
Gene Acquisition Convergence between Entomopoxviruses and Baculoviruses


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Viral Orthologous Clustering
2.2. Phylogenetic Analyses
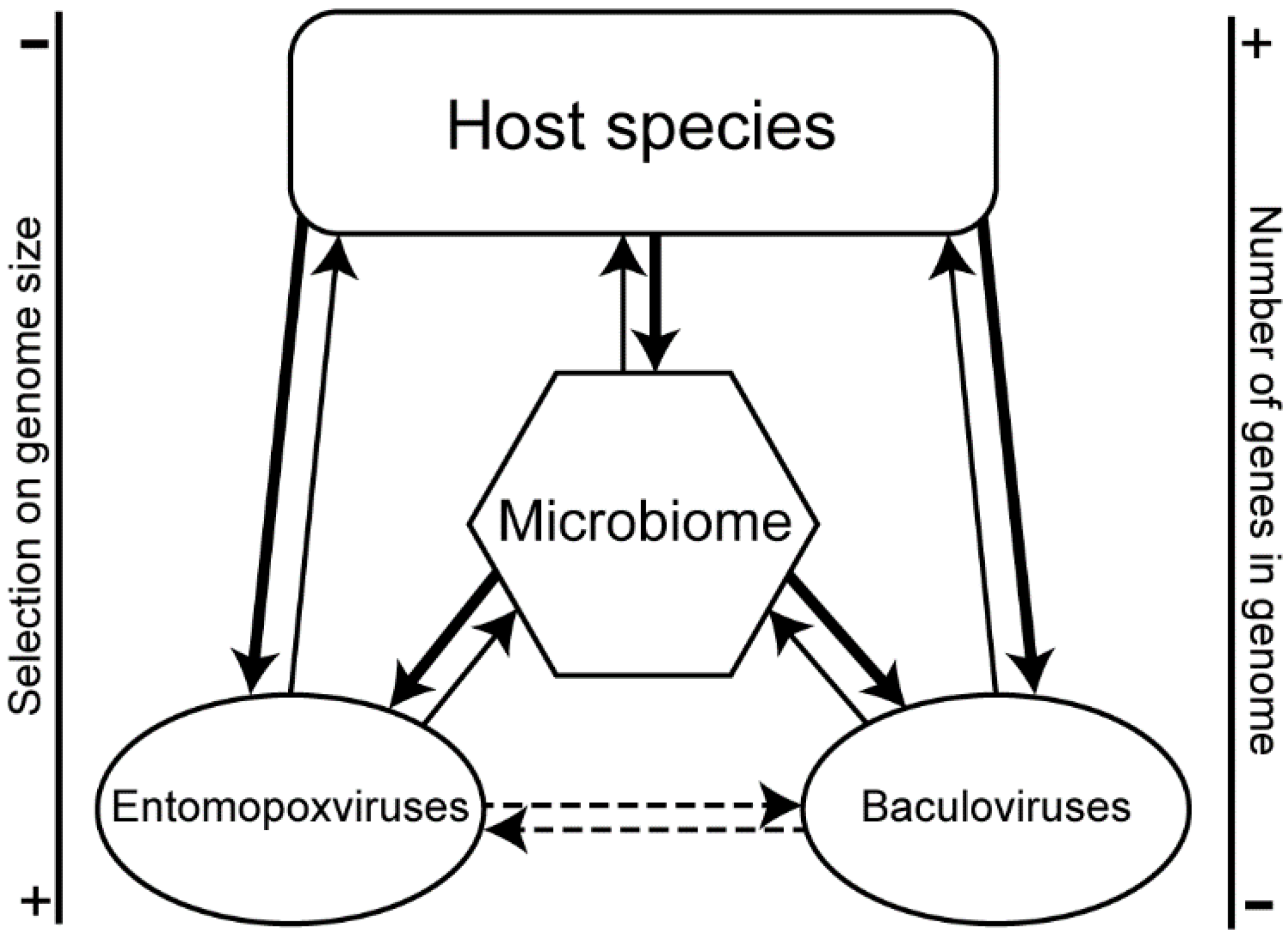
3. Results
3.2. Viruses Infecting the Same Hosts Share Homologous Genes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viral homologous gene cluster name | Best blastp hit between EPVs and BVs (e-value) | Presence in other large dsDNA viruses | Putative origins of EPV/BV genes | Phylogeny (Figure) |
|---|---|---|---|---|
| DNA polymerase | 6 × 10−9 | X | - | - |
| bro gene family protein | 9 × 10−26 | X | Bacteria | S1 |
| helicase 2 | 1 × 10−67 | X | Bacteria | S2 |
| matrixin metalloproteinase | 1 × 10−9 | X | Bacteria | S3 |
| nla gene he65 (AcMNPV orf105) | 8 × 10−97 | - | Bacteria | S4 |
| protein tyrosine phosphatase 2 (ptp-2) | 2 × 10−33 | X | Bacteria | S5 |
| putative phage antirepressor | 9 × 10−26 | X | Bacteria | S6 |
| unknown LdMNPV orf129 | 2 × 10−57 | - | Bacteria | S7 |
| chitin binding protein (AcMNPV orf145) | 1 × 10−17 | X | Insecta | S8 |
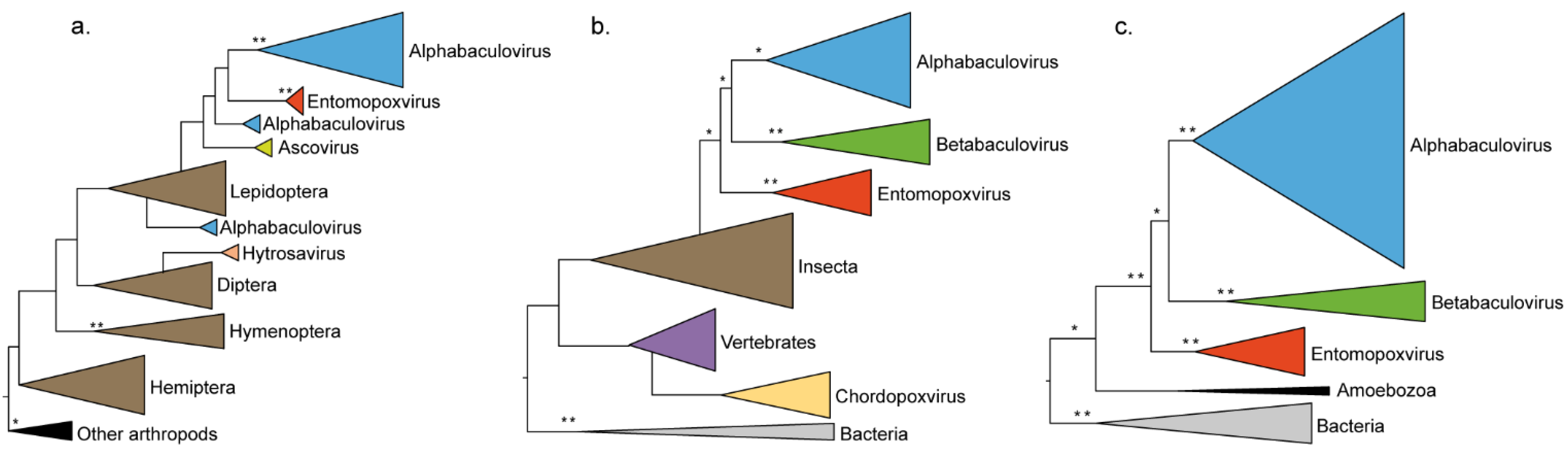
| Cu/Zn superoxide dismutase | 4 × 10−56 | X | Insecta | 2b/S9 |
| inhibitor of apoptosis | 4 × 10−79 | X | Insecta | S10 |
| MTG motif gene family protein | 7 × 10−10 | X | Insecta | S11 |
| ubiquitin | 6 × 10−38 | X | Insecta | S12 |
| unknown AcMNPV orf7 | 5 × 10−9 | - | Insecta | S13 |
| unknown XecnGV orf106 | 3 × 10−13 | X | Insecta | S14 |
| unknown XecnGV orf22 | 6 × 10−103 | X | Insecta | S15 |
| acetyltransferase | 7 × 10−13 | - | Insecta (Lepidoptera) | S16 |
| protein phosphatase 1, regulary subunit 15A | 5 × 10−15 | X | Insecta (Lepidoptera) | S17 |
| ribonucleotide reductase small subunit homolog | 8 × 10−134 | X | Insecta (Lepidoptera) | 2a/S18 |
| unknown XecnGV orf72 | 8 × 10−133 | - | Insecta (Lepidoptera) | S19 |
| protein tyrosine phosphatase 1 (ptp-1) | 4 × 10−41 | X | Insecta (Lepidoptera) & Eukaryote | S20 |
| DNA photolyase | 3 × 10−140 | X | Bilateria/Insecta (Lepidoptera) | S21 |
| dUTPase | 1 × 10−43 | X | Insecta/Unknown | S22 |
| unknown ClanGV orf085 | 9 × 10−17 | X | Eukaryote | S23 |
| leucine rich gene family protein | 4 × 10−27 | - | Unicellular eukaryote | S24 |
| fusolin/spindlin/gp37 (AcMNPV orf64) | 4 × 10−67 | - | Unicellular eukaryote (Amoebozoa) | 2c/S25 |
| conotoxin-like protein | 3 × 10−20 | - | Virus | S26 |
| p35/p49 apoptosis inhibitor | 4 × 10−30 | - | Virus | S27 |
| unknown AcMNPV orf18 | 8 × 10−11 | - | Virus | S28 |
| unknown AdorNPV orf110 | 8 × 10−11 | X | Virus | S29 |
| unknown AgseGV orf4 | 7 × 10−103 | - | Virus | NA |
| unknown ChocGV orf11 | 3 × 10−14 | - | Virus | NA |
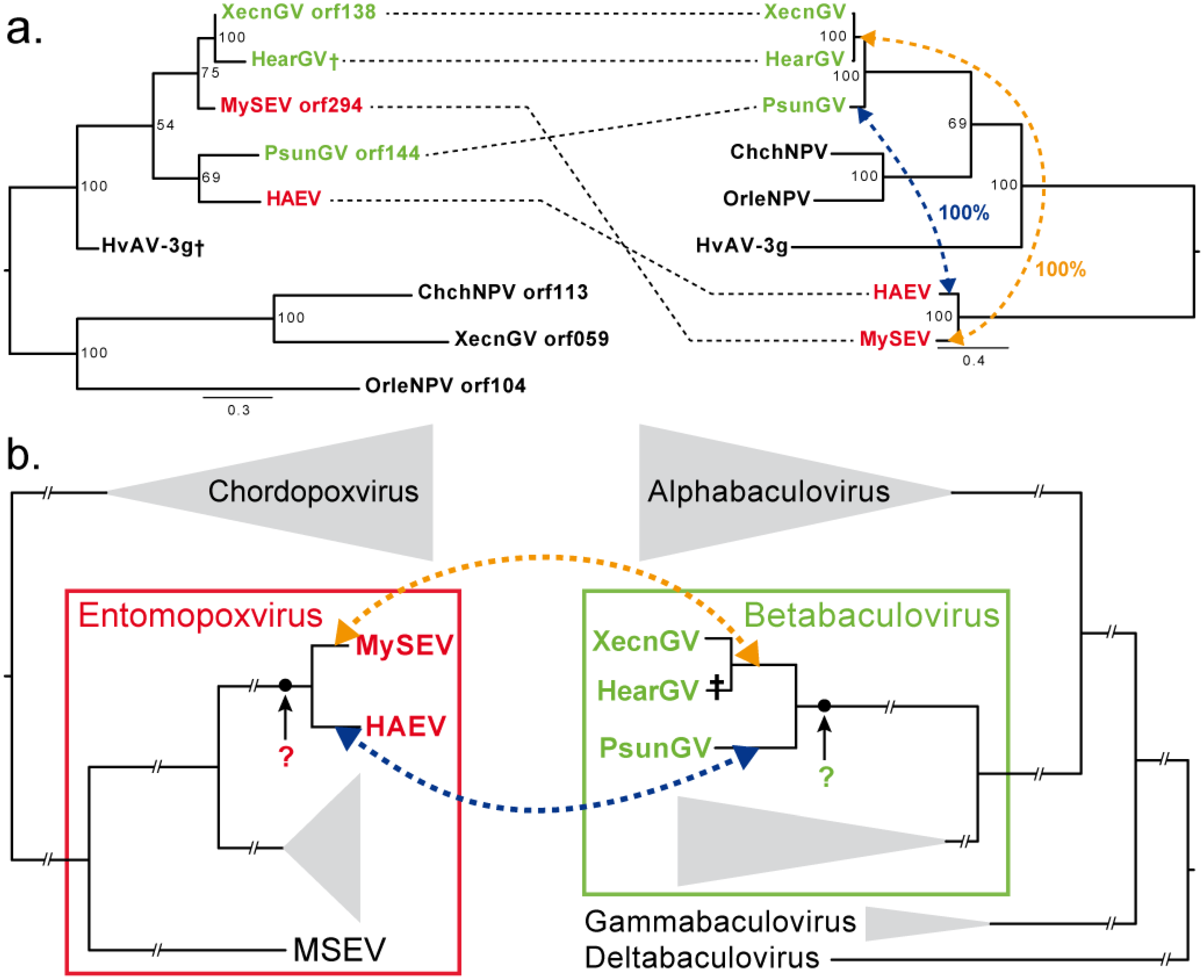
| unknown XecnGV orf138 | 0 | X | Virus | 1a |

3.3. Convergence between Viruses Infecting Different Hosts

4. Discussion

Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar]
- Cox, F.E.G. Concomitant infections, parasites and immune responses. Parasitology 2011, 122, S23–S38. [Google Scholar] [CrossRef]
- Tenaillon, O.; Rodríguez-Verdugo, A.; Gaut, R.L.; McDonald, P.; Bennett, A.F.; Long, A.D.; Gaut, B.S. The molecular diversity of adaptive convergence. Science 2012, 335, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Smillie, C.S.; Smith, M.B.; Friedman, J.; Cordero, O.X.; David, L.A.; Alm, E.J. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 2011, 480, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.; Tsagkogeorga, G.; Cotton, J.A.; Liu, Y.; Provero, P.; Stupka, E.; Rossiter, S.J. Genome-wide signatures of convergent evolution in echolocating mammals. Nature 2013, 502, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Herniou, E.A.; Huguet, E.; Thézé, J.; Bézier, A.; Periquet, G.; Drezen, J.-M. When parasitic wasps hijacked viruses: Genomic and functional evolution of polydnaviruses. Phil. Trans. R. Soc. B 2013, 368, e20130051. [Google Scholar] [CrossRef]
- Combes, C. Parasitism: The Ecology and Evolution of Intimate Interactions; University of Chicago Press: Chicago, IL, USA, 2001. [Google Scholar]
- Shackelton, L.A.; Holmes, E.C. The evolution of large DNA viruses: Combining genomic information of viruses and their hosts. Trends Microbiol. 2004, 12, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Sabehi, G.; Shaulov, L.; Silver, D.H.; Yanai, I.; Harel, A.; Lindell, D. A novel lineage of myoviruses infecting cyanobacteria is widespread in the oceans. Proc. Natl. Acad. Sci. USA 2012, 109, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, K.; Duhaime, M.B.; Breier, J.A.; Wendt, K.A.; Toner, B.M.; Dick, G.J. Sulfur oxidation genes in diverse deep-sea viruses. Science 2014, 344, 757–760. [Google Scholar] [CrossRef] [PubMed]
- May, R.M. How many species are there on earth? Science 1988, 241, 1441. [Google Scholar] [CrossRef] [PubMed]
- Cory, J. Use of baculoviruses as biological insecticides. Mol. Biotechnol. 1997, 7, 303–313. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.; Lefkowitz, E.; Adams, M.J.; Carstens, E.B. Virus Taxonomy: Ninth Report of the International Committee of Taxonomy of Viruses; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Thézé, J.; Takatsuka, J.; Li, Z.; Gallais, J.; Doucet, D.; Arif, B.; Nakai, M.; Herniou, E.A. New insights into the evolution of Entomopoxvirinae from the complete genome sequences of four entomopoxviruses infecting Adoxophyes honmai, Choristoneura biennis, Choristoneura rosaceana, and Mythimna separata. J. Virol. 2013, 87, 7992–8003. [Google Scholar] [CrossRef] [PubMed]
- Forterre, P.; Prangishvili, D. The origin of viruses. Res. Microbiol. 2009, 160, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Upton, C.; Slack, S.; Hunter, A.L.; Ehlers, A.; Roper, R.L. Poxvirus orthologous clusters: Toward defining the minimum essential poxvirus genome. J. Virol. 2003, 77, 7590–7600. [Google Scholar] [CrossRef] [PubMed]
- Garavaglia, M.J.; Miele, S.A.B.; Iserte, J.A.; Belaich, M.N.; Ghiringhelli, P.D. The ac53, ac78, ac101, and ac103 genes are newly discovered core genes in the family Baculoviridae. J. Virol. 2012, 86, 12069–12079. [Google Scholar] [CrossRef] [PubMed]
- De Andrade Zanotto, P.M.; Krakauer, D.C. Complete genome viral phylogenies suggests the concerted evolution of regulatory cores and accessory satellites. PLoS ONE 2008, 3, e3500. [Google Scholar] [CrossRef] [PubMed]
- Dall, D.; Luque, T.; O’Reilly, D. Insect-virus relationships: Sifting by informatics. Bioessays 2001, 23, 184–193. [Google Scholar] [PubMed]
- Hughes, A.L.; Friedman, R. Genome-wide survey for genes horizontally transferred from cellular organisms to baculoviruses. Mol. Biol. Evol. 2003, 20, 979–987. [Google Scholar] [CrossRef] [PubMed]
- McLysaght, A.; Baldi, P.F.; Gaut, B.S. Extensive gene gain associated with adaptive evolution of poxviruses. Proc. Natl. Acad. Sci. USA 2003, 100, 15655–15660. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L.; Friedman, R. Poxvirus genome evolution by gene gain and loss. Mol. Phylogenet. Evol. 2005, 35, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Bratke, K.A.; McLysaght, A. Identification of multiple independent horizontal gene transfers into poxviruses using a comparative genomics approach. BMC Evol. Biol. 2008, 8, e67. [Google Scholar] [CrossRef]
- Insect Virology; Asgari, S.; Johnson, K.N. (Eds.) Caister Academic Press: Norfolk, UK, 2010.
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, e539. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Mukawa, S.; Goto, C. In vivo characterization of two granuloviruses in larvae of Mythimna separata (Lepidoptera: Noctuidae). J. Gen. Virol. 2008, 89, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Boc, A.; Philippe, H.; Makarenkov, V. Inferring and validating horizontal gene transfer events using bipartition dissimilarity. Syst. Biol. 2010, 59, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Thézé, J.; Bézier, A.; Periquet, G.; Drezen, J.-M.; Herniou, E.A. Paleozoic origin of insect large dsDNA viruses. Proc. Natl. Acad. Sci. USA 2011, 108, 15931–15935. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H. The not so universal tree of life or the place of viruses in the living world. Phil. Trans. R. Soc. B 2009, 364, 2263–2274. [Google Scholar] [CrossRef] [PubMed]
- Filée, J.; Pouget, N.; Chandler, M. Phylogenetic evidence for extensive lateral acquisition of cellular genes by Nucleocytoplasmic large DNA viruses. BMC Evol. Biol. 2008, 8, e320. [Google Scholar] [CrossRef]
- Possee, R.D.; Rohrmann, G.F. Baculovirus genome organization and evolution. In The Baculoviruses (The Viruses); Miller, L.K., Ed.; Springer: Berlin, Germany, 1997; pp. 109–134. [Google Scholar]
- Xu, J.; Hukuhara, T. Enhanced infection of a nuclear polyhedrosis virus in larvae of the armyworm, Pseudaletia separata, by a factor in the spheroids of an entomopoxvirus. J. Invertebr. Pathol. 1992, 60, 259–264. [Google Scholar] [CrossRef]
- Mitsuhashi, W.; Kawakita, H.; Murakami, R.; Takemoto, Y.; Saiki, T.; Miyamoto, K.; Wada, S. Spindles of an entomopoxvirus facilitate its infection of the host insect by disrupting the peritrophic membrane. J. Virol. 2007, 81, 4235–4243. [Google Scholar] [CrossRef] [PubMed]
- Takatsuka, J.; Okuno, S.; Ishii, T.; Nakai, M.; Kunimi, Y. Fitness-related traits of entomopoxviruses isolated from Adoxophyes honmai (Lepidoptera: Tortricidae) at three localities in Japan. J. Invertebr. Pathol. 2010, 105, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Nakai, M.; Shiotsuki, T.; Kunimi, Y. An entomopoxvirus and a granulovirus use different mechanisms to prevent pupation of Adoxophyes honmai. Virus Res. 2004, 101, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Katzourakis, A.; Gifford, R.J. Endogenous viral elements in animal genomes. PLoS Genet. 2010, 6, e1001191. [Google Scholar] [CrossRef] [PubMed]
- Dunning Hotopp, J.C.; Clark, M.E.; Oliveira, D.C.S.G.; Foster, J.M.; Fischer, P.; Muñoz Torres, M.C.; Giebel, J.D.; Kumar, N.; Ishmael, N.; Wang, S.; et al. Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science 2007, 317, 1753–1756. [Google Scholar] [CrossRef] [PubMed]
- Sharon, I.; Alperovitch, A.; Rohwer, F.; Haynes, M.; Glaser, F.; Atamna-Ismaeel, N.; Pinter, R.Y.; Partensky, F.; Koonin, E.V.; Wolf, Y.I.; et al. Photosystem I gene cassettes are present in marine virus genomes. Nature 2009, 461, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Zilber-Rosenberg, I.; Rosenberg, E. Role of microorganisms in the evolution of animals and plants: The hologenome theory of evolution. FEMS Microbiol. Rev. 2008, 32, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Chateigner, A.; Ernenwein, L.; Barbe, V.; Bézier, A.; Herniou, E.A.; Cordaux, R. Population genomics supports baculoviruses as vectors of horizontal transfer of insect transposons. Nat. Commun. 2014, 5, 3348. [Google Scholar] [PubMed]
- Pace, J.K.; Gilbert, C.; Clark, M.S.; Feschotte, C. Repeated horizontal transfer of a DNA transposon in mammals and other tetrapods. Proc. Natl. Acad. Sci. USA 2008, 105, 17023–17028. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C.; Pritham, E.J. A cornucopia of Helitrons shapes the maize genome. Proc. Natl. Acad. Sci. USA 2009, 106, 19747–19748. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Elde, N.C.; Child, S.J.; Eickbush, M.T.; Kitzman, J.O.; Rogers, K.S.; Shendure, J.; Geballe, A.P.; Malik, H.S. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell 2012, 150, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Bergin, D.; Reeves, E.P.; Renwick, J.; Wientjes, F.B.; Kavanagh, K. Superoxide production in Galleria mellonella hemocytes: Identification of proteins homologous to the NADPH oxidase complex of human neutrophils. Infect. Immun. 2005, 73, 4161–4170. [Google Scholar] [CrossRef] [PubMed]
- Clem, R.J. Baculoviruses and apoptosis: A diversity of genes and responses. Curr. Drug Targets 2007, 8, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L. Evolution of inhibitors of apoptosis in baculoviruses and their insect hosts. Infect. Genet. Evol. 2002, 2, 3–10. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thézé, J.; Takatsuka, J.; Nakai, M.; Arif, B.; Herniou, E.A. Gene Acquisition Convergence between Entomopoxviruses and Baculoviruses. Viruses 2015, 7, 1960-1974. https://doi.org/10.3390/v7041960
Thézé J, Takatsuka J, Nakai M, Arif B, Herniou EA. Gene Acquisition Convergence between Entomopoxviruses and Baculoviruses. Viruses. 2015; 7(4):1960-1974. https://doi.org/10.3390/v7041960
Chicago/Turabian StyleThézé, Julien, Jun Takatsuka, Madoka Nakai, Basil Arif, and Elisabeth A. Herniou. 2015. "Gene Acquisition Convergence between Entomopoxviruses and Baculoviruses" Viruses 7, no. 4: 1960-1974. https://doi.org/10.3390/v7041960
APA StyleThézé, J., Takatsuka, J., Nakai, M., Arif, B., & Herniou, E. A. (2015). Gene Acquisition Convergence between Entomopoxviruses and Baculoviruses. Viruses, 7(4), 1960-1974. https://doi.org/10.3390/v7041960