
Modeling Influenza Virus Infection: A Roadmap for Influenza Research


Abstract
:
1. Introduction
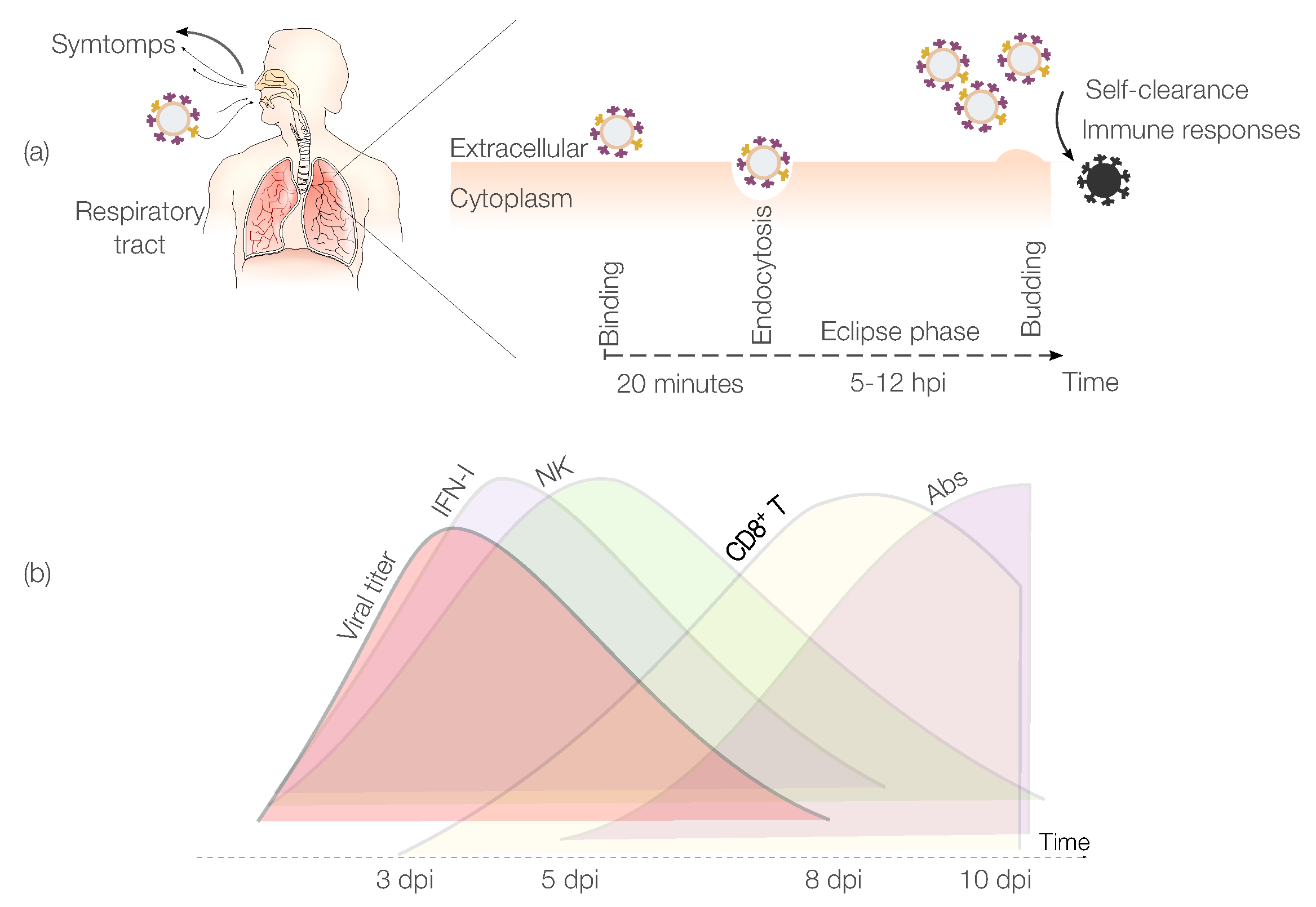
IAV Pathogenesis

2. Mathematical Models of IAV Infections
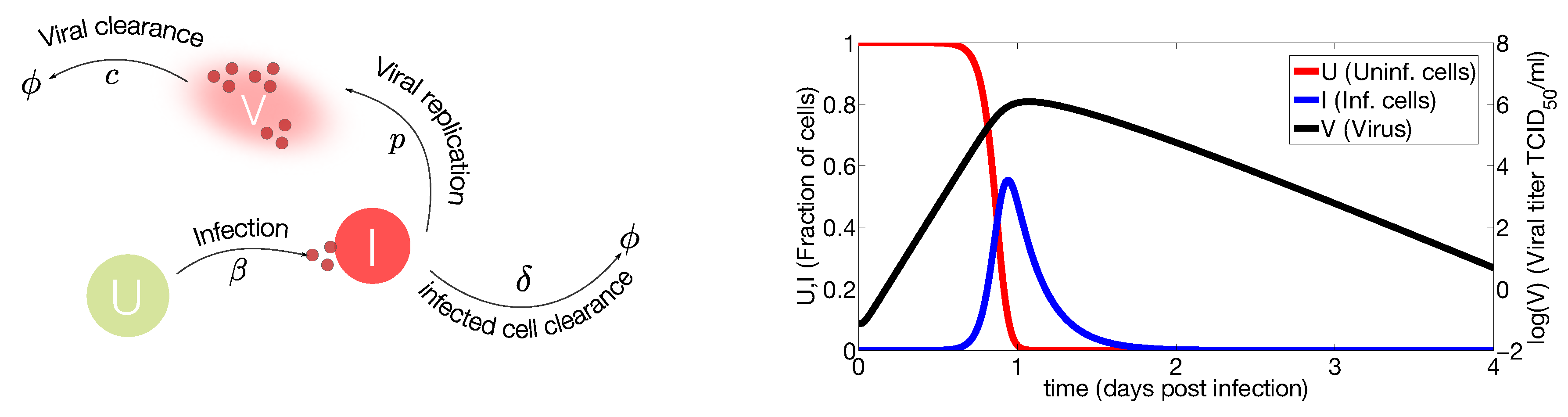
2.1. In Vivo Systems

2.2. Mathematical Models Including the Immune Response
2.3. In Vitro Systems
2.4. Data for Modeling: Scarce and Diverse

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| References | In Vitro | In Vivo | Host | Coinfection | Aging | |
|---|---|---|---|---|---|---|
| Innate | Adaptive | |||||
| Antia et al. [48] | √ |  | ||||
| Baccam et al. [26] | √ | | ||||
| Beauchemin et al. [31] | √ |  | ||||
| Bocharov and Romanyukha [38] | √ | | ||||
| Canini and Carrat [45] | √ | | ||||
| Cao et al. [43] | √ |  | ||||
| Chen et al. [60] | √ | | ||||
| Dobrovolny et al. [35] | √ | Various | ||||
| Hancioglu et al. [39] | √ | | ||||
| Handel et al. [33] | √ | √ | | |||
| Handel and Antia [49] | √ | | ||||
| [61] | √ | | ||||
| Hernandez-Vargas et al. [42] | √ | √ |  | √ | ||
| Holder et al. [57] | √ | | ||||
| Holder and Beauchemin [32] | √ | | ||||
| Le et al. [50] | √ | | ||||
| Lee et al. [52] | √ | √ | | |||
| Miao et al. [25] | √ | √ | | |||
| Mitchell et al. [62] | √ | | ||||
| Moehler et al. [55] | √ | | ||||
| Paradis et al. [58] | √ | | ||||
| Pawelek et al. [40] | √ |  | ||||
| Petrie et al. [36] | √ | | ||||
| Pinilla et al. [21] | √ | | ||||
| Price et al. [51] | √ | √ | | |||
| Reperant et al. [63] | √ | √ | | |||
| Saenz et al. [41] | √ | | ||||
| Schulze-Horsel et al. [56] | √ | | ||||
| Smith et al. [64] | √ | | √ | |||
| Tridane and Kuang [54] | √ | | ||||
2.5. Parameter Estimation: A Continuous Challenge

2.6. Case Study: Identification of a Mathematical Model of IAV Infection Including the Immune Response

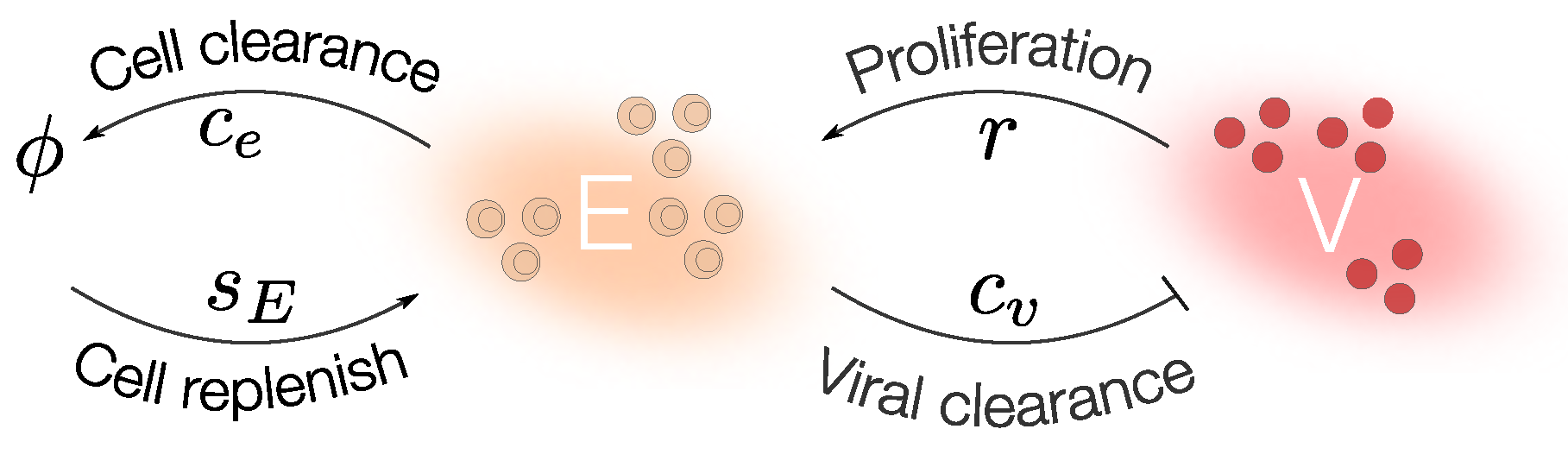
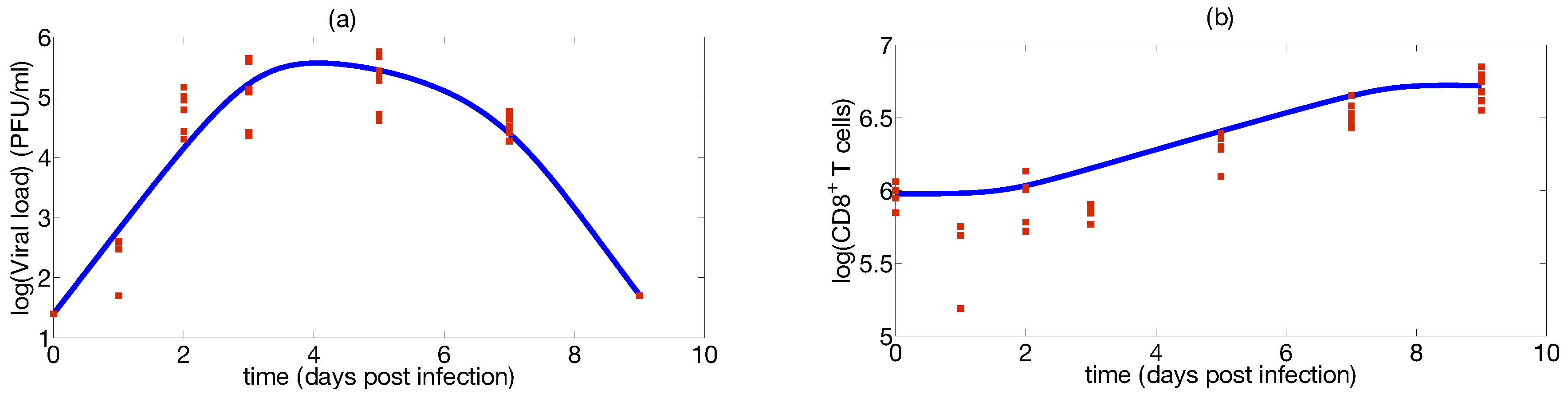
Step 1: Mathematical Modeling
Step 2: Identifiability Analysis
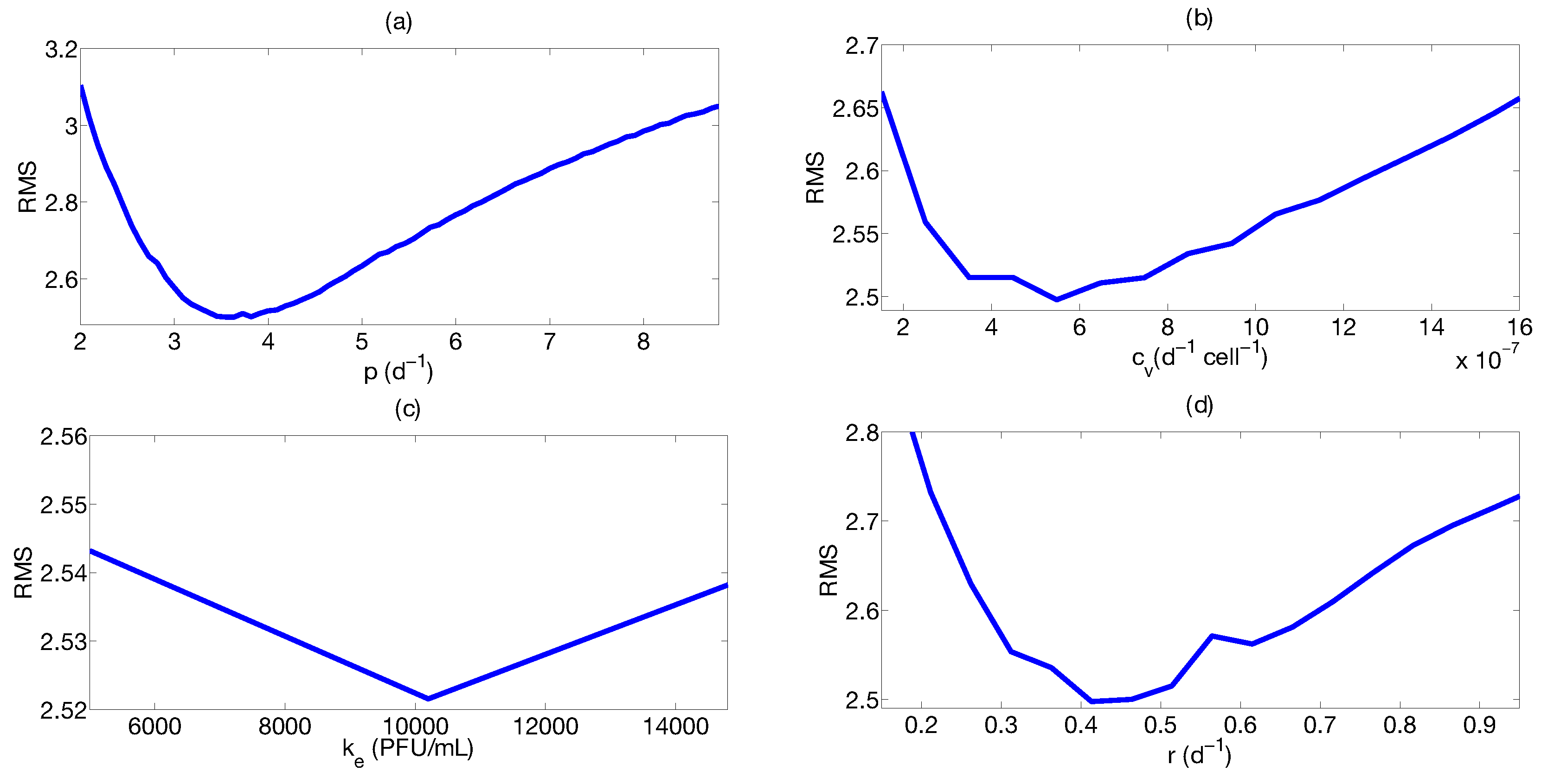
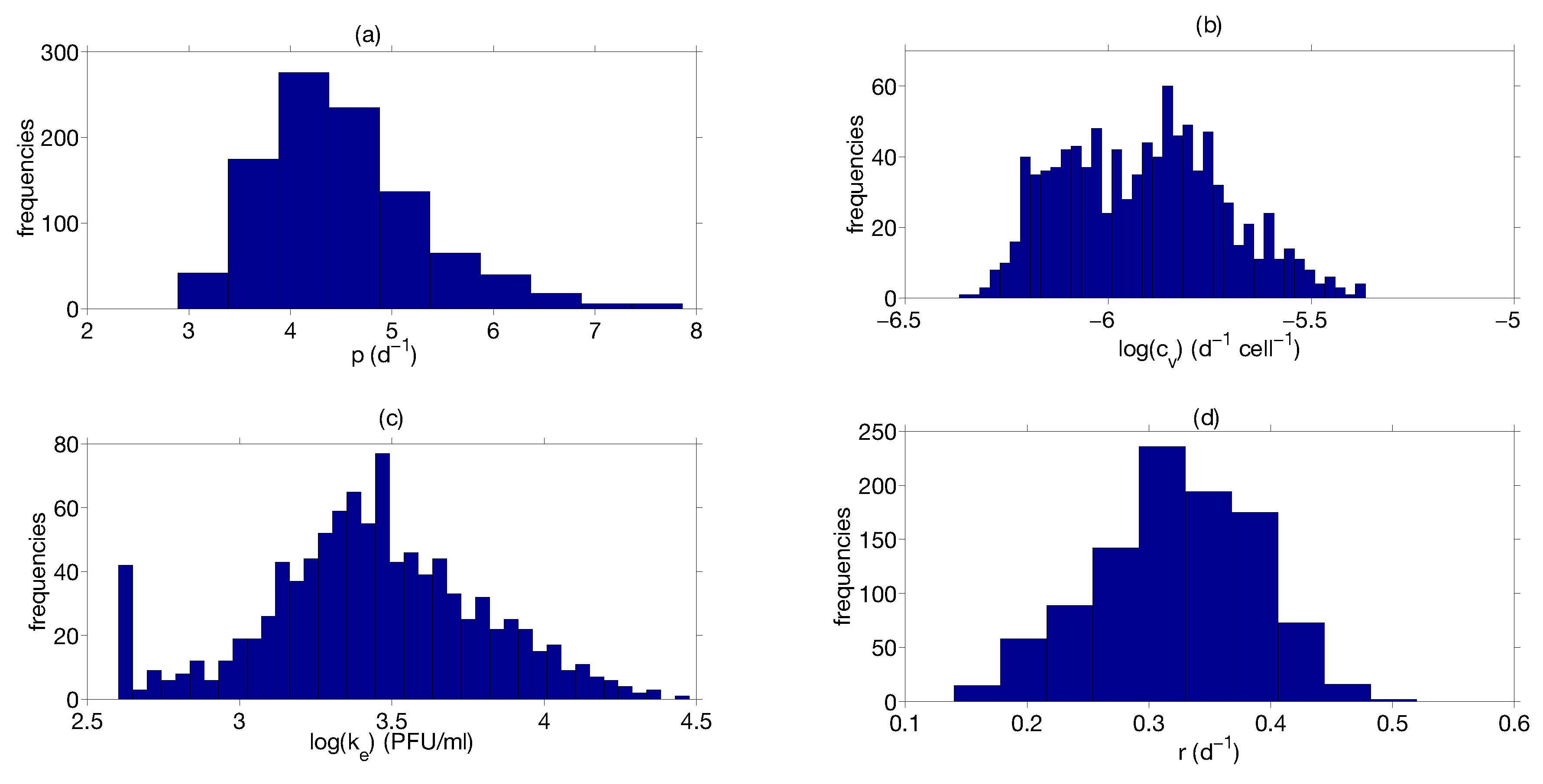
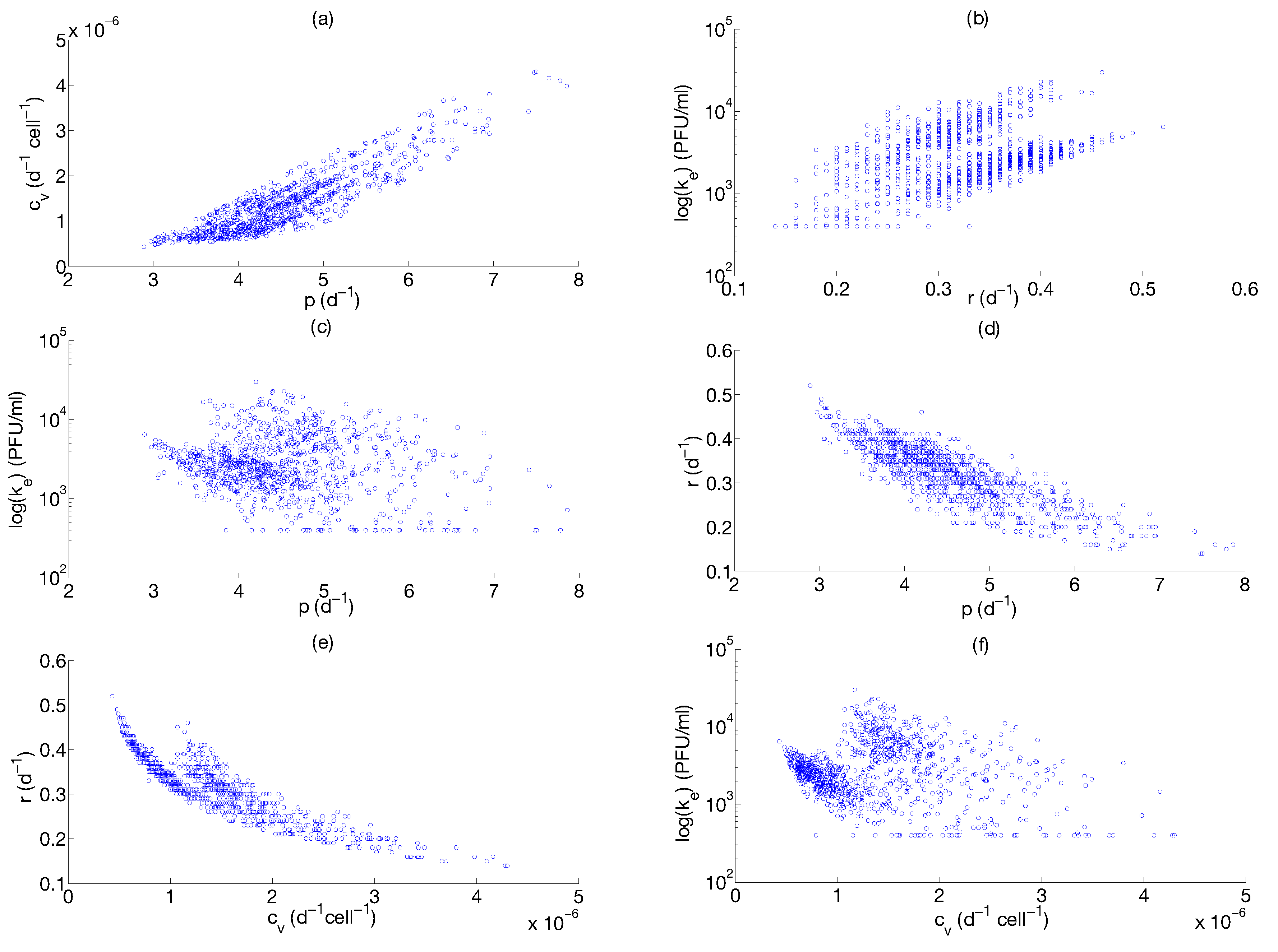
Step 3: Parameter Uncertainty



| Parameter | Median | Confidence Interval (95%) | Constraints for Optimization Algorithm |
|---|---|---|---|
| 4.4 | [3.43 ; 6.08] | [1; 8] | |
| [ ; ] | [; ] | ||
| 0.33 | [0.20 ; 0.42] | [0.01; 1] | |
| [ ; ] | [; ] |

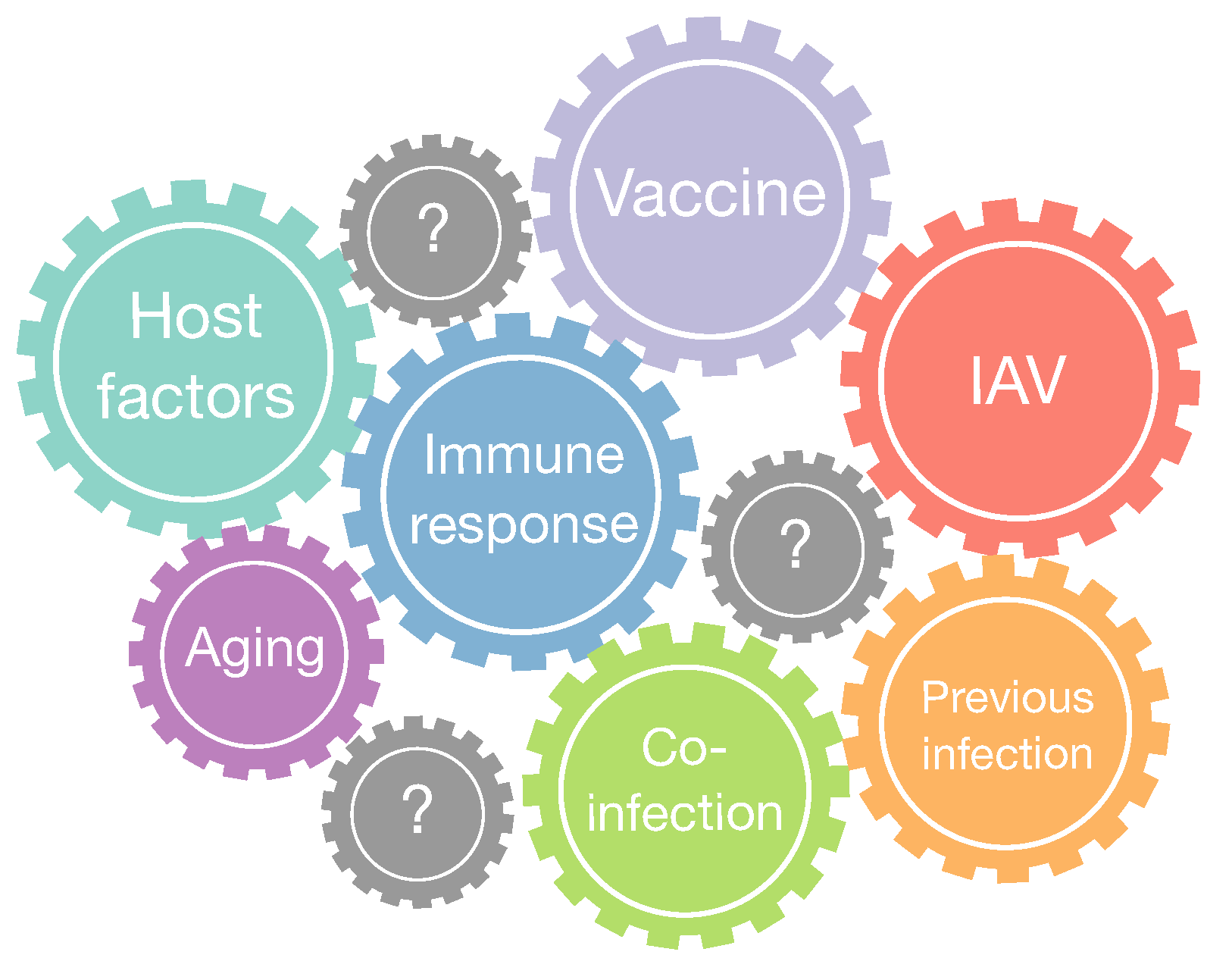
3. Discussion and Future Perspectives

3.1. Bacterial Coinfection
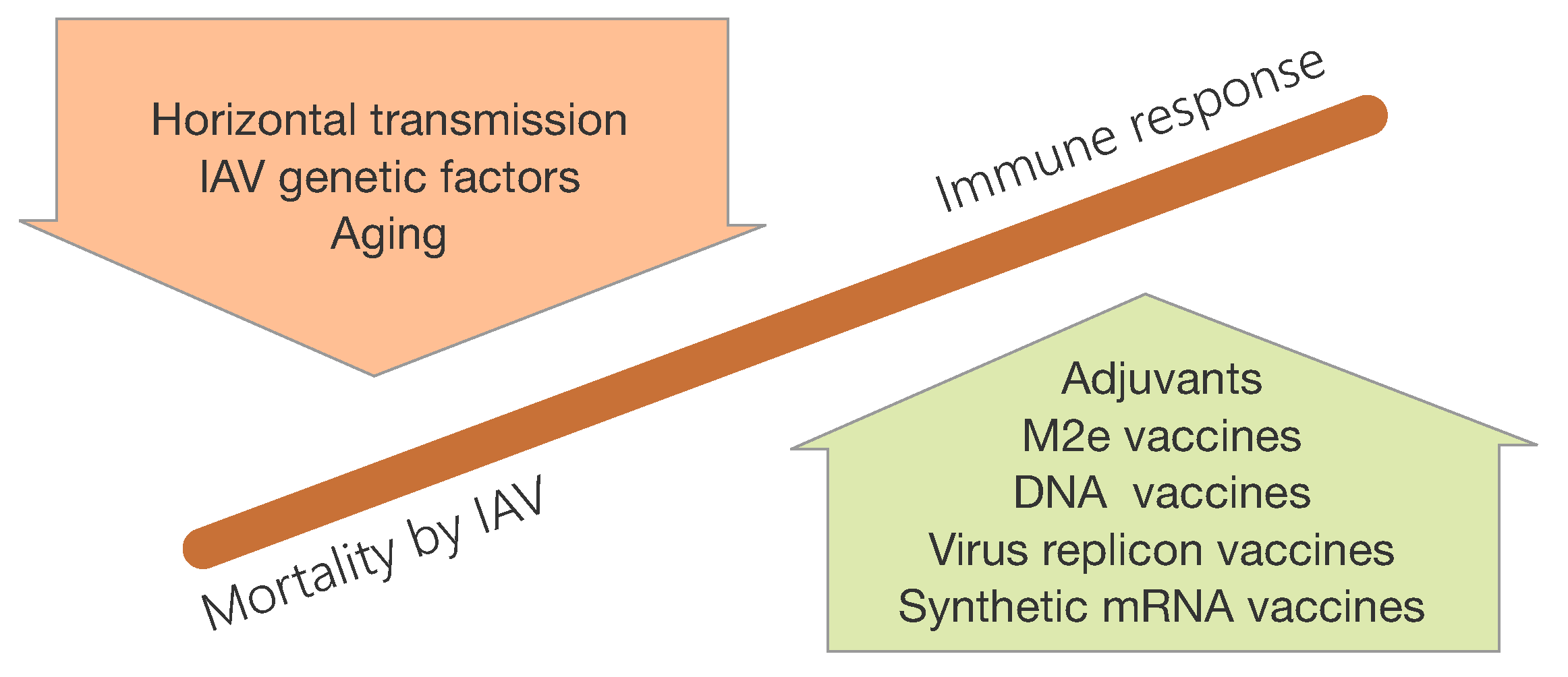
3.2. Aging of the Immune System and the Role in IAV Infections
3.3. Challenges for Influenza Vaccination

3.4. Host and IAV Genetic Factors
| Host Factors | Role |
|---|---|
| IFITM3 | Restrict morbidity and mortality of IAV infection [161,162,163] |
| CPT2 | Related complication as influenza-associated encephalopathy [164] |
| TMPRSS2 | Resistance to IAV infection [165,166,167] |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization (WHO). Influenza (Seasonal) Factsheet N 211; WHO: Geneva, Switzerland, 2009. [Google Scholar]
- Lang, P.O.; Mendes, A.; Socquet, J.; Assir, N.; Govind, S.; Aspinall, R. Effectiveness of influenza vaccine in aging and older adults: Comprehensive analysis of the evidence. Clin. Interv. Aging 2012, 7, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, C.W. A history of influenza. J. Appl. Microbiol. 2001, 91, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Kilbourne, E.D. Influenza pandemics of the 20th century. Emerg. Infect. Dis. 2006, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Influenza (Seasonal): Fact Sheet N∘211; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- Carrat, F.; Flahault, A. Influenza vaccine: The challenge of antigenic drift. Vaccine 2007, 25, 6852–6862. [Google Scholar] [CrossRef] [PubMed]
- Hensley, S.E. Challenges of selecting seasonal influenza vaccine strains for humans with diverse pre-exposure histories. Curr. Opin. Virol. 2014, 8, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Madhi, S.A.; Cutland, C.L.; Kuwanda, L.; Weinberg, A.; Hugo, A.; Jones, S.; Adrian, P.V.; van Niekerk, N.; Treurnicht, F.; Ortiz, J.R.; et al. Influenza Vaccination of Pregnant Women and Protection of Their Infants. Obstet. Gynecol. Survey 2015, 70, 3–5. [Google Scholar] [CrossRef]
- Beauchemin, C.A.A.; Handel, A. A review of mathematical models of influenza A infections within a host or cell culture: Lessons learned and challenges ahead. BMC Public Health 2011, 11, S7. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Perelson, A.S. Influenza A virus infection kinetics: Quantitative data and models. Syst. Biol. Med. 2011, 3, 429–445. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolny, H.M.; Reddy, M.B.; Kamal, M.A.; Rayner, C.R.; Beauchemin, C.A. Assessing Mathematical Models of Influenza Infections Using Features of the Immune Response. PLoS ONE 2013, 8, e57088. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; Rasmussen, A.L.; Feldmann, F.; Bushmaker, T.; Martellaro, C.; Haddock, E.; Okumura, A.; Proll, S.C.; Chang, J.; Gardner, D.; et al. Influenza virus A/Anhui/1/2013 (H7N9) replicates efficiently in the upper and lower respiratory tracts of cynomolgus macaques. mBio 2014, 5, e01331-14. [Google Scholar] [CrossRef] [PubMed]
- Welliver, T.P.; Garofalo, R.P.; Hosakote, Y.; Hintz, K.H.; Avendano, L.; Sanchez, K.; Velozo, L.; Jafri, H.; Chavez-Bueno, S.; Ogra, P.L.; et al. Severe Human Lower Respiratory Tract Illness Caused by Respiratory Syncytial Virus and Influenza Virus Is Characterized by the Absence of Pulmonary Cytotoxic Lymphocyte Responses. J. Infect. Dis. 2007, 195, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Van Riel, D.; Munster, V.J.; de Wit, E.; Rimmelzwaan, G.F.; Fouchier, R.A.M.; Osterhaus, A.D.M.E.; Kuiken, T. H5N1 Virus Attachment to Lower Respiratory Tract. Science 2006, 312, 399. [Google Scholar] [CrossRef] [PubMed]
- Reeth, K.V. Cytokines in the pathogenesis of influenza. Vet. Microbiol. 2000, 74, 109–116. [Google Scholar] [CrossRef]
- Valkenburg, S.A.; Rutigliano, J.A.; Ellebedy, A.H.; Doherty, P.C.; Thomas, P.G.; Kedzierska, K. Immunity to seasonal and pandemic influenza A viruses. Microbes Infect. 2011, 13, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, W.G.; Noti, J.D.; Blachere, F.M.; Thewlis, R.E.; Martin, S.B.; Othumpangat, S.; Noorbakhsh, B.; Goldsmith, W.T.; Vishnu, A.; Palmer, J.E.; et al. Viable Influenza A Virus in Airborne Particles from Human Coughs. J. Occup. Environ. Hyg. 2015, 12, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Oldstone, M.B.A.; Compans, R.W. Influenza Pathogenesis and Control; Current Topics in Microbiology and Immunology 386; Springer International Publishing: Berlin, Germany, 2015. [Google Scholar]
- Oguin, T.H.; Sharma, S.; Stuart, A.D.; Duan, S.; Scott, S.A.; Jones, C.K.; Daniels, J.S.; Lindsley, C.W.; Thomas, P.G.; Brown, H.A. Phospholipase D facilitates efficient entry of influenza virus, allowing escape from innate immune inhibition. J. Biol. Chem. 2014, 289, 25405–25417. [Google Scholar] [CrossRef] [PubMed]
- White, D.O.; Cheyne, I.M. Early events in the eclipse phase of influenza and parainfluenza virus infection. Virology 1966, 29, 49–59. [Google Scholar] [CrossRef]
- Pinilla, L.T.; Holder, B.P.; Abed, Y.; Boivin, G.; Beauchemin, C.A.A. The H275Y neuraminidase mutation of the pandemic A/H1N1 influenza virus lengthens the eclipse phase and reduces viral output of infected cells, potentially compromising fitness in ferrets. J. Virol. 2012, 86, 10651–10660. [Google Scholar] [CrossRef] [PubMed]
- Horsfall, F.L. On the reproduction of influenza virus quantitative studies with procedures which enumerate infective and hemagglutinating virus particles. J. Exp. Med. 1954, 100, 135–161. [Google Scholar] [CrossRef] [PubMed]
- Banatvala, J.E.; Griffiths, P.; Schoub, B.; Mortimer, P. Principles and Practice of Clinical Virology; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Tamura, S.i.; Kurata, T. Defense mechanisms against influenza virus infection in the respiratory tract mucosa. Jpn. J. Infect. Dis. 2004, 57, 236–247. [Google Scholar] [PubMed]
- Miao, H.; Hollenbaugh, J.A.; Zand, M.S.; Holden-Wiltse, J.; Mosmann, T.R.; Perelson, A.S.; Wu, H.; Topham, D.J. Quantifying the early immune response and adaptive immune response kinetics in mice infected with influenza A virus. J. Virol. 2010, 84, 6687–6698. [Google Scholar] [CrossRef] [PubMed]
- Baccam, P.; Beauchemin, C.A.A.; Macken, C.A.A.; Hayden, F.G.; Perelson, A.S. Kinetics of influenza A virus infection in humans. J. Virol. 2006, 80, 7590–7599. [Google Scholar] [CrossRef] [PubMed]
- Larson, E.W.; Dominik, J.W.; Rowberg, A.H.; Higbee, G.A. Influenza virus population dynamics in the respiratory tract of experimentally infected mice. Infect. Immun. 1976, 13, 438–447. [Google Scholar] [PubMed]
- Nowak, M.A.; May, R. Virus Dynamics: Mathematical Principles of Immunology and Virology; Oxford University Press: Oxford, UK, 2000; Volume 291, pp. 1–256. [Google Scholar]
- Perelson, A.S. Modelling viral and immune system dynamics. Nat. Rev. Immunol. 2002, 2, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.M.; Lo, A.; Perelson, A.S. Dynamics of hepatitis B virus infection. Microbes Infect. 2002, 4, 829–835. [Google Scholar] [CrossRef]
- Beauchemin, C.A.A.; McSharry, J.J.; Drusano, G.L.; Nguyen, J.T.; Went, G.T.; Ribeiro, R.M.; Perelson, A.S. Modeling amantadine treatment of influenza A virus in vitro. J. Theor. Biol. 2008, 254, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Holder, B.P.; Beauchemin, C.A.A. Exploring the effect of biological delays in kinetic models of influenza within a host or cell culture. BMC Public Health 2011, 11, S10. [Google Scholar] [CrossRef] [PubMed]
- Handel, A.; Longini, I.M., Jr.; Antia, R. Neuraminidase inhibitor resistance in influenza: Assessing the danger of its generation and spread. PLoS Comput. Biol. 2007, 3, e240. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolny, H.M.; Gieschke, R.; Davies, B.E.; Jumbe, N.L.; Beauchemin, C.A.A. Neuraminidase inhibitors for treatment of human and avian strain influenza: A comparative modeling study. J. Theor. Biol. 2011, 269, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolny, H.M.; Baron, M.J.; Gieschke, R.; Davies, B.E.; Jumbe, N.L.; Beauchemin, C.A.A. Exploring cell tropism as a possible contributor to influenza infection severity. PLoS ONE 2010, 5, e13811–e13826. [Google Scholar] [CrossRef] [PubMed]
- Petrie, S.M.; Guarnaccia, T.; Laurie, K.L.; Hurt, A.C.; McVernon, J.; McCaw, J.M. Reducing Uncertainty in Within-Host Parameter Estimates of Influenza Infection by Measuring Both Infectious and Total Viral Load. PLoS ONE 2013, 8, e64098. [Google Scholar] [CrossRef] [PubMed]
- Doherty, P.C.; Turner, S.J.; Webby, R.G.; Thomas, P.G. Influenza and the challenge for immunology. Nat. Immunol. 2006, 7, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, G.; Romanyukha, A. Mathematical model of antiviral immune response III. Influenza A virus infection. J. Theor. Biol. 1994, 167, 323–360. [Google Scholar] [CrossRef] [PubMed]
- Hancioglu, B.; Swigon, D.; Clermont, G. A dynamical model of human immune response to influenza A virus infection. J. Theor. Biol. 2007, 246, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, K.A.; Huynh, G.T.; Quinlivan, M.; Cullinane, A.; Rong, L.; Perelson, A.S. Modeling within-host dynamics of influenza virus infection including immune responses. PLoS Comput. Biol. 2012, 8, e1002588. [Google Scholar] [CrossRef] [PubMed]
- Saenz, R.A.; Quinlivan, M.; Elton, D.; Macrae, S.; Blunden, A.S.; Mumford, J.A.; Daly, J.M.; Digard, P.; Cullinane, A.; Grenfell, B.T.; et al. Dynamics of influenza virus infection and pathology. J. Virol. 2010, 84, 3974–3983. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Vargas, E.A.; Wilk, E.; Canini, L.; Toapanta, F.R.; Binder, S.C.; Uvarovskii, A.; Ross, T.M.; Guzmán, C.A.; Perelson, A.S.; Meyer-Hermann, M. Effects of aging on influenza virus infection dynamics. J. Virol. 2014, 88, 4123–4131. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Yan, A.W.; Heffernan, J.M.; Petrie, S.; Moss, R.G.; Carolan, L.A.; Guarnaccia, T.A.; Kelso, A.; Barr, I.G.; McVernon, J.; et al. Innate Immunity and the Inter-exposure Interval Determine the Dynamics of Secondary Influenza Virus Infection and Explain Observed Viral Hierarchies. PLoS Comput. Biol. 2015, 11, e1004334. [Google Scholar] [CrossRef] [PubMed]
- Laurie, K.L.; Guarnaccia, T.A.; Carolan, L.A.; Yan, A.W.; Aban, M.; Petrie, S.; Cao, P.; Heffernan, J.M.; McVernon, J.; Mosse, J.; et al. The time-interval between infections and viral hierarchies are determinants of viral interference following influenza virus infection in a ferret model. J. Infect. Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Canini, L.; Carrat, F. Population modeling of influenza A/H1N1 virus kinetics and symptom dynamics. J. Virol. 2011, 85, 2764–2770. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Vargas, E.A.; Meyer-Hermann, M. Innate immune system dynamics to influenza virus. In Proceedings of the 8th IFAC Symposium on Biological and Medical Systems, Budapest, Hungary, 29–31 August 2012; Volume 8, pp. 1–6.
- Handel, A.; Longini, I.M.; Antia, R. Towards a quantitative understanding of the within-host dynamics of influenza A infections. J. Royal Soc. Interface Royal Soc. 2010, 7, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Antia, R.; Bergstrom, C.T.; Pilyugin, S.S.; Kaech, S.M.; Ahmed, R. Models of CD8+ responses: 1. What is the antigen-independent proliferation program. J. Theor. Biol. 2003, 221, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Handel, A.; Antia, R. A simple mathematical model helps to explain the immunodominance of CD8 T cells in influenza A virus infections. J. Virol. 2008, 82, 7768–7772. [Google Scholar] [CrossRef] [PubMed]
- Le, D.; Miller, J.D.; Ganusov, V.V. Mathematical modeling provides kinetic details of the human immune response to vaccination. Front. Cell. Infect. Microbiol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Price, I.; Mochan-Keef, E.D.; Swigon, D.; Ermentrout, G.B.; Lukens, S.; Toapanta, F.R.; Ross, T.M.; Clermont, G. The inflammatory response to influenza A virus (H1N1): An experimental and mathematical study. J. Theor. Biol. 2015, 374, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Topham, D.J.; Park, S.Y.; Hollenbaugh, J.; Treanor, J.; Mosmann, T.R.; Jin, X.; Ward, B.M.; Miao, H.; Holden-Wiltse, J.; et al. Simulation and prediction of the adaptive immune response to influenza A virus infection. J. Virol. 2009, 83, 7151–7165. [Google Scholar] [CrossRef] [PubMed]
- De Boor, C. A Practical Guide to Splines, revised Ed.; Applied Mathematical Sciences; Springer: Berlin, Germany, 2001; Volume 27. [Google Scholar]
- Tridane, A.; Kuang, Y. Modeling the interaction of cytotoxic T lymphocytes and influenza virus infected epithelial cells. Math. Biosci. Eng. 2010, 7, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Moehler, L.; Flockerzi, D.; Sann, H.; Reichl, U. Mathematical model of influenza A virus production in large-scale microcarrier culture. Biotechnol. Bioeng. 2005, 90, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Horsel, J.; Schulze, M.; Agalaridis, G.; Genzel, Y.; Reichl, U. Infection dynamics and virus-induced apoptosis in cell culture-based influenza vaccine production Flow cytometry and mathematical modeling. Vaccine 2009, 27, 2712–2722. [Google Scholar] [CrossRef] [PubMed]
- Holder, B.P.; Simon, P.; Liao, L.E.; Abed, Y.; Bouhy, X.; Beauchemin, C.A.A.; Boivin, G. Assessing the in vitro fitness of an oseltamivir-resistant seasonal A/H1N1 influenza strain using a mathematical model. PLoS ONE 2011, 6, e14767. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.G.; Pinilla, L.T.; Holder, B.P.; Abed, Y.; Boivin, G.; Beauchemin, C.A.A. Impact of the H275Y and I223V Mutations in the Neuraminidase of the 2009 Pandemic Influenza Virus in Vitro and Evaluating Experimental Reproducibility. PLoS ONE 2015, 10, e0126115. [Google Scholar] [PubMed]
- Reperant, L.A.; Kuiken, T.; Osterhaus, A.D. Adaptive pathways of zoonotic influenza viruses: From exposure to establishment in humans. Vaccine 2012, 30, 4419–4434. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; You, S.; Liu, C.; Chio, C.; Liao, C. Using experimental human influenza infections to validate a viral dynamic model and the implications for prediction. Epidemiol. Infect. 2012, 140, 1557–1568. [Google Scholar] [CrossRef] [PubMed]
- Heldt, F.S.; Frensing, T.; Pflugmacher, A.; Gröpler, R.; Peschel, B.; Reichl, U. Multiscale modeling of influenza A virus infection supports the development of direct-acting antivirals. PLoS Comput. Biol. 2013, 9, e1003372. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, H.; Levin, D.; Forrest, S.; Beauchemin, C.A.A.; Tipper, J.; Knight, J.; Donart, N.; Layton, R.C.; Pyles, J.; Gao, P.; et al. Higher level of replication efficiency of 2009 (H1N1) pandemic influenza virus than those of seasonal and avian strains: Kinetics from epithelial cell culture and computational modeling. J. Virol. 2011, 85, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Reperant, L.A.; Kuiken, T.; Grenfell, B.T.; Osterhaus, A.D. The immune response and within-host emergence of pandemic influenza virus. Lancet 2014, 384, 2077–2081. [Google Scholar] [CrossRef]
- Smith, A.M.; Adler, F.R.; Ribeiro, R.M.; Gutenkunst, R.N.; McAuley, J.L.; McCullers, J.A.; Perelson, A.S. Kinetics of coinfection with influenza A virus and Streptococcus pneumoniae. PLoS Pathog. 2013, 9, e1003238. [Google Scholar] [CrossRef] [PubMed]
- Burnham, K.P.; Anderson, D.R. Multimodel inference understanding AIC and BIC in model selection. Sociol. Methods Res. 2004, 33, 261–304. [Google Scholar] [CrossRef]
- Li, P.; Vu, Q.D. Identification of parameter correlations for parameter estimation in dynamic biological models. BMC Syst. Biol. 2013, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Moog, C. Identifiability of nonlinear systems with application to HIV/AIDS models. IEEE Trans. Autom. Control 2003, 48, 330–336. [Google Scholar] [CrossRef]
- Miao, H.; Xia, X.; Perelson, A.S.; Wu, H. On Identifiability of Nonlinear Ode Models and Applications in Viral Dynamics. SIAM Rev. Soc. Ind. Appl. Math. 2011, 53, 3–39. [Google Scholar] [CrossRef] [PubMed]
- Chis, O.T.; Banga, J.R.; Balsa-Canto, E. Structural identifiability of systems biology models: A critical comparison of methods. PLoS ONE 2011, 6, e27755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brun, R.; Reichert, P.; Kuensch, H.R. Practical identifiability analysis of large environmental simulation models. Water Resour. Res. 2001, 37, 1015–1030. [Google Scholar] [CrossRef]
- Prill, R.J.; Marbach, D.; Saez-Rodriguez, J.; Sorger, P.K.; Alexopoulos, L.G.; Xue, X.; Clarke, N.D.; Altan-Bonnet, G.; Stolovitzky, G. Towards a rigorous assessment of systems biology models: The DREAM3 challenges. PLoS ONE 2010, 5, e9202. [Google Scholar] [CrossRef] [PubMed]
- Raue, A.; Kreutz, C.; Maiwald, T.; Bachmann, J.; Schilling, M.; Klingmüller, U.; Timmer, J. Structural and practical identifiability analysis of partially observed dynamical models by exploiting the profile likelihood. Bioinform. (Oxf. Engl.) 2009, 25, 1923–1929. [Google Scholar] [CrossRef] [PubMed]
- Efron, B.; Tibshirani, R.J. An Introduction to the Bootstrap; CRC Press: Boca Raton, FL, USA, 1994. [Google Scholar]
- Nguyen, V.K.; Binder, S.C.; Boianelli, A.; Meyer-Hermann, M.; Hernandez-Vargas, E.A. Ebola Virus Infection Modelling and Identifiability Problems. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Kosorok, M.R. Robust semiparametric M-estimation and the weighted bootstrap. J. Multivar. Anal. 2005, 96, 190–217. [Google Scholar] [CrossRef]
- Xue, H.; Miao, H.; Wu, H. Sieve estimation of constant and time-varying coefficients in nonlinear ordinary differential equation models by considering both numerical error and measurement error. Ann. Stat. 2010, 38, 2351–2387. [Google Scholar] [CrossRef] [PubMed]
- Vinod, H.D. Maximum Entropy Bootstrap Algorithm Enhancements. Available at SSRN 2285041 2013. [Google Scholar] [CrossRef]
- Lesaffre, E.; Lawson, A.B. Bayesian Biostatistics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Raue, A.; Kreutz, C.; Joachim Theis, F.; Timmer, J. Joining forces of Bayesian and frequentist methodology: A study for inference in the presence of non-identifiability. Philos. Trans. Ser. A 2013, 371. [Google Scholar] [CrossRef] [PubMed]
- Chou, I.C.; Voit, E.O. Recent developments in parameter estimation and structure identification of biochemical and genomic systems. Math. Biosci. 2009, 219, 57–83. [Google Scholar] [CrossRef] [PubMed]
- Toapanta, F.R.; Ross, T.M. Impaired immune responses in the lungs of aged mice following influenza infection. Respir. Res. 2009, 10, 112–131. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, M.; Bell, E. The survival and turnover of mature and immature CD8 T cells. Immunology 1995, 84, 514–520. [Google Scholar] [PubMed]
- Smith, A.M.; Adler, F.R.; McAuley, J.L.; Gutenkunst, R.N.; Ribeiro, R.M.; McCullers, J.A.; Perelson, A.S. Effect of 1918 PB1-F2 expression on influenza A virus infection kinetics. PLoS Comput. Biol. 2011, 7, e1001081. [Google Scholar] [CrossRef] [PubMed]
- Storn, R.; Price, K. Differential evolution—A simple and efficient heuristic for global optimization over continuous spaces. J. Global Optim. 1997, 11, 341–359. [Google Scholar] [CrossRef]
- Murillo, L.N.; Murillo, M.S.; Perelson, A.S. Towards multiscale modeling of influenza infection. J. Theor. Biol. 2013, 332, 267–290. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: Implications for pandemic influenza preparedness. J. Infect. Dis. 2008, 198, 962–970. [Google Scholar] [CrossRef] [PubMed]
- McCullers, J.A. Insights into the interaction between influenza virus and pneumococcus. Clin. Microbiol. Rev. 2006, 19, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Louie, J.; Jean, C.; Chen, T.; Park, S.; Ueki, R.; Harper, T.; Chmara, E.; Myers, J.; Stoppacher, R.; Catanese, C.; et al. Bacterial coinfections in lung tissue specimens from fatal cases of 2009 pandemic influenza A (H1N1)-United States, May-August 2009. Morb. Mortal. Wkly. Rep. 2009, 58, 1071–1074. [Google Scholar]
- Chertow, D.S.; Memoli, M.J. Bacterial coinfection in influenza: A grand rounds review. JAMA 2013, 309, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Louria, D.B.; Blumenfeld, H.L.; Ellis, J.T.; Kilbourne, E.D.; Rogers, D.E. Studies on influenza in the pandemic of 1957-1958. II. Pulmonary complications of influenza. J. Clin. Investig. 1959, 38, 213–265. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.M.; Kunin, C.M.; Gottlieb, L.S.; Barnes, M.W.; Liu, C.; Finland, M. Asian Influenza A in Boston. 1957–1958: I. Observations in Thirty-Two Influenza-Associated Fatal Cases. AMA Arch. Intern. Med. 1959, 103, 515–531. [Google Scholar] [CrossRef] [PubMed]
- McCullers, J.A. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev. Microbiol. 2014, 12, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Kolls, J.K.; Alcorn, J.F. The immunology of influenza virus-associated bacterial pneumonia. Curr. Opin. Immunol. 2015, 34, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Didierlaurent, A.; Goulding, J.; Patel, S.; Snelgrove, R.; Low, L.; Bebien, M.; Lawrence, T.; van Rijt, L.S.; Lambrecht, B.N.; Sirard, J.C.; et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolutionof respiratory influenza infection. J. Exp. Med. 2008, 205, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Metzger, D.W. Inhibition of pulmonary antibacterial defense by interferon-γ during recovery from influenza infection. Nat. Med. 2008, 14, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Stegemann-Koniszewski, S.; Gereke, M.; Orrskog, S.; Lienenklaus, S.; Pasche, B.; Bader, S.R.; Gruber, A.D.; Akira, S.; Weiss, S.; Henriques-Normark, B.; et al. TLR7 contributes to the rapid progression but not to the overall fatal outcome of secondary pneumococcal disease following influenza A virus infection. J. Innate Immun. 2013, 5, 84–96. [Google Scholar] [CrossRef] [PubMed]
- McNamee, L.A.; Harmsen, A.G. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect. Immun. 2006, 74, 6707–6721. [Google Scholar] [CrossRef] [PubMed]
- Small, C.L.; Shaler, C.R.; McCormick, S.; Jeyanathan, M.; Damjanovic, D.; Brown, E.G.; Arck, P.; Jordana, M.; Kaushic, C.; Ashkar, A.A.; et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J. Immunol. 2010, 184, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moltedo, B.; Moran, T.M. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of γδ T cells. J. Virol. 2012, 86, 12304–12312. [Google Scholar] [CrossRef] [PubMed]
- Shahangian, A.; Chow, E.K.; Tian, X.; Kang, J.R.; Ghaffari, A.; Liu, S.Y.; Belperio, J.A.; Cheng, G.; Deng, J.C. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Investig. 2009, 119, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Kash, J.C.; Walters, K.A.; Davis, A.S.; Sandouk, A.; Schwartzman, L.M.; Jagger, B.W.; Chertow, D.S.; Qi, L.; Kuestner, R.E.; Ozinsky, A.; et al. Lethal synergism of 2009 pandemic H1N1 influenza virus and Streptococcus pneumoniae coinfection is associated with loss of murine lung repair responses. mBio 2011, 2, e00172-11. [Google Scholar] [CrossRef] [PubMed]
- Goulding, J.; Godlee, A.; Vekaria, S.; Hilty, M.; Snelgrove, R.; Hussell, T. Lowering the threshold of lung innate immune cell activation alters susceptibility to secondary bacterial superinfection. J. Infect. Dis. 2011, 204, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- McCullers, J.A.; McAuley, J.L.; Browall, S.; Iverson, A.R.; Boyd, K.L.; Normark, B.H. Influenza enhances susceptibility to natural acquisition of and disease due to Streptococcus pneumoniae in ferrets. J. Infect. Dis. 2010, 202, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Siegel, S.J.; Roche, A.M.; Weiser, J.N. Influenza promotes pneumococcal growth during coinfection by providing host sialylated substrates as a nutrient source. Cell Host Microbe 2014, 16, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Mina, M.J.; Klugman, K.P. The role of influenza in the severity and transmission of respiratory bacterial disease. Lancet Respir. Med. 2014, 2, 750–763. [Google Scholar] [CrossRef]
- Mina, M.J.; McCullers, J.A.; Klugman, K.P. Live attenuated influenza vaccine enhances colonization of Streptococcus pneumoniae and Staphylococcus aureus in mice. mBio 2014, 5, e01040-13. [Google Scholar] [CrossRef] [PubMed]
- Department of Economic, U.N. World Population Ageing, 1950–2050; Number 207; United Nations: New York, NY, USA, 2002. [Google Scholar]
- Goronzy, J.J.; Weyand, C.M. Understanding immunosenescence to improve responses to vaccines. Nat. Immunol. 2013, 14, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A. The aging immune system: Primer and prospectus. Science 1996, 273, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Wick, G.; Grubeck-Loebenstein, B. The aging immune system: Primary and secondary alterations of immune reactivity in the elderly. Exp. Gerontol. 1997, 32, 401–413. [Google Scholar] [CrossRef]
- Shaw, A.C.; Joshi, S.; Greenwood, H.; Panda, A.; Lord, J.M. Aging of the innate immune system. Curr. Opin. Immunol. 2010, 22, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Mahbub, S.; Brubaker, A.L.; Kovacs, E.J. Aging of the innate immune system: An update. Curr. Immunol. Rev. 2011, 7, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Solana, R.; Tarazona, R.; Gayoso, I.; Lesur, O.; Dupuis, G.; Fulop, T. Innate immunosenescence: Effect of aging on cells and receptors of the innate immune system in humans. Semin. Immunol. 2012, 24, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Goronzy, J.J.; Lee, W.W.; Weyand, C.M. Aging and T-cell diversity. Exp. Gerontol. 2007, 42, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Bi, R.; Su, K.; Yel, L.; Chiplunkar, S.; Gollapudi, S. Characterization of naive, memory and effector CD8+ T cells: Effect of age. Exp. Gerontol. 2004, 39, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, A.N. CD28 extinction in human T cells: Altered functions and the program of T-cell senescence. Immunol. Rev. 2005, 205, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Le Page, A.; Fortin, C.; Witkowski, J.M.; Dupuis, G.; Larbi, A. Cellular signaling in the aging immune system. Curr. Opin. Immunol. 2014, 29, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Ling, W.; Yan, X.; Li-Jing, Z.; Chang, T.T.; Jie, L. An association between immunosenescence and CD4+ CD25+ regulatory T cells: A systematic review. Biomed. Environ. Sci. 2010, 23, 327–332. [Google Scholar]
- Poland, G.A.; Kennedy, R.B.; McKinney, B.A.; Ovsyannikova, I.G.; Lambert, N.D.; Jacobson, R.M.; Oberg, A.L. Vaccinomics, adversomics, and the immune response network theory: Individualized vaccinology in the 21st century. Semin. Immunol. 2013, 25, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Poland, G.A.; Ovsyannikova, I.G.; Kennedy, R.B.; Lambert, N.D.; Kirkland, J.L. A systems biology approach to the effect of aging, immunosenescence and vaccine response. Curr. Opin. Immunol. 2014, 29, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Durando, P.; Iudici, R.; Alicino, C.; Alberti, M.; de Florentis, D.; Ansaldi, F.; Icardi, G. Adjuvants and alternative routes of administration towards the development of the ideal influenza vaccine. Hum. Vaccines 2011, 7, 29–40. [Google Scholar] [CrossRef]
- Monto, A.S.; Ansaldi, F.; Aspinall, R.; McElhaney, J.E.; Montano, L.F.; Nichol, K.L.; Puig-Barberà, J.; Schmitt, J.; Stephenson, I. Influenza control in the 21st century: Optimizing protection of older adults. Vaccine 2009, 27, 5043–5053. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.C.; Wu, T.Z.; Liu, D.P.; Shao, P.L.; Chang, L.Y.; Lu, C.Y.; Lee, C.Y.; Huang, F.Y.; Huang, L.M. Influenza pandemics: Past, present and future. J. Formos. Med. Assoc. 2006, 105, 1–6. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, L. Possible origin of current influenza A H1N1 viruses. Lancet Infect. Dis. 2009, 9, 456–457. [Google Scholar] [CrossRef]
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and genetic characteristics of swine-origin 2009 A (H1N1) influenza viruses circulating in humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Monto, A.S.; Black, S.; Plotkin, S.A.; Orenstein, W.A. Response to the 2009 pandemic: Effect on influenza control in wealthy and poor countries. Vaccine 2011, 29, 6427–6431. [Google Scholar] [CrossRef] [PubMed]
- Clegg, C.H.; Rininger, J.A.; Baldwin, S.L. Clinical vaccine development for H5N1 influenza. Expert Rev. Vaccines 2013, 12, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, G.; Weinberger, B.; Grubeck-Loebenstein, B. Vaccines for the Elderly. Gerontology 2015, 61, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Amorij, J.P.; Hinrichs, W.L.; Frijlink, H.W.; Wilschut, J.C.; Huckriede, A. Needle-free influenza vaccination. Lancet Infect. Dis. 2010, 10, 699–711. [Google Scholar] [CrossRef]
- Neutra, M.R.; Kozlowski, P.A. Mucosal vaccines: The promise and the challenge. Nat. Rev. Immunol. 2006, 6, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Lycke, N. Recent progress in mucosal vaccine development: Potential and limitations. Nat. Rev. Immunol. 2012, 12, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Fujihashi, K.; Sato, S.; Kiyono, H. Mucosal adjuvants for vaccines to control upper respiratory infections in the elderly. Exp. Gerontol. 2014, 54, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Van Ginkel, F.W.; Jackson, R.J.; Yuki, Y.; McGhee, J.R. Cutting edge: The mucosal adjuvant cholera toxin redirects vaccine proteins into olfactory tissues. J. Immunol. 2000, 165, 4778–4782. [Google Scholar] [CrossRef]
- Mutsch, M.; Zhou, W.; Rhodes, P.; Bopp, M.; Chen, R.T.; Linder, T.; Spyr, C.; Steffen, R. Use of the inactivated intranasal influenza vaccine and the risk of Bell’s palsy in Switzerland. N. Engl. J. Med. 2004, 350, 896–903. [Google Scholar] [CrossRef]
- Wegmann, F.; Gartlan, K.H.; Harandi, A.M.; Brinckmann, S.A.; Coccia, M.; Hillson, W.R.; Kok, W.L.; Cole, S.; Ho, L.P.; Lambe, T.; et al. Polyethyleneimine is a potent mucosal adjuvant for viral glycoprotein antigens. Nat. Biotechnol. 2012, 30, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Stanberry, L.; Simon, J.; Johnson, C.; Robinson, P.; Morry, J.; Flack, M.; Gracon, S.; Myc, A.; Hamouda, T.; Baker, J. Safety and immunogenicity of a novel nanoemulsion mucosal adjuvant W 80 5EC combined with approved seasonal influenza antigens. Vaccine 2012, 30, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Huleatt, J.W.; Nakaar, V.; Desai, P.; Huang, Y.; Hewitt, D.; Jacobs, A.; Tang, J.; McDonald, W.; Song, L.; Evans, R.K.; et al. Potent immunogenicity and efficacy of a universal influenza vaccine candidate comprising a recombinant fusion protein linking influenza M2e to the TLR5 ligand flagellin. Vaccine 2008, 26, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, S.M.; Tebianian, M. Influenza A viruses: Why focusing on M2e-based universal vaccines. Virus Genes 2011, 42, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Atsmon, J.; Kate-Ilovitz, E.; Shaikevich, D.; Singer, Y.; Volokhov, I.; Haim, K.Y.; Ben-Yedidia, T. Safety and immunogenicity of multimeric-001-a novel universal influenza vaccine. J. Clin. Immunol. 2012, 32, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yedidia, T.; Arnon, R. Epitope-based vaccine against influenza. Expert Rev. Vaccines 2007, 6, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Sullivan, M.; Wilson, P.C. Targeting B cell responses in universal influenza vaccine design. Trends Immunol. 2011, 32, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.J.; Boyington, J.C.; McTamney, P.M.; Kong, W.P.; Pearce, M.B.; Xu, L.; Andersen, H.; Rao, S.; Tumpey, T.M.; Yang, Z.Y.; et al. Induction of broadly neutralizing H1N1 influenza antibodies by vaccination. Science 2010, 329, 1060–1064. [Google Scholar] [CrossRef] [PubMed]
- Price, G.E.; Soboleski, M.R.; Lo, C.Y.; Misplon, J.A.; Quirion, M.R.; Houser, K.V.; Pearce, M.B.; Pappas, C.; Tumpey, T.M.; Epstein, S.L. Single-dose mucosal immunization with a candidate universal influenza vaccine provides rapid protection from virulent H5N1, H3N2 and H1N1 viruses. PLoS ONE 2010, 5, e13162. [Google Scholar] [CrossRef] [PubMed]
- Poon, L.L.; Leung, Y.C.; Nicholls, J.M.; Perera, P.Y.; Lichy, J.H.; Yamamoto, M.; Waldmann, T.A.; Peiris, J.M.; Perera, L.P. Vaccinia virus-based multivalent H5N1 avian influenza vaccines adjuvanted with IL-15 confer sterile cross-clade protection in mice. J. Immunol. 2009, 182, 3063–3071. [Google Scholar] [CrossRef] [PubMed]
- Suter, R.; Summerfield, A.; Thomann-Harwood, L.J.; McCullough, K.C.; Tratschin, J.D.; Ruggli, N. Immunogenic and replicative properties of classical swine fever virus replicon particles modified to induce IFN-α/β and carry foreign genes. Vaccine 2011, 29, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.C.; Bassi, I.; Démoulins, T.; Thomann-Harwood, L.J.; Ruggli, N. Functional RNA delivery targeted to dendritic cells by synthetic nanoparticles. Ther. Deliv. 2012, 3, 1077–1099. [Google Scholar] [CrossRef] [PubMed]
- Pica, N.; Palese, P. Toward a universal influenza virus vaccine: Prospects and challenges. Annu. Rev. Med. 2013, 64, 189–202. [Google Scholar] [CrossRef]
- Bolton, K.J.; McCaw, J.M.; Brown, L.; Jackson, D.; Kedzierska, K.; McVernon, J. Prior Population Immunity Reduces the Expected Impact of CTL-Inducing Vaccines for Pandemic Influenza Control. PLoS ONE 2015, 10, e0120138. [Google Scholar] [CrossRef]
- Baguelin, M.; Flasche, S.; Camacho, A.; Demiris, N.; Miller, E.; Edmunds, W.J. Assessing optimal target populations for influenza vaccination programmes: An evidence synthesis and modelling study. PLoS Med. 2013, 10, e1001527. [Google Scholar] [CrossRef] [PubMed]
- Pettini, E.; Prota, G.; Ciabattini, A.; Boianelli, A.; Fiorino, F.; Pozzi, G.; Vicino, A.; Medaglini, D. Vaginal Immunization to Elicit Primary T-Cell Activation and Dissemination. PLoS ONE 2013, 8, e80545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boianelli, A.; Pettini, E.; Prota, G.; Medaglini, D.; Vicino, A. Identification of a branching process model for adaptive immune response. In Proceedings of the 2013 IEEE 52nd Annual Conference on Decision and Control (CDC), Firenze, Italy, 10–13 December 2013; pp. 7205–7210.
- Boianelli, A.; Pettini, E.; Prota, G.; Medaglini, D.; Vicino, A. A Stochastic Model for CD4+ T Cell Proliferation and Dissemination Network in Primary Immune Response. PLoS ONE 2015, 10, e0135787. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Hermann, M.; Mohr, E.; Pelletier, N.; Zhang, Y.; Victora, G.D.; Toellner, K.M. A theory of germinal center B cell selection, division, and exit. Cell Rep. 2012, 2, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Victora, G.D.; Nussenzweig, M.C. Germinal centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Hermann, M. Overcoming the Dichotomy of Quantity and Quality in Antibody Responses. J. Immunol. 2014, 193, 5414–5419. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, S.; Reifman, J.; Wallqvist, A. Simulation of B Cell Affinity Maturation Explains Enhanced Antibody Cross-Reactivity Induced by the Polyvalent Malaria Vaccine AMA1. J. Immunol. 2014, 193, 2073–2086. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, B.; Błażejewska, P.; Heßmann, M.; Bruder, D.; Geffers, R.; Mauel, S.; Gruber, A.D.; Schughart, K. Host genetic background strongly influences the response to influenza a virus infections. PLoS ONE 2009, 4, e4857. [Google Scholar] [CrossRef] [PubMed]
- Consortium, C.C. The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics 2012, 190, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Bottomly, D.; Ferris, M.T.; Aicher, L.D.; Rosenzweig, E.; Whitmore, A.; Aylor, D.L.; Haagmans, B.L.; Gralinski, L.E.; Bradel-Tretheway, B.G.; Bryan, J.T.; et al. Expression quantitative trait Loci for extreme host response to influenza a in pre-collaborative cross mice. G3 Genes Genomes Genet. 2012, 2, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Ferris, M.T.; Aylor, D.L.; Bottomly, D.; Whitmore, A.C.; Aicher, L.D.; Bell, T.A.; Bradel-Tretheway, B.; Bryan, J.T.; Buus, R.J.; Gralinski, L.E.; et al. Modeling host genetic regulation of influenza pathogenesis in the collaborative cross. PLoS Pathog. 2013, 9, e1003196. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.C.; Huang, I.C.; Kam, C.; Farzan, M. Ifitm3 limits the severity of acute influenza in mice. PLoS Pathog. 2012, 8, e1002909. [Google Scholar] [CrossRef]
- Everitt, A.R.; Clare, S.; Pertel, T.; John, S.P.; Wash, R.S.; Smith, S.E.; Chin, C.R.; Feeley, E.M.; Sims, J.S.; Adams, D.J.; et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 2012, 484, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.; Wang, L.; Li, W.; Zi, H.; Guo, Y.; Yan, W.; Chen, X.; Wei, P. IFITM3 rs12252 T > C polymorphism is associated with the risk of severe influenza: A meta-analysis. Epidemiol. Infect. 2015, 143, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mak, C.M.; Lam, C.W.; Fong, N.C.; Siu, W.K.; Lee, H.C.H.; Siu, T.S.; Lai, C.K.; Law, C.Y.; Tong, S.F.; Poon, W.T.; et al. Fatal viral infection-associated encephalopathy in two Chinese boys: A genetically determined risk factor of thermolabile carnitine palmitoyltransferase II variants. J. Hum. Genet. 2011, 56, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Hatesuer, B.; Bertram, S.; Mehnert, N.; Bahgat, M.M.; Nelson, P.S.; Pöhlman, S.; Schughart, K. TMPRSS2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog. 2013, 9, e1003774. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Ami, Y.; Tahara, M.; Kubota, T.; Anraku, M.; Abe, M.; Nakajima, N.; Sekizuka, T.; Shirato, K.; Suzaki, Y.; et al. The host protease TMPRSS2 plays a major role in in vivo replication of emerging H7N9 and seasonal influenza viruses. J. Virol. 2014, 88, 5608–5616. [Google Scholar] [CrossRef]
- Tarnow, C.; Engels, G.; Arendt, A.; Schwalm, F.; Sediri, H.; Preuss, A.; Nelson, P.S.; Garten, W.; Klenk, H.D.; Gabriel, G.; et al. TMPRSS2 is a host factor that is essential for pneumotropism and pathogenicity of H7N9 influenza A virus in mice. J. Virol. 2014, 88, 4744–4751. [Google Scholar] [CrossRef]
- Kollmus, H.; Wilk, E.; Schughart, K. Systems biology and systems genetics-novel innovative approaches to study host—pathogen interactions during influenza infection. Curr. Opin. Virol. 2014, 6, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic gene expression in a single cell. Science 2002, 297, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- El Samad, H.; Khammash, M.; Petzold, L.; Gillespie, D. Stochastic modelling of gene regulatory networks. Int. J. Robust Nonlinear Control 2005, 15, 691–711. [Google Scholar] [CrossRef]
- Paulsson, J. Summing up the noise in gene networks. Nature 2004, 427, 415–418. [Google Scholar] [CrossRef]
- Eldar, A.; Elowitz, M.B. Functional roles for noise in genetic circuits. Nature 2010, 467, 167–173. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boianelli, A.; Nguyen, V.K.; Ebensen, T.; Schulze, K.; Wilk, E.; Sharma, N.; Stegemann-Koniszewski, S.; Bruder, D.; Toapanta, F.R.; Guzmán, C.A.; et al. Modeling Influenza Virus Infection: A Roadmap for Influenza Research. Viruses 2015, 7, 5274-5304. https://doi.org/10.3390/v7102875
Boianelli A, Nguyen VK, Ebensen T, Schulze K, Wilk E, Sharma N, Stegemann-Koniszewski S, Bruder D, Toapanta FR, Guzmán CA, et al. Modeling Influenza Virus Infection: A Roadmap for Influenza Research. Viruses. 2015; 7(10):5274-5304. https://doi.org/10.3390/v7102875
Chicago/Turabian StyleBoianelli, Alessandro, Van Kinh Nguyen, Thomas Ebensen, Kai Schulze, Esther Wilk, Niharika Sharma, Sabine Stegemann-Koniszewski, Dunja Bruder, Franklin R. Toapanta, Carlos A. Guzmán, and et al. 2015. "Modeling Influenza Virus Infection: A Roadmap for Influenza Research" Viruses 7, no. 10: 5274-5304. https://doi.org/10.3390/v7102875