google Scholar] [CrossRef]
- Reina, R.; Barbezange, C.; Niesalla, H.; de Andres, X.; Arnarson, H.; Biescas, E.; Mazzei, M.; Fraisier, C.; McNeilly, T.; Liu, C.; et al. Mucosal immunization against ovine lentiviruses using PEI-DNA complexes and modified vaccine Ankara encoding the gag and/or env genes. Vaccine 2008, 26, 4494–4505. [Google Scholar]
- Cheevers, W.; Hotzel, I.; Beyer, J.; Kumpula-McWhirter, N. Immune response to caprine arthritis-encephalitis virus surface protein induced by coimmuization with recombinant vaccine viruses expressing the caprine arthritis-encephalitis virus envelope gene and caprine interlukin-12. Vaccine 2000, 18, 2494–2503. [Google Scholar]
- Cheevers, W.; Hotzel, I. Plasmid DNA encoding caprine interferon gamma inhibits antibody response to caprine arthritis encephalitis virus (CAEV) surface protein encoded by a co-administered plasmid expressing CAEV env and tat genes. Vaccine 2001, 19, 3209–3215. [Google Scholar] [CrossRef]
- Perry, L.; Wilkerson, M.; Hullinger, G.; Cheevers, W. Depressed CD4+ T lymphocyte proliferative response and enhanced antibody response to viral antigen in chronic lentivirus-induced arthritis. J. Infect. Dis. 1995, 171, 328–334. [Google Scholar] [CrossRef]
- Reina, R.; Glaria, I.; Benavides, J.; de Andrés, X.; Crespo, H.; Solano, C.; Perez, V.; lujan, L.; Perez, M.; Perez, J.; et al. Association of CD80 and CD86 expression levels with disease status of Visna/Maedi virus infected sheep. 2007. Viral Immunol. 2007, 20, 609–622. [Google Scholar]
- Murphy, B.; Hotzel, I.; Jasmer, D.; Davis, W.; Knowles, D. TNFα and GM-CSF-induced activation of the CAEV promoter is independent of AP-1. Virology 2006, 352, 188–199. [Google Scholar] [CrossRef]
- Murphy, B.; Jasmer, D.; Stephen, W.; Knowles, D. Localization of a TNF-activated transcpriton site and interactions with the gamma activated site within the CAEV U3 70 base pair repeat. Virology 2007, 634, 196–207. [Google Scholar]
- Murphy, B.; Hillman, C.; Castillo, D.; Vapniarsky, N.; Rowe, J. The presence or absence of the gamma-mediated transcriptional activation in CAEV prometers cloned from the mammary gland and joint synovium of a single CAEV-infected goat. Virus Res. 2012, 163, 537–545. [Google Scholar] [CrossRef]
- Juganaru, M.; Reina, R.; Grego, E.; Profiti, M.; Rosati, S. LTR promoter activity of SRLV genotype E, strain Roccaverano. Vet. Res. Commun. 2010, 34, 47–51. [Google Scholar] [CrossRef]
- Bertoni, G.; Hertig, C.; Zahno, M.; Vogt, H.; Dufour, S.; Cordano, P.; Peterhans, E.; Cheevers, W.; Sonigo, P.; Pancino, G. B-Cell epitopes of the envelope glycoprotein of caprine arthritis encephalitis virus and antibody response in infected goats. J. Gen. Virol. 2000, 81, 2929–2940. [Google Scholar]
- Rachid, A.; Croise, B.; Russo, P.; Vignoni, M.; Lacerenza, D.; Rosati, S.; Kuzmak, J.; Valas, S. Diverse host-pathogen interactions following caprine arthritis encephalitis virus infection in sheep and goats. J. Gen. Virol. 2013, 94, 634–642. [Google Scholar] [CrossRef]
- Narayan, O.; Clements, J.; Griffin, D.; Wolinsky, J. Neutralizing antibody spectrum determines the antigenic profiles of emerging mutants of visna virus. Infect. Immun. 1891, 32, 1045–1050. [Google Scholar]
- Narayan, O.; Griffin, D.; Clements, J. Virus mutation during slow infection: Temporal development and characterization of mutants of visna virus recovered form sheep. J. Gen. Virol. 1978, 41, 343–352. [Google Scholar] [CrossRef]
- Haflidadóttir, B.; Matthíasdóttir, S.; Agnarsdóttir, G.; Torsteinsdóttir, S.; Pétursson, G.; Andrésson, Ó.; Andrésdóttir, V. Mutational analysis of a principal neutralization domain of visna/maedi virus envelope glycoprotein. J. Gen. Virol. 2008, 89, 716–721. [Google Scholar] [CrossRef]
- Trujillo, J.; Hotzel, K.; Snekvik, K.; Cheevers, W. Antibody response to the surface glycoprotein of caprine arthritis-encephalitis lentivirus: Disease status is predicted by SU antibody isotype. Virology 2004, 325, 129–136. [Google Scholar] [CrossRef]
- González, B.; Reina, R.; García, I.; Andrés, S.; Glaria, I.; Alzueta, M.; Mora, M.; Jugo, B.; Arrieta-Aguirre, A.; Perez, J.; et al. Mucosal immunization of sheep with a Maedi-Visna virus (MVV) env DNA vaccine protects against early MVV productive infection. Vaccine 2005, 23, 4342–4352. [Google Scholar]
- Pérez, M.; Biescas, E.; Reina, R.; Glaria, I.; Marín, B.; Marquina, A.; Salazar, E.; Alvarez, N.; de Andres, D.; Fantova, E.; et al. Small ruminant lentivirus–induced arthritis clinicopathologic findings in sheep infected by a highly replicative SRLV B2 genotype. Vet. Pathol. Online 2014. [Google Scholar] [CrossRef]
- Bodungen, U.; Lechner, F.; Pifster, H.; Vogt, H.; Cheevers, W.; Bertoni, T.; Jungi, W.; Peterhans, E. Immunohistology of the early course of lentivirus-induced arthritis. Clin. Exp. Immunol. 1998, 111, 384–390. [Google Scholar] [CrossRef]
- Lechner, F.; Machado, J.; Bertoni, G.; Seow, H.; Dobbelaere, D.; Peterhans, E. Caprine arthritis encephalitis virus disregulates the expression of cytokines in macrophages. J. Virol. 1997, 71, 7488–7497. [Google Scholar]
- Zhang, Z.; Harkiss, G.; Hopkins, J.; Woodall, C. Granulocyte macrophage colony stimulating factor is elevated in alveolar macrophages from sheep naturally infected with maedi-visna virus and stimulates maedi-visna virus replication in macrophages in vitro. Clin. Exp. Immunol. 2002, 129, 240–246. [Google Scholar] [CrossRef]
- Crespo, H.; Bertolotti, L.; Juganaru, M.; Glaria, I.; de Andrés, D.; Amorena, B.; Rosati, S.; Reina, R. Small ruminant macrophage polarization may play a pivotal role on lentiviral infection. Vet. Res. 2013, 44, 83. [Google Scholar] [CrossRef]
- Fluri, A.; Nenci, C.; Zahno, M.; Vogt, H.; Charan, S.; Busato, A.; Pancino, G.; Peterhans, E.; Obexer-Ruff, G.; Bertoni, G. The MHC-haplotype influence primary, but not memory, immune responses to an immunodominant peptide containing T- and B-cell epitopes of the caprine arthritis encephalitis virus gag protein. Vaccine 2006, 24, 597–606. [Google Scholar]
- Pyrah, I.; Watt, N. Immunohistological study of the depressed cutaneous DTH response in sheep naturally infected with an ovine lentivirus (Maedi-Visna virus). Clin. Exp. Immunol. 1996, 104, 32–36. [Google Scholar]
- Kuniholm, M.; Gao, X.; Xue, X.; Kovacs, A.; Anastos, K.; Marti, D.; Greenblatt, R.; Cohen, M.; Minkoff, H.; Gange, S.; et al. Human leukocyte antigen genotype and risk of HIV disease progression before and after initiation of antiretroviral therapy. J. Virol. 2011, 85, 10826–10833. [Google Scholar] [CrossRef]
- Naruto, T.; Gatanaga, H.; Nelson, G.; Sakai, K.; Carrington, M.; Oka, S.; Takiguchi, M. HLA class I-mediated control of HIV-1 in the Japanese population, in which the protective HLA-B*57 and HLA-B*27 alleles are absent. J. Virol. 2012, 86, 10870–10872. [Google Scholar] [CrossRef]
- An, P.; Johnson, R.; Phair, J.; Kirk, G.D.; Yu, X.F.; Donfield, S.; Buchbinder, S.; Goedert, J.; Winkler, C.A. APOBEC3B deletion and risk of HIV-1 acquisition. J. Infect. Dis. 2009, 200, 1054–1058. [Google Scholar] [CrossRef]
- Marmor, M.; Sheppard, H.; Donell, D.; Bozeman, S.; Celum, C.; Buchbinder, S.; Koblin, B.; Seage, G. Homozygous and heterozygous CCR5-Δ32 genotypes are associated with resistance to HIV infection. J. Acquir. Immunodefic. Syndr. 2001, 27, 472–481. [Google Scholar] [CrossRef]
- Novembre, J.; Galvani, A.; Slatkin, M. The geographic spread of CCR5 Δ32 HIV-resistance allele. PLoS Biol. 2005, 3, e339. [Google Scholar] [CrossRef]
- Mikula, I.; Bhide, M.; Pastorekova, S.; Mikula, I. Characterization of ovine TLR7 and TLR8 protein coding regions, detection of mutations and Maedi Visna virus infection. Vet. Immunol. Immunopathol. 2010, 138, 51–59. [Google Scholar] [CrossRef]
- Larruskain, A.; Bernales, I.; Luján, L.; de Andrés, D.; Amorena, B.; Jugo, B.M. Expression analysis of 13 ovine immune response candidate genes in Visna/Maedi disease progression. Comp. Immunol. Microbiol. Infect. Dis. 2013, 36, 405–413. [Google Scholar]
- Bowles, D.; Carson, A.; Isaac, P. Genetic distinctiveness of the herdwick sheep breed and two other locally adapted hill breeds of the UK. PLoS One 2014, 9, e87823. [Google Scholar] [CrossRef]
- White, S.; Mousel, M.; Reynolds, J.; Lewis, G.; Herrmann-Hoesing, L. Common promoter deletion is associated with 3.9-fold differential transcription of ovine CCR5 and reduced proviral level of ovine progressive pneumonia virus. Anim. Gen. 2009, 40, 583–589. [Google Scholar] [CrossRef]
- Brajon, G.; Mandas, D.; Liciardi, M.; Taccori, F.; Meloni, M.; Corrias, F.; Montaldo, C.; Coghe, F.; Casciari, C.; Giammarioli, M.; et al. Development and field testing of a real-time PCR assayfor caprine arthritis encephalitis virus (CAEV). Open Virol. J. 2012, 6, 82–90. [Google Scholar]
- Herrmann-Hoesing, L.; Broughton-Neiswanger, L.; Gouine, K.; White, S.; Mousel, M.; Lewis, G.; Marshall, K.; Knowles, D. Evaluation of caprine arthritis encephalitis virus/maedi-visna virus indirect enzyme-linked immunosorbent assay in the serological diagnosis of ovine progressive pneumonia virus in U.S. sheep. Clin. Vacc. Immunol. 2010, 17, 307–310. [Google Scholar]
- Santry, L.A.; de Jong, J.; Gold, A.C.; Walsh, S.R.; Menzies, P.I.; Wootton, S.K. Genetic characterization of small ruminant lentiviruses circulating in naturally infected sheep and goats in Ontario, Canada. Virus Res. 2013, 175, 30–44. [Google Scholar] [CrossRef]
- Heaton, M.; Clawson, M.; Chitko-McKown, C.; Leymaster, K.; Smith, T.; Harhay, G.; White, S.; Herrmann-Hoesing, L.; Mousel, M.; Lewis, G.; et al. Reduced lentivirus susceptibility in sheep with TEMEM154 mutations. PLoS Gen. 2012, 8, 1–12. [Google Scholar]
- White, S.; Mousel, M.; Herrmann-Hoesing, L.; Reynolds, J.; Leymaster, K.; Neibergs, H.; Lewis, G.; Knowles, D. Genome-Wide association identifies multiple genomic regions associated with susceptibility to and the control of ovine lentivirus. PLoS One 2012, 7, e47829. [Google Scholar] [CrossRef]
- Petrovski, S.; Fellay, J.; Shianna, K.; Carpenetti, N.; Kumwenda, J.; Kamanga, G.; Kamwendo, D.; Letvin, N.; McMicheal, A.; Haynes, B.; et al. Common human genetic variants and HIV-1 susceptibility: A genome-wide survey in a homogenous African population. AIDS 2011, 25, 513–518. [Google Scholar] [CrossRef]
- Luo, M.; Sainsbury, J.; Tuff, J.; Lacap, P.; Yuan, X.; Hirbod, T.; Kimani, J.; Wachihi, C.; Ramdahin, S.; Bielawny, T.; et al. A genetic polymorphism of FREM1 is associated with resistance against HIV infection in the Pumwani sex worker cohort. J. Virol. 2012, 86, 11899–11905. [Google Scholar] [CrossRef]
- Bishop, S. Possibilities to breed for resistance to nematode parasite infections in small ruminants in tropical production systems. Animal 2012, 6, 741–747. [Google Scholar] [CrossRef]
- Thompson-Crispi, K.; University of Guelph, Department of Pathobiology, Guelph, Ontario, Canada. Personal Communication 2013.
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Stonos, N.; Wootton, S.K.; Karrow, N. Immunogenetics of Small Ruminant Lentiviral Infections. Viruses 2014, 6, 3311-3333. https://doi.org/10.3390/v6083311
AMA Style
Stonos N, Wootton SK, Karrow N. Immunogenetics of Small Ruminant Lentiviral Infections. Viruses. 2014; 6(8):3311-3333. https://doi.org/10.3390/v6083311
Chicago/Turabian StyleStonos, Nancy, Sarah K. Wootton, and Niel Karrow. 2014. "Immunogenetics of Small Ruminant Lentiviral Infections" Viruses 6, no. 8: 3311-3333. https://doi.org/10.3390/v6083311