The Role of Ubiquitin and Ubiquitin-Like Modification Systems in Papillomavirus Biology

{kind=link}
{kind=link}
Abstract
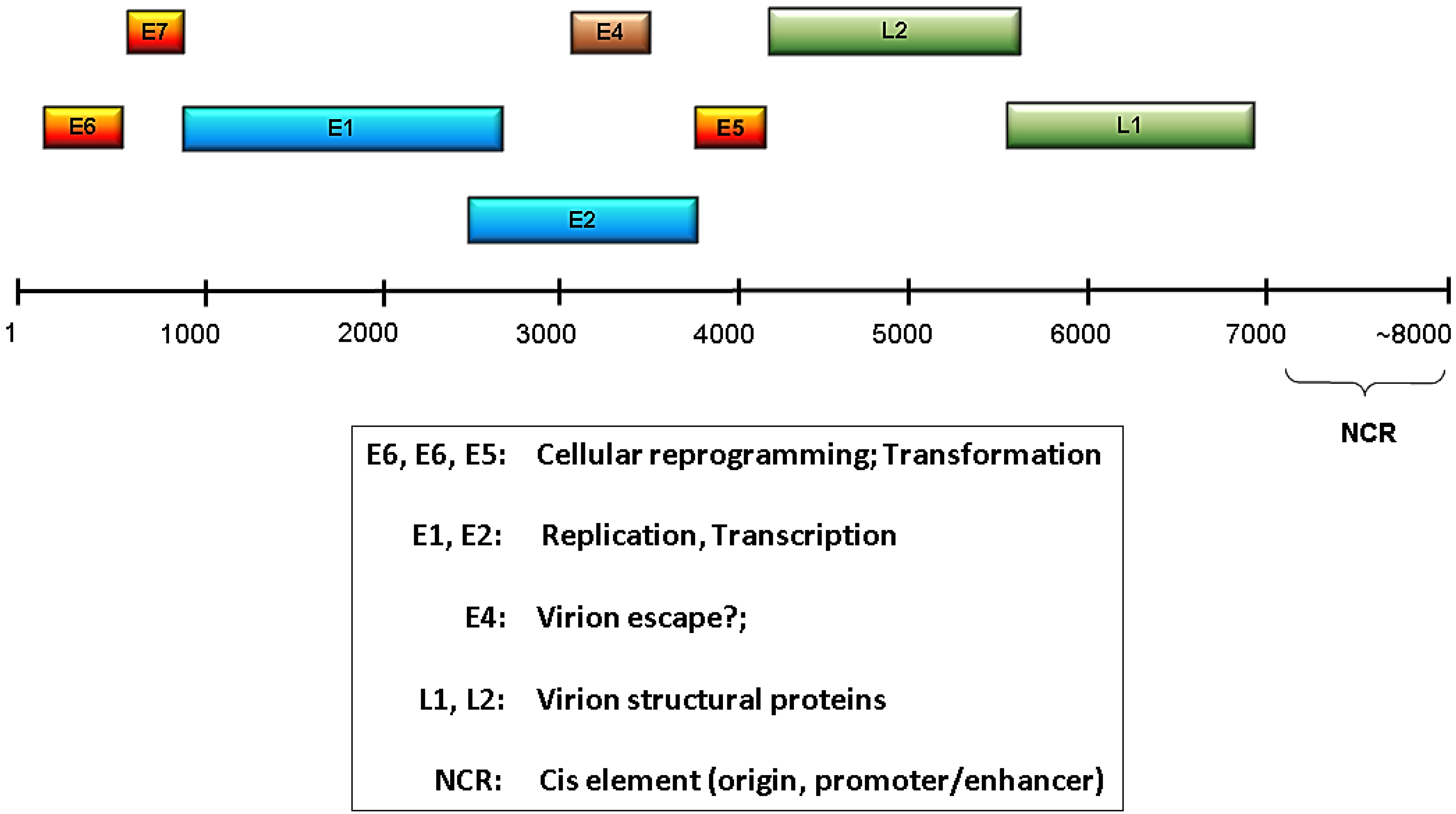
:1. Introduction –— Human Papillomaviruses

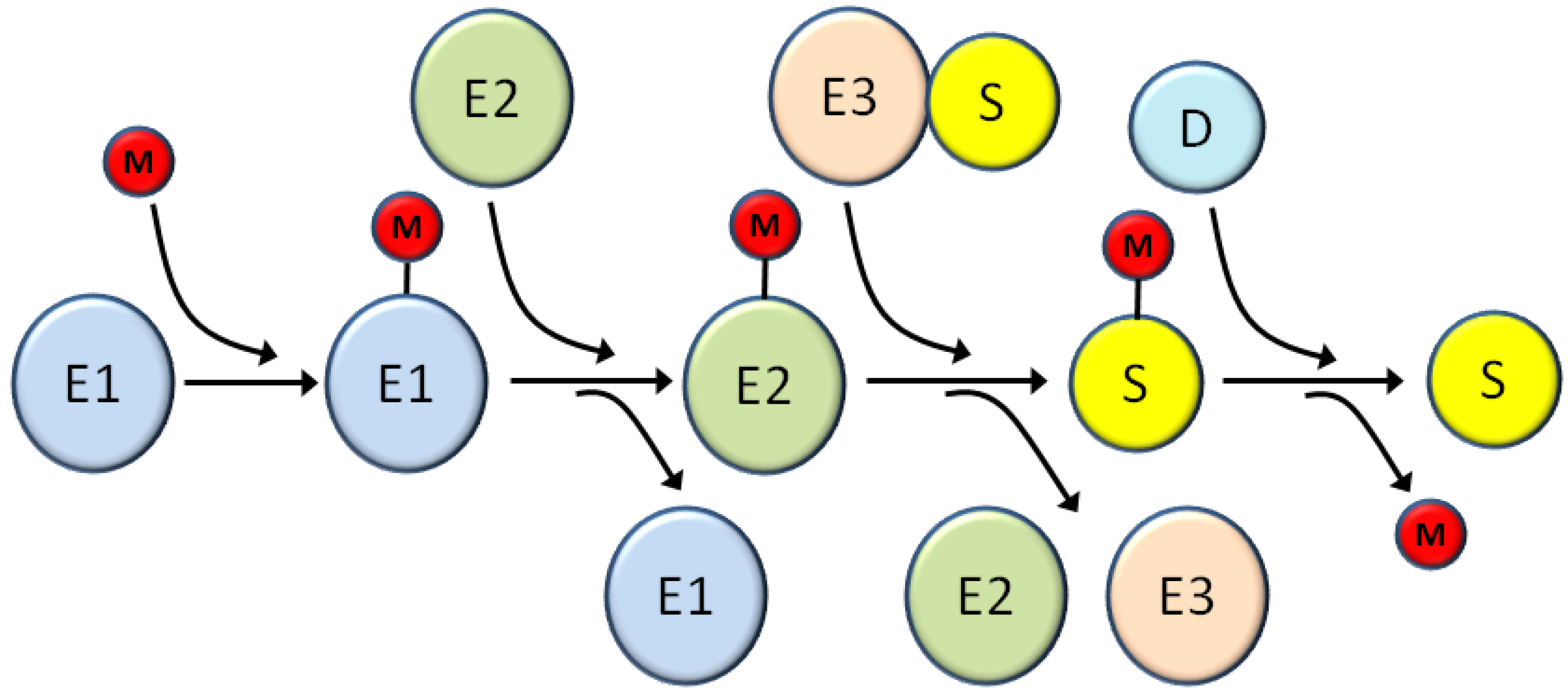
2. Overview of the Ubiquitin Superfamily

3. Modification of HPV Viral Proteins by Ubiquitin and Ubiquitin-Like Modifiers (Ubls)
3.1. The E1 Proteins
3.2. The E2 Proteins
3.3. The E4 Proteins
3.4. The E5 Proteins
3.5. The E6 Proteins
3.6. The E7 Proteins
3.7. L1 and L2 Proteins
4. Modulation of the Host Environment Mediated Through HPV-Ubl Interactions
4.1. The E1 Proteins
4.2. The E2 Proteins
4.3. The E4 Proteins
4.4. The E5 Proteins
4.5. The E6 Proteins
4.6. The E7 Proteins
4.7. L1 and L2 Proteins
5. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Bhatia, N.; Lynde, C.; Vender, R.; Bourcier, M. Understanding genital warts: epidemiology, pathogenesis, and burden of disease of human papillomavirus. J. Cutan. Med. Surg. 2013, 17, S47–S54. [Google Scholar] [PubMed]
- Bzhalava, D.; Guan, P.; Franceschi, S.; Dillner, J.; Clifford, G. A systematic review of the prevalence of mucosal and cutaneous human papillomavirus types. Virology 2013, 445, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Chouhy, D.; Bolatti, E.M.; Perez, G.R.; Giri, A.A. Analysis of the genetic diversity and phylogenetic relationships of putative human papillomavirus types. J. Gen. Virol. 2013, 94, 2480–2488. [Google Scholar] [CrossRef] [PubMed]
- Dubina, M.; Goldenberg, G. Viral-associated nonmelanoma skin cancers: A review. Am. J. Dermatopathol. 2009, 31, 561–573. [Google Scholar] [CrossRef] [PubMed]
- White, E.A.; Sowa, M.E.; Tan, M.J. A.; Jeudy, S.; Hayes, S.D.; Santha, S.; Munger, K.; Harper, J.W.; Howley, P.M. Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc. Natl. Acad. Sci. USA 2012, 109, E260–E267. [Google Scholar] [CrossRef] [PubMed]
- White, E.A.; Kramer, R.E.; Tan, M.J.; Hayes, S.D.; Harper, J.W.; Howley, P.M. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J. Virol. 2012, 86, 13174–13186. [Google Scholar] [PubMed]
- Rozenblatt-Rosen, O.; Deo, R.C.; Padi, M.; Adelmant, G.; Calderwood, M.A.; Rolland, T.; Grace, M.; Dricot, A.; Askenazi, M.; Tavares, M.; et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 2012, 487, 491–495. [Google Scholar] [PubMed]
- Bernard, H.U. Regulatory elements in the viral genome. Virology 2013, 445, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Roden, R.B. L2, the minor capsid protein of papillomavirus. Virology 2013, 445, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Day, P.M.; Trus, B.L. The papillomavirus major capsid protein L1. Virology 2013, 445, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Bergvall, M.; Melendy, T.; Archambault, J. The E1 proteins. Virology 2013, 445, 35–56. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, D.; Petti, L.M. The E5 proteins. Virology 2013, 445, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nature Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar]
- Munger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Frazer, I.H. Interaction of human papillomaviruses with the host immune system: A well evolved relationship. Virology 2009, 384, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Hughes, F.J.; Romanos, M.A. E1 protein of human papillomavirus is a DNA helicase/ATPase. Nucleic Acids Res. 1993, 21, 5817–5823. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.S.; Müller, F.; Lusky, M.; Hurwitz, J. Bovine papilloma virus (BPV)-encoded E1 protein contains multiple activities required for BPV DNA replication. Proc. Natl. Acad. Sci. USA 1993, 90, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, O.; Earnshaw, D.; Sarginson, G.; Delvecchio, A.; Tsai, J.; Kallender, H.; Amegadzie, B.; Browne, M. Characterization of the helicase and atpase activity of human papillomavirus type 6b E1 protein. J. Gen. Virol. 1996, 77, 1805–1809. [Google Scholar] [CrossRef]
- Calistri, A.; Munegato, D.; Carli, I.; Parolin, C.; Palu, G. The ubiquitin-conjugating system: Multiple roles in viral replication and infection. Cells 2014, 3, 386–417. [Google Scholar] [CrossRef] [PubMed]
- Hannoun, Z.; Greenhough, S.; Jaffray, E.; Hay, R.T.; Hay, D.C. Post-translational modification by SUMO. Toxicology 2010, 278, 288–293. [Google Scholar] [PubMed]
- Zhang, D.; Zhang, D.E. Interferon-stimulated gene 15 and the protein ISGylation system. J. Interferon Cytokine Res. 2011, 31, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Watson, I.R.; Irwin, M.S.; Ohh, M. NEDD8 pathways in cancer, Sine Quibus Non. Cancer Cell 2011, 19, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, G.; Aichem, A.; Groettrup, M. FAT10ylation as a signal for proteasomal degradation. Biochim Biophys Acta 2014, 1843, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Van der Veen, A.G.; Ploegh, H.L. Ubiquitin-like proteins. Annu. Rev. Biochem. 2012, 81, 323–357. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Ann. Rev. Cell Develop. Biol. 2006, 22, 159–180. [Google Scholar]
- Wenzel, D.M.; Stoll, K.E.; Klevit, R.E. E2s: Structurally economical and functionally replete. Biochem. J. 2011, 433, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, C.E.; Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Desterro, J.M.; Rodriguez, M.S.; Hay, R.T. SUMO-1 modification of IkappaBalpha inhibits NF‑kappaB activation. Mol. Cell 1998, 2, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Klenk, C.; Humrich, J.; Quitterer, U.; Lohse, M.J. SUMO-1 controls the protein stability and the biological function of phosducin. J. Biol. Chem. 2006, 281, 8357–8364. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, C.; Wang, E.; Wang, J. PolyUbiquitin chain linkage topology selects the functions from the underlying binding landscape. PLoS Comput. Biol. 2014, 10, e1003691. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Wang, H.; Wang, S.; Quon, D.; Liu, Y.W.; Cordell, B. Positive and negative regulation of APP amyloidogenesis by sumoylation. Proc. Natl. Acad. Sci. USA 2003, 100, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.; Kim, K.B.; Crews, C.M. The ubiquitin-proteasome pathway and proteasome inhibitors. Medicinal Res. Revv 2001, 21, 245–273. [Google Scholar] [CrossRef]
- Isaacson, M.K.; Ploegh, H.L. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 2009, 5, 559–570. [Google Scholar] [PubMed]
- Wilson, V.G. SUMOylation at the host-pathogen interface. Biomolecules 2012, 2, 203–227. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, P.; Schreiner, S.; Dobner, T. Human pathogens and the host cell SUMOylation system. J. Virol. 2012, 86, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Lenschow, D.J. Antiviral properties of ISG15. Viruses 2010, 2, 2154–2168. [Google Scholar] [CrossRef] [PubMed]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Li, M.; Zhang, Y.; Yang, P.; Eckenrode, S.; Hopkins, D.; Zheng, W.; Purohit, S.; Podolsky, R.H.; Muir, A.; et al. A functional variant of SUMO4, a new I kappa B alpha modifier, is associated with type 1 diabetes. Nat. Genet. 2004, 36, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Cheng, C.Y.; Campbell, M.; Yang, Y.C.; Hsu, H.W.; Chang, T.Y.; Chu, C.H.; Lee, Y.W.; Hung, C.L.; Lai, S.M.; et al. The chromatin modification by SUMO-2/3 but not SUMO-1 prevents the epigenetic activation of key immune-related genes during Kaposi's sarcoma associated herpesvirus reactivation. BMC Genomics 2013, 14, 824. [Google Scholar] [CrossRef] [PubMed]
- Vertegaal, A.C.O.; Andersen, J.S.; Ogg, S.C.; Hay, R.T.; Mann, M.; Lamond, A.I. Distinct and overlapping sets of SUMO-1 and SUMO-2 target proteins revealed by quantitative proteomics. Mol. Cell. Proteomics 2006, 5, 2298–2310. [Google Scholar] [CrossRef] [PubMed]
- Ayaydin, F.; Dasso, M. Distinct in vivo dynamics of vertebrate SUMO paralogues. Mol. Biol. Cell. 2004, 15, 5208–5218. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Arnaoutov, A.; Dasso, M. SUMO-2/3 regulates topoisomerase II in mitosis. J. Cell. Biol. 2003, 163, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.T.H. SUMOylation and de-SUMOylation: Wrestling with life's processes. J. Biol. Chem. 2009, 284, 8223–8227. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, J.; Zhao, W.M.; Zhao, K.N. Expression of HPV 58 long and short L1 capsid proteins in primary mouse keratinocyte cultures. Protein Pept. Lett. 2009, 16, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Marusic, M.B.; Mencin, N.; Licen, M.; Banks, L.; Grm, H.S.T. Modification of human papillomavirus minor capsid protein L2 by sumoylation. J. Virol. 2010, 84, 11585–11589. [Google Scholar] [CrossRef] [PubMed]
- Malcles, M.H.; Cueille, N.; Mechali, F.; Coux, O.; Bonne-Andrea, C. Regulation of bovine papillomavirus replicative helicase E1 by the ubiquitin-proteasome pathway. J. Virol. 2002, 76, 11350–11358. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, S.; Demeret, C.; Goyat, S.; Thierry, F. Stability of the human papillomavirus type 18 E2 protein is regulated by a proteasome degradation pathway through its amino-terminal transactivation domain. J. Virol. 2001, 75, 7244–7251. [Google Scholar] [CrossRef] [PubMed]
- Kehmeier, E.; Ruhl, H.; Voland, B.; Stoppler, M.C.; Androphy, E.; Stoppler, H. Cellular steady-state levels of "high risk" but not "low risk" human papillomavirus (HPV) E6 proteins are increased by inhibition of proteasome-dependent degradation independent of their p53-and E6AP-binding capabilities. Virology 2002, 299, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Reinstein, E.; Scheffner, M.; Oren, M.; Ciechanover, A.; Schwartz, A. Degradation of the E7 human papillomavirus oncoprotein by the ubiquitin-proteasome system: Targeting via ubiquitination of the N-terminal residue. Oncogene 2000, 19, 5944–5950. [Google Scholar] [CrossRef] [PubMed]
- Bassermann, F.; Eichner, R.; Pagano, M. The ubiquitin proteasome system—Implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta 2014, 1843, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.K.; Reed, S.I. Ubiquitin ligases and cell cycle control. Annu. Rev. Biochem. 2013, 82, 387–414. [Google Scholar] [CrossRef] [PubMed]
- Strikoudis, A.; Guillamot, M.; Aifantis, I. Regulation of stem cell function by protein ubiquitylation. EMBO Rep. 2014, 15, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Lomeli, H.; Vazquez, M. Emerging roles of the SUMO pathway in development. Cell. Mol. Life Sci. 2011, 68, 4045–4064. [Google Scholar] [CrossRef]
- Mechali, F.; Hsu, C.Y.; Castro, A.; Lorca, T.; Bonne-Andrea, C. Bovine papillomavirus replicative helicase E1 is a target of the ubiquitin ligase APC. J. Virol. 2004, 78, 2615–2619. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, D.; Wilson, V.G. Bovine papillomavirus E1 protein is sumoylated by the host cell Ubc9 protein. J. Biol. Chem. 2000, 275, 30487–30495. [Google Scholar] [CrossRef]
- Rosas-Acosta, G.; Langereis, M.A.; Deyrieux, A.; Wilson, V.G. Proteins of the PIAS family enhance the sumoylation of the papillomavirus E1 protein. Virology 2005, 331, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Brault, K.; Titolo, S.; Howley, P.M.; Archambault, J. Characterization of papillomavirus E1 helicase mutants defective for interaction with the SUMO-conjugating enzyme Ubc9. Virology 2009, 395, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, D.; Woytek, K.; Khan, S.A.; Wilson, V.G. SUMO-1 modification of bovine papillomavirus E1 protein is required for intranuclear accumulation. J. Biol. Chem. 2000, 275, 37999–38004. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Acosta, G.; Wilson, V.G. Identification of a nuclear export signal sequence for bovine papillomavirus E1 protein. Virology 2008, 373, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Gali, H.; Juhasz, S.; Morocz, M.; Hajdu, I.; Fatyol, K.; Szukacsov, V.; Burkovics, P.; Haracska, L. Role of SUMO modification of human PCNA at stalled replication fork. Nucleic Acids Res. 2012, 40, 6049–6059. [Google Scholar] [CrossRef] [PubMed]
- Penrose, K.J.; McBride, A.A. Proteasome-mediated degradation of the papillomavirus E2-TA protein is regulated by phosphorylation and can modulate viral genome copy number. J. Virol. 2000, 74, 6031–6038. [Google Scholar] [CrossRef] [PubMed]
- Blachon, S.; Bellanger, S.; Demeret, C.; Thierry, G. Nucleo-cytoplasmic shuttling of high risk human Papillomavirus E2 proteins induces apoptosis. J. Biol. Chem. 2005, 280, 36088–36098. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, D.; Joubert, S.; Senechal, H.; Fradet-Turcotte, A.; Torre, S.; Archambault, J. Proteasomal degradation of the papillomavirus E2 protein is inhibited by overexpression of bromodomain-containing protein 4. J. Virol. 2009, 83, 4127–4139. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, S.; Tan, C.L.; Nei, W.; He, P.P.; Thierry, F. The human papillomavirus type 18 E2 protein is a cell cycle-dependent target of the SCFSkp2 ubiquitin ligase. J. Virol. 2010, 84, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Gardiol, D.; Massimi, P.; Banks, L. The Mdm2 ubiquitin ligase enhances transcriptional activity of human papillomavirus E2. J. Virol. 2009, 83, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, S.; Blachon, S.; Mechali, F.; Bonne-Andrea, C.; Thierry, F. High-risk but not low-risk HPV E2 proteins bind to the APC activators Cdh1 and Cdc20 and cause genomic instability. Cell Cycle 2005, 4, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Maitland, N.J.; Conway, S.; Wilkinson, N.S.; Ramsdale, J.; Morris, J.R.; Sanders, C.M.; Burns, J.E.; Stern, P.L.; Wells, M. Expression patterns of the human papillomavirus type 16 transcription factor E2 in low- and high-grade cervical intraepithelial neoplasia. J. Pathol. 1998, 186, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Schweiger, M.R.; Martinez-Noel, G.; Zheng, L.; Smith, J.A.; Harper, J.W.; Howley, P.M. Brd4 regulation of papillomavirus protein E2 stability. J. Virol. 2009, 83, 8683–8692. [Google Scholar] [CrossRef] [PubMed]
- Ilves, I.; Maemets, K.; Silla, T.; Janikson, K.; Ustav, M. Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle. J. Virol. 2006, 80, 3660–3665. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Chiang, C.M. Chromatin adaptor Brd4 modulates E2 transcription activity and protein stability. J. Biol. Chem. 2009, 284, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Q.; Diaz, J.; You, J. Brd4-mediated nuclear retention of the papillomavirus E2 protein contributes to its stabilization in host cells. Viruses 2014, 6, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Naidu, S.R.; Sverdrup, F.; Androphy, E.J. Tax1BP1 interacts with papillomavirus E2 and regulates E2-dependent transcription and stability. J. Virol. 2009, 83, 2274–2284. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.W.; Tsao, Y.P.; Lin, C.Y.; Chen, S.L. NRIP, a novel calmodulin binding protein, activates calcineurin to dephosphorylate human papillomavirus E2 protein. J. Virol. 2011, 85, 6750–6763. [Google Scholar] [CrossRef] [PubMed]
- King, L.E.; Dornan, E.S.; Donaldson, M.M.; Morgan, I.M. Human papillomavirus 16 E2 stability and transcriptional activation is enhanced by E1 via a direct protein-protein interaction. Virology 2011, 414, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Davy, C.; McIntosh, P.; Jackson, D.J.; Sorathia, R.; Miell, M.; Wang, Q.; Khan, J.; Soneji, Y.; Doorbar, J. A novel interaction between the human papillomavirus type 16 E2 and E1^E4 proteins leads to stabilization of E2. Virology 2009, 394, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Harhaj, N.S.; Liebl, D.J.; Harhaj, E.W. Essential role for TAX1BP1 in the termination of TNF-alpha-, IL-1- and LPS-mediated NF-kappaB and JNK signaling. EMBO J. 2007, 26, 3910–3922. [Google Scholar] [PubMed]
- Johansson, C.; Graham, S.V.; Dornan, E.S.; Morgan, I.M. The human papillomavirus 16 E2 protein is stabilised in S phase. Virology 2009, 394, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, V.; McBride, A.A. Phosphorylation regulates binding of the human papillomavirus type 8 E2 protein to host chromosomes. J. Virol. 2012, 86, 10047–10058. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Deyrieux, A.F.; Wilson, V.G. Papillomaviruses and the host SUMOylation system. Biochem. Soc. Trans. 2007, 35, 1433–1435. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Roark, A.A.; Bian, X.L.; Wilson, V.G. Modification of papillomavirus E2 proteins by the small ubiquitin-like modifier family members (SUMOs). Virology 2008, 378, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Bian, X.L.; Heaton, P.H.; Deyrieux, A.F.; Wilson, V.G. Host cell sumoylation level influences papillomavirus E2 protein stability. Virology 2009, 387, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Deyrieux, A.F.; Rosas-Acosta, G.; Ozbun, M.A.; Wilson, V.G. Sumoylation dynamics during keratinocyte differentiation. J. Cell. Sci. 2007, 120, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Maglennon, G.A.; McIntosh, P.; Doorbar, J. Persistence of viral DNA in the epithelial basal layer suggests a model for papillomavirus latency following immune regression. Virology 2011, 414, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Foo, C.; Coleman, N.; Medcalf, L.; Hartley, O.; Prospero, T.; Napthine, S.; Sterling, J.; Winter, G.; Griffin, H. Characterization of events during the late stages of HPV16 infection in vivo using high-affinity synthetic fabs to E4. Virology 1997, 238, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Kennedy, A.; Das, P.; McIntosh, P.B.; Howell, S.A.; Isaacson, E.R.; Hinz, S.A.; Davy, C.; Doorbar, J. Phosphorylation of the human papillomavirus type 16 E1^E4 Protein at T57 by ERK triggers a structural change that enhances keratin binding and protein stability. J. Virol. 2009, 83, 3668–3683. [Google Scholar] [CrossRef] [PubMed]
- Davy, C.E.; Ayub, M.; Jackson, D.J.; Das, P.; McIntosh, P.; Doorbar, J. HPV16 E1^E4 protein is phosphorylated by Cdk2/cyclin A and relocalizes this complex to the cytoplasm. Virology 2006, 349, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Li, L.; Whyte, P. Human papillomavirus 18 E1^E4 protein interacts with cyclin A/CDK 2 through an RXL motif. Mol. Cell. Biochem. 2013, 373, 29–40. [Google Scholar] [CrossRef]
- McIntosh, P.B.; Martin, S.R.; Jackson, D.J.; Khan, J.; Isaacson, E.R.; Calder, L.; Raj, K.; Griffin, H.M.; Wang, Q.; Laskey, P.; Eccleston, J.F.; Doorbar, J. Structural analysis reveals an amyloid form of the human papillomavirus type 16 E1^E4 protein and provides a molecular basis for its accumulation. J. Virol. 2008, 82, 8196–8203. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.; Davy, C.E.; McIntosh, P.B.; Jackson, D.J.; Hinz, S.; Wang, Q.; Doorbar, J. Role of calpain in the formation of human papillomavirus type 16 E1^E4 amyloid fibers and reorganization of the keratin network. J. Virol. 2011, 85, 9984–9997. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.R.; Mora, R.; Laimins, L.A. Intracellular localization and DNA-binding properties of human papillomavirus type 18 E6 protein expressed with a baculovirus vector. J. Virol. 1989, 63, 366–374. [Google Scholar] [PubMed]
- Androphy, E.J.; Hubbert, N.L.; Schiller, J.T.; Lowy, D.R. Identification of the HPV-16 E6 protein from transformed mouse cells and human cervical carcinoma cell lines. EMBO J. 1987, 6, 989–992. [Google Scholar]
- Stewart, D.; Kazemi, S.; Li, S.Y.; Massimi, P.; Banks, L.; Koromilas, A.E.; Matlashewski, G. Ubiquitination and proteasome degradation of the E6 proteins of human papillomavirus types 11 and 18. J. Gen. Virol. 2004, 85, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991, 10, 4129–4135. [Google Scholar] [PubMed]
- Tomaic, V.; Pim, D.; Banks, L. The stability of the human papillomavirus E6 oncoprotein is E6AP dependent. Virology 2009, 393, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Tomaic, V.; Pim, D.; Myers, M.P.; Tommasino, M.; Banks, L. Interactions between E6AP and E6 proteins from alpha and beta HPV types. Virology 2013, 435, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Vos, R.M.; Altreuter, J.; White, E.A.; Howley, P.M. The Ubiquitin-Specific Peptidase USP15 Regulates Human Papillomavirus Type 16 E6 Protein Stability. J. Virol. 2009, 83, 8885–8892. [Google Scholar] [CrossRef] [PubMed]
- Tomaic, V.; Pim, D.; Thomas, M.; Massimi, P.; Myers, M.P.; Banks, L. Regulation of the human papillomavirus type 18 E6/E6AP ubiquitin ligase complex by the HECT domain-containing protein EDD. J. Virol. 2011, 85, 3120–3127. [Google Scholar] [CrossRef] [PubMed]
- Kuhnle, S.; Kogel, U.; Glockzin, S.; Marquardt, A.; Ciechanover, A.; Matentzoglu, K.; Scheffner, M. Physical and functional interaction of the HECT ubiquitin-protein ligases E6AP and HERC2. J. Biol. Chem. 2011, 286, 19410–19416. [Google Scholar] [CrossRef]
- Martinez-Noel, G.; Galligan, J.T.; Sowa, M.E.; Arndt, V.; Overton, T.M.; Harper, J.W.; Howley, P.M. Identification and proteomic analysis of distinct UBE3A/E6AP protein complexes. Mol. Cell. Biol. 2012, 32, 3095–3106. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, L.; Davy, C.; Raj, K.; Kranjec, C.; Banks, L.; Doorbar, J. Stabilization of HPV16 E6 protein by PDZ proteins, and potential implications for genome maintenance. Virology 2011, 414, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Gardiol, D.; Kuhne, C.; Glaunsinger, B.; Lee, S.S.; Javier, R.; Banks, L. Oncogenic human papillomavirus E6 proteins target the discs large tumour suppressor for proteasome-mediated degradation. Oncogene 1999, 18, 5487–5496. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Huibregtse, J.M. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol. Cell. Biol. 2000, 20, 8244–8253. [Google Scholar] [CrossRef] [PubMed]
- Selvey, L.A.; Dunn, L.A.; Tindle, R.W.; Park, D.S.; Frazer, I.H. Human papillomavirus (HPV) type 18 E7 protein is a short-lived steroid-inducible phosphoprotein in HPV-transformed cell lines. J. Gen. Virol. 1994, 75, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sampath, A.; Raychaudhuri, P.; Bagchi, S. Both Rb and E7 are regulated by the ubiquitin proteasome pathway in HPV-containing cervical tumor cells. Oncogene 2001, 20, 4740–4749. [Google Scholar] [CrossRef] [PubMed]
- Ben-Saadon, R.; Fajerman, I.; Ziv, T.; Hellman, U.; Schwartz, A.L.; Ciechanover, A. The tumor suppressor protein p16(INK4a) and the human papillomavirus oncoprotein-58 E7 are naturally occurring lysine-less proteins that are degraded by the ubiquitin system—Direct evidence for ubiquitination at the N-terminal residue. J. Biol. Chem. 2004, 279, 41414–41421. [Google Scholar] [CrossRef] [PubMed]
- Kamio, M.; Yoshida, T.; Ogata, H.; Douchi, T.; Nagata, Y.; Inoue, M.; Hasegawa, M.; Yonemitsu, Y.; Yoshimura, A. SOC1 inhibits HPV-E7-mediated transformation by inducing degradation of E7 protein. Oncogene 2004, 23, 9449. [Google Scholar] [CrossRef]
- Zhang, J.G.; Farley, A.; Nicholson, S.E.; Willson, T.A.; Zugaro, L.M.; Simpson, R.J.; Moritz, R.L.; Cary, D.; Richardson, R.; Hausmann, G.; et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl. Acad. Sci. USA 1999, 96, 2071–2076. [Google Scholar]
- Oh, K.J.; Kalinina, A.; Wang, J.; Nakayama, K.; Nakayama, K.I.; Bagchi, S. The papillomavirus E7 oncoprotein is ubiquitinated by UbcH7 and Cullin 1- and Skp2-containing E3 ligase. J. Virol. 2004, 78, 5338–5346. [Google Scholar] [CrossRef] [PubMed]
- Huh, K.; Zhou, X.; Hayakawa, H.; Cho, J.Y.; Libermann, T.A.; Jin, J.; Harper, J.W.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J. Virol. 2007, 81, 9737–9747. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Chang, H.S.; Yu, W.C.Y. USP11 stabilizes HPV-16E7 and further modulates the E7 biological activity. J. Biol. Chem. 2008, 283, 15681–15688. [Google Scholar] [CrossRef] [PubMed]
- Bund, T.; Spoden, G.A.; Koynov, K.; Hellmann, N.; Boukhallouk, F.; Arnold, P.; Hinderberger, D.; Florin, L. An L2 SUMO interacting motif is important for PML localization and infection of human papillomavirus type 16. Cell Microbiol. 2014, 16, 1179–1200. [Google Scholar] [CrossRef] [PubMed]
- Durfee, L.A.; Lyon, N.; Seo, K.; Huibregtse, J.M. The ISG15 conjugation system broadly targets newly synthesized proteins: Implications for the antiviral function of ISG15. Mol. Cell 2010, 38, 722–732. [Google Scholar] [CrossRef]
- Wilson, V.G.; Heaton, P.R. Ubiquitin proteolytic system: Focus on SUMO. Expert Rev. Proteomics 2008, 5, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Porras-Yakushi, T.R.; Hess, S. Recent advances in defining the ubiquitylome. Expert Rev. Proteomics 2014, 11, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Wang, S. E3 ubiquitin ligases and human papillomavirus-induced carcinogenesis. J. Int. Med. Res. 2014, 42, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Cote-Martin, A.; Moody, C.; Fradet-Turcotte, A.; D'Abramo, C.M.; Lehoux, M.; Joubert, S.; Poirier, G.G.; Coulombe, B.; Laimins, L.A.; Archambault, J. Human papillomavirus E1 helicase interacts with the WD repeat protein p80 to promote maintenance of the viral genome in keratinocytes. J. Virol. 2008, 82, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Lehoux, M.; Fradet-Turcotte, A.; Lussier-Price, M.; Omichinski, J.G.; Archambault, J. Inhibition of human papillomavirus DNA replication by an E1-derived p80/UAF1-binding peptide. J. Virol. 2012, 86, 3486–3500. [Google Scholar] [CrossRef] [PubMed]
- Lehoux, M.; Gagnon, D.; Archambault, J. E1-mediated recruitment of a UAF1-USP deubiquitinase complex facilitates human papillomavirus DNA replication. J. Virol. 2014, 88, 8545–8555. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.Y.; Jones, A.; Yang, C.; Zhai, L.; Smith, A.D.T.; Zhang, Z.; Chandrasekharan, M.B.; Sun, Z.W.; Renfrow, M.B.; Wang, Y.; et al. Regulation of histone H2A and H2B deubiquitination and Xenopus development by USP12 and USP46. J. Biol. Chem. 2011, 286, 7190–7201. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Nijman, S.M.; Mirchandani, K.D.; Galardy, P.J.; Cohn, M.A.; Haas, W.; Gygi, S.P.; Ploegh, H.L.; Bernards, R.; D'Andrea, A.D. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 2006, 8, 339–347. [Google Scholar] [PubMed]
- Nijman, S.M.; Huang, T.T.; Dirac, A.M.; Brummelkamp, T.R.; Kerkhoven, R.M.; D'Andrea, A.D.; Bernards, R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol. Cell 2005, 17, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Wenzel, S.; Tansey, W.P. Ubiquitin and proteasomes in transcription. Annu. Rev. Biochem. 2012, 81, 177–201. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Demeret, C. The HPV E2-host protein-protein interactions: A complex hijacking of the cellular network. Open Virol. J. 2012, 6, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Jacob, Y.; Jones, L.; Weiss, A.; Brino, L.; Chantier, T.; Lotteau, V.; Favre, M.; Demeret, C. Large scale genotype comparison of human papillomavirus E2-host interaction networks provides new insights for E2 molecular functions. PLoS Pathog. 2012, 8, e1002761. [Google Scholar] [CrossRef]
- Palvimo, J.J. PIAS proteins as regulators of small ubiquitin-related modifier (SUMO) modifications and transcription. Biochem. Soc. Trans. 2007, 35, 1405–1408. [Google Scholar] [CrossRef] [PubMed]
- Bryan, J.T.; Brown, D.R. Association of the human papillomavirus type 11 E1^E4 protein with cornified cell envelopes derived from infected genital epithelium. Virology 2000, 277, 262–269. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, P.B.; Laskey, P.; Sullivan, K.; Davy, C.; Wang, Q.A.; Jackson, D.J.; Griffin, H.M.; Doorbar, J. E1^E4-mediated keratin phosphorylation and ubiquitylation: A mechanism for keratin depletion in HPV16-infected epithelium. J. Cell. Sci. 2010, 123, 2810–2822. [Google Scholar] [CrossRef] [PubMed]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The smallest oncoprotein with many functions. Mol. Cancer 2011, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.Y.; Srirangam, A.; Potter, D.A.; Roman, A. HPV16 E5 protein disrupts the c-Cbl-EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene 2005, 24, 2585–2588. [Google Scholar] [CrossRef] [PubMed]
- Belleudi, F.; Leone, L.; Purpura, V.; Cannella, F.; Scrofani, C.; Torrisi, M.R. HPV16 E5 affects the KGFR/FGFR2b-mediated epithelial growth through alteration of the receptor expression, signaling and endocytic traffic. Oncogene 2011, 30, 4963–4976. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Kawana, K.; Schust, D.J.; Fujii, T.; Yokoyama, T.; Iwasawa, Y.; Nagamatsu, T.; Adachi, K.; Tomio, A.; Tomio, K.; et al. CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: A possible mechanism for immune evasion by HPV. J. Virol. 2010, 84, 11614–11623. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Kim, S.H.; Cho, E.A.; Song, Y.S.; Kim, W.H.; Juhnn, Y.S. Human papillomavirus type 16 E5 protein inhibits hydrogen-peroxide-induced apoptosis by stimulating ubiquitin-proteasome-mediated degradation of Bax in human cervical cancer cells. Carcinogenesis 2010, 31, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Pedroza-Saavedra, A.; Lam, E.W.F.; Esquivel-Guadarrama, F.; Gutierrez-Xicotencatl, L. The human papillomavirus type 16 E5 oncoprotein synergizes with EGF-receptor signaling to enhance cell cycle progression and the down-regulation of p27(Kip1). Virology 2010, 400, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, K.; Alonso, A. The human papillomavirus type 16 E5 protein impairs TRAIL- and FasL-mediated apoptosis in HaCaT cells by different mechanisms. J. Virol. 2002, 76, 12162–12172. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.L.; Keiger, K.E.; Lee, C.J.; Huibregtse, J.M. The global transcriptional effects of the human papillomavirus E6 protein in cervical carcinoma cell lines are mediated by the E6AP ubiquitin ligase. J. Virol. 2005, 79, 3737–3747. [Google Scholar] [PubMed]
- Kuhnle, S.; Mothes, B.; Matentzoglu, K.; Scheffner, M. Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein. Proc. Natl. Acad. Sci. USA 2013, 110, 8888–8893. [Google Scholar] [PubMed]
- Shai, A.; Pitot, H.C.; Lambert, P.F. E6-associated protein is required for human papillomavirus type 16 E6 to cause cervical cancer in mice. Cancer Res. 2010, 70, 5064–5073. [Google Scholar] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Beaudenon, S.; Huibregtse, J.M. HPV E6, E6AP and cervical cancer. BMC Biochem. 2008, 9, S4. [Google Scholar] [PubMed]
- Kiyono, T.; Hiraiwa, A.; Fujita, M.; Hayashi, Y.; Akiyama, T.; Ishibashi, M. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 11612–11616. [Google Scholar] [CrossRef]
- Glaunsinger, B.A.; Lee, S.S.; Thomas, M.; Banks, L.; Javier, R. Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene 2000, 19, 5270–5280. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Glaunsinger, B.; Mantovani, F.; Banks, L.; Javier, R.T. Multi-PDZ domain protein MUPP1 is a cellular target for both adenovirus E4-ORF1 and high-risk papillomavirus type 18 E6 oncoproteins. J. Virol. 2000, 74, 9680–9693. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Zheng, J.J. PDZ domains and their binding partners: Structure, specificity, and modification. Cell Commun. Signal. 2010, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Kuballa, P.; Matentzoglu, K.; Scheffner, M. The role of the ubiquitin ligase E6-AP in human papillomavirus E6-mediated degradation of PDZ domain-containing proteins. J. Biol. Chem. 2007, 282, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Brimer, N.; Lyons, C.; Pol, S.B.V. Association of E6AP (UBE3A) with human papillomavirus type 11 E6 protein. Virology 2007, 358, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol. Cell. Biol. 1993, 13, 4918–4927. [Google Scholar]
- Tong, X.; Boll, W.; Kirchhausen, T.; Howley, P.M. Interaction of the bovine papillomavirus E6 Protein with the clathrin adaptor complex AP-1. J. Virol. 1998, 72, 476–482. [Google Scholar] [PubMed]
- Chen, J.J.; Hong, Y.H.; Rustamzadeh, E.; Baleja, J.D.; Androphy, E.J. Identification of an alpha helical motif sufficient for association with papillomavirus E6. J. Biol. Chem. 1998, 273, 13537–13544. [Google Scholar] [CrossRef] [PubMed]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.J.A.; White, E.A.; Sowa, M.E.; Harper, J.W.; Aster, J.C.; Howley, P.M. Cutaneous beta-human papillomavirus E6 proteins bind Mastermind-like coactivators and repress Notch signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1473–E1480. [Google Scholar] [CrossRef] [PubMed]
- Brimer, N.; Lyons, C.; Wallberg, A.E.; Vande Pol, S.B. Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene 2012, 31, 4639–4646. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Bergant, M.; Boon, S.S.; Ganti, K.; Kranjec, C.; Massimi, P.; Subbaiah, V.K.; Thomas, M.; Tomaic, V.; Banks, L. Human papillomaviruses and the specificity of PDZ domain targeting. FEBS J. 2012, 279, 3530–3537. [Google Scholar] [CrossRef] [PubMed]
- Klingelhutz, A.J.; Roman, A. Cellular transformation by human papillomaviruses: Lessons learned by comparing high- and low-risk viruses. Virology 2012, 424, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Mighty, K.K.; Laimins, L.A. The role of human papillomaviruses in oncogenesis. Recent Results Cancer Res. 2014, 193, 135–148. [Google Scholar] [PubMed]
- Howie, H.L.; Katzenellenbogen, R.A.; Galloway, D.A. Papillomavirus E6 proteins. Virology 2009, 384, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.M.; Underbrink, M.P.; Howie, H.L.; Galloway, D.A. The E6 oncoproteins from human betapapillomaviruses differentially activate telomerase through an E6AP-dependent mechanism and prolong the lifespan of primary keratinocytes. J. Virol. 2008, 82, 3894–3902. [Google Scholar] [CrossRef] [PubMed]
- Underbrink, M.P.; Howie, H.L.; Bedard, K.M.; Koop, J.I.; Galloway, D.A. E6 proteins from multiple human betapapillomavirus types degrade Bak and protect keratinocytes from apoptosis after UVB irradiation. J. Virol. 2008, 82, 10408–10417. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Banks, L. Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene 1998, 17, 2943–2954. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Banks, L. Human papillomavirus (HPV) E6 interactions with Bak are conserved amongst E6 proteins from high and low risk HPV types. J. Gen. Virol. 1999, 80, 1513–1517. [Google Scholar] [PubMed]
- Westphal, D.; Dewson, G.; Czabotar, P.E.; Kluck, R.M. Molecular biology of Bax and Bak activation and action. Biochim. Biophys. Acta 2011, 1813, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Tomaic, V.; Ganti, K.; Pim, D.; Bauer, C.; Blattner, C.; Banks, L. Interaction of HPV E6 oncoproteins with specific proteasomal subunits. Virology 2013, 446, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Bischof, O.; Schwamborn, K.; Martin, N.; Werner, A.; Sustmann, C.; Grosschedl, R.; Dejean, A. The E3 SUMO ligase PIASy is a regulator of cellular senescence and apoptosis. Mol. Cell 2006, 22, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Heaton, P.R.; Deyrieux, A.F.; Bian, X.L.; Wilson, V.G. HPV E6 proteins target Ubc9, the SUMO conjugating enzyme. Virus Res. 2011, 158, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Heaton, P.R.; Santos, A.; Rosas-Acosta, G.; Wilson, V.G. Analysis of global sumoylation changes occurring during keratinocyte differentiation. PLoS One 2012, 7, e30165. [Google Scholar] [CrossRef] [PubMed]
- Chand, V.; John, R.; Jaiswal, N.; Johar, S.S.; Nag, A. High-risk HPV16E6 stimulates hADA3 degradation by enhancing its SUMOylation. Carcinogenesis 2014, 35, 1830–1839. [Google Scholar]
- Berezutskaya, E.; Yu, B.; Morozov, A.; Raychaudhuri, P.; Bagchi, S. Differential regulation of the pocket domains of the retinoblastoma family proteins by the HPV16 E7 oncoprotein. Cell Growth Differen. 1997, 8, 1277–1286. [Google Scholar]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar] [PubMed]
- Demers, G.W.; Foster, S.A.; Halbert, C.L.; Galloway, D.A. Growth arrest by induction of p53 in DNA damaged keratinocytes is bypassed by human papillomavirus 16 E7. Proc. Natl. Acad. Sci. USA 1994, 91, 4382–4386. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Munger, K. Analysis of the p53-mediated G1 growth arrest pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J. Virol. 1997, 71, 2905–2912. [Google Scholar] [PubMed]
- Oh, K.J.; Kalinina, A.; Bagchi, S. Destabilization of Rb by human papillomavirus E7 is cell cycle dependent: E2–25K is involved in the proteolysis. Virology 2010, 396, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Classon, M.; Dyson, N. p107 and p130: Versatile proteins with interesting pockets. Exp. Cell. Res. 2001, 264, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.L.; Stremlau, M.; He, X.; Basile, J.R.; Munger, K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J. Virol. 2001, 75, 7583–7591. [Google Scholar] [PubMed]
- Helt, A.M.; Galloway, D.A. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J. Virol. 2001, 75, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.Y.; Chen, W.; Roman, A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Westphal, K.; Akgul, B.; Storey, A.; Nindl, I. Cutaneous human papillomavirus E7 type-specific effects on differentiation and proliferation of organotypic skin cultures. Cell. Oncol. 2009, 31, 213–226. [Google Scholar] [PubMed]
- Spardy, N.; Covella, K.; Cha, E.; Hoskins, E.E.; Wells, S.I.; Duensing, A.; Duensing, S. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009, 69, 7022–7029. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Munger, K. Human papillomavirus type 16 E7 oncoprotein inhibits the anaphase promoting complex/cyclosome activity by dysregulating EMI1 expression in mitosis. Virology 2013, 446, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.Y.; Munger, K. Human papillomavirus type 16 E7 oncoprotein engages but does not abrogate the mitotic spindle assembly checkpoint. Virology 2012, 432, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Mo, M.; Shahar, S.; Fleming, S.B.; Mercer, A.A. How viruses affect the cell cycle through manipulation of the APC/C. Trends Microbiol. 2012, 20, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Berezutskaya, E.; Bagchi, S. The human papillomavirus E7 oncoprotein functionally interacts with the S4 subunit of the 26S proteasome. J. Biol. Chem. 1997, 272, 30135–30140. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Shim, J.H.; Kho, C.W.; Park, S.G.; Park, B.C.; Kim, J.W.; Lim, J.S.; Choe, Y.K.; Paik, S.G.; Yoon, D.Y. Protein profiling and identification of modulators regulated by the E7 oncogene in the C33A cell line by proteomics and genomics. Proteomics 2004, 4, 839–848. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wilson, V.G. The Role of Ubiquitin and Ubiquitin-Like Modification Systems in Papillomavirus Biology. Viruses 2014, 6, 3584-3611. https://doi.org/10.3390/v6093584
Wilson VG. The Role of Ubiquitin and Ubiquitin-Like Modification Systems in Papillomavirus Biology. Viruses. 2014; 6(9):3584-3611. https://doi.org/10.3390/v6093584
Chicago/Turabian StyleWilson, Van G. 2014. "The Role of Ubiquitin and Ubiquitin-Like Modification Systems in Papillomavirus Biology" Viruses 6, no. 9: 3584-3611. https://doi.org/10.3390/v6093584
APA StyleWilson, V. G. (2014). The Role of Ubiquitin and Ubiquitin-Like Modification Systems in Papillomavirus Biology. Viruses, 6(9), 3584-3611. https://doi.org/10.3390/v6093584