
The Subcellular Localisation of the Human Papillomavirus (HPV) 16 E7 Protein in Cervical Cancer Cells and Its Perturbation by RNA Aptamers



 and
and 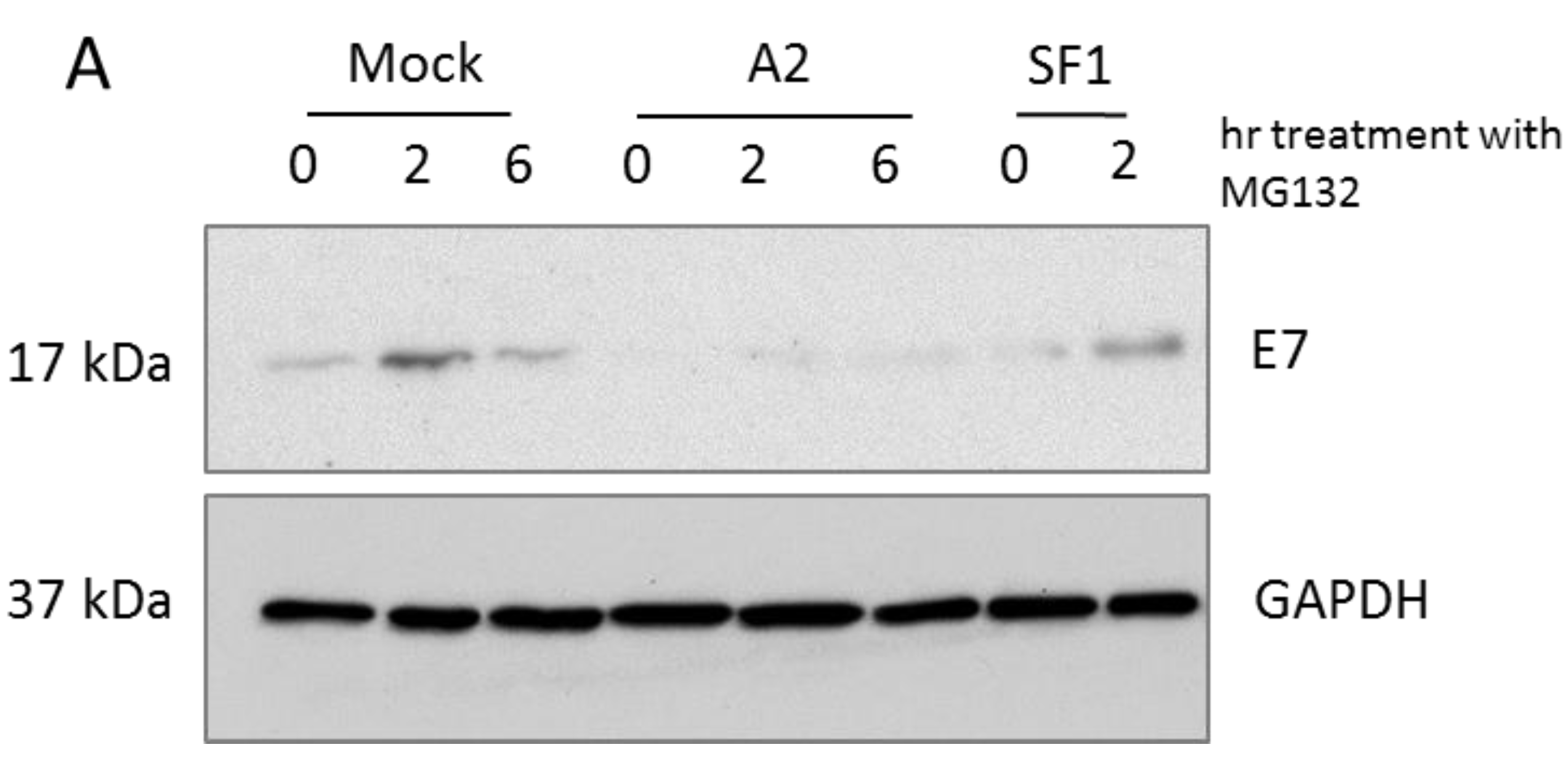{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Generation of 2′-Fluoro-Modified RNA Molecules
2.3. Transfection of Cells with Aptamers Using Oligofectamine
2.4. Collection of Cells and Protein Extraction
2.5. SDS-PAGE and Western Blotting
2.6. Immunostaining of Cells and Fluorescence Microscopy
2.7. Quantitation of Co-Localisation by Imeris Software
3. Results
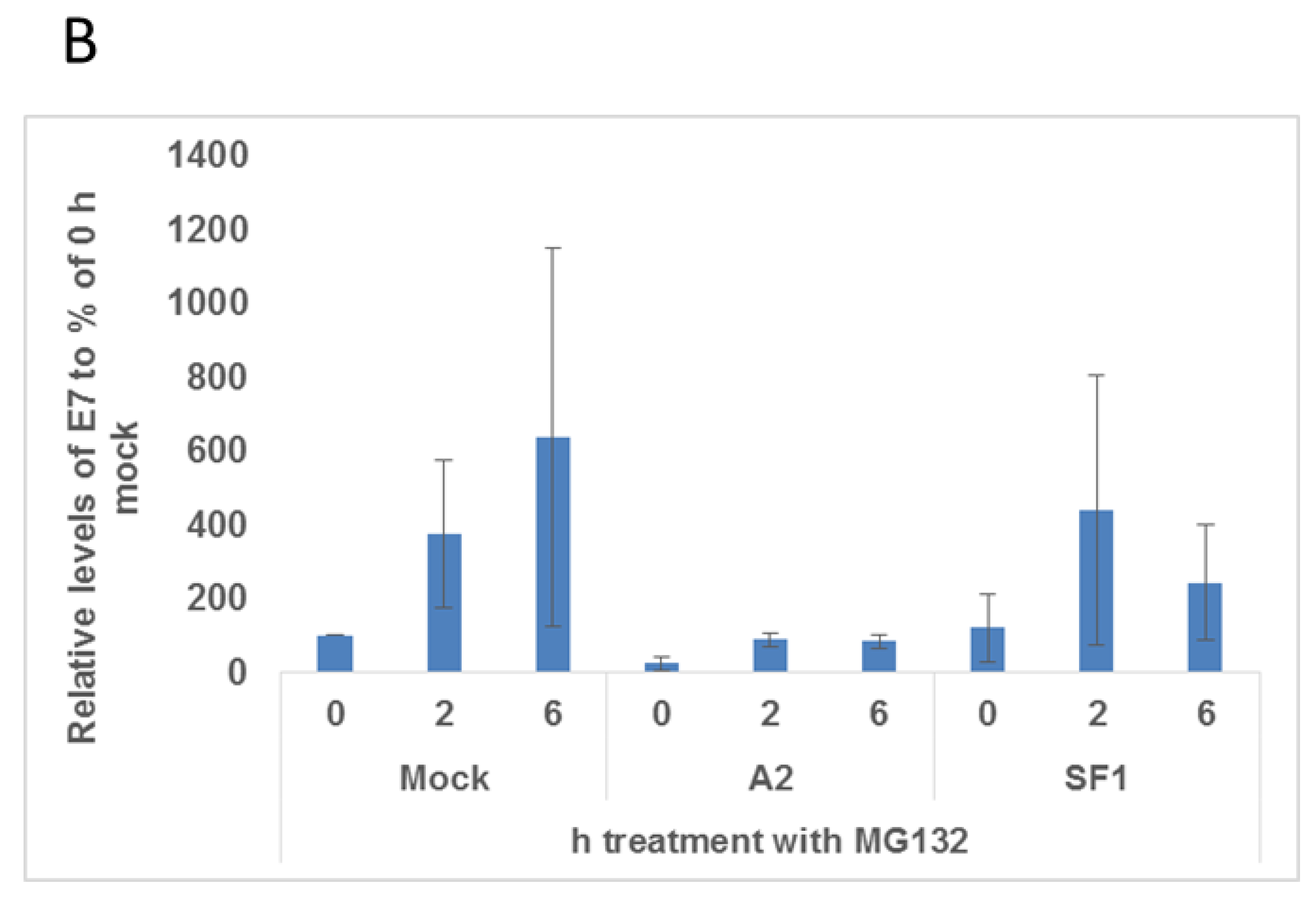
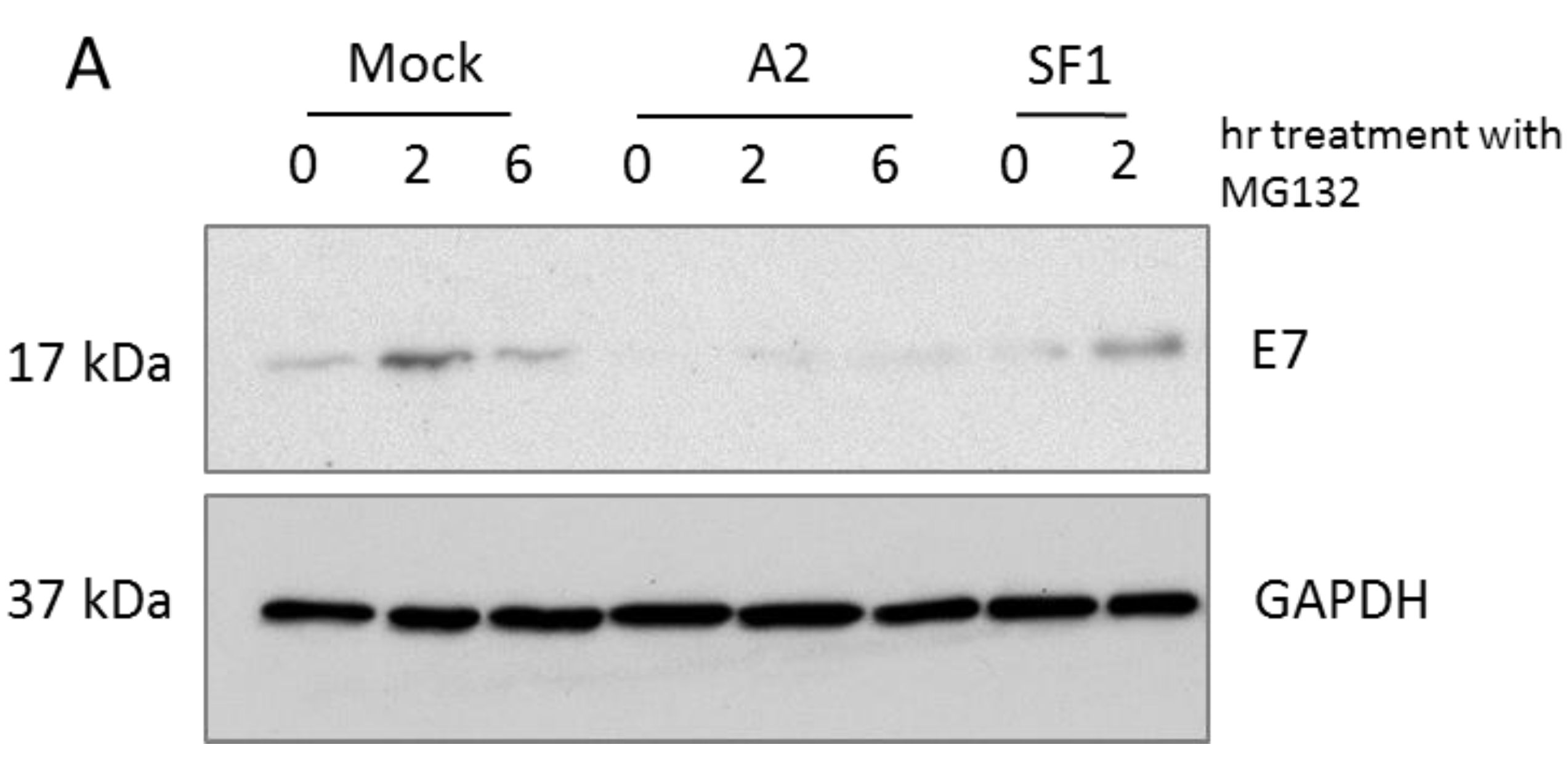
3.1. A2-Mediated Degradation of E7 Is Not Mediated via Proteasomal Pathways

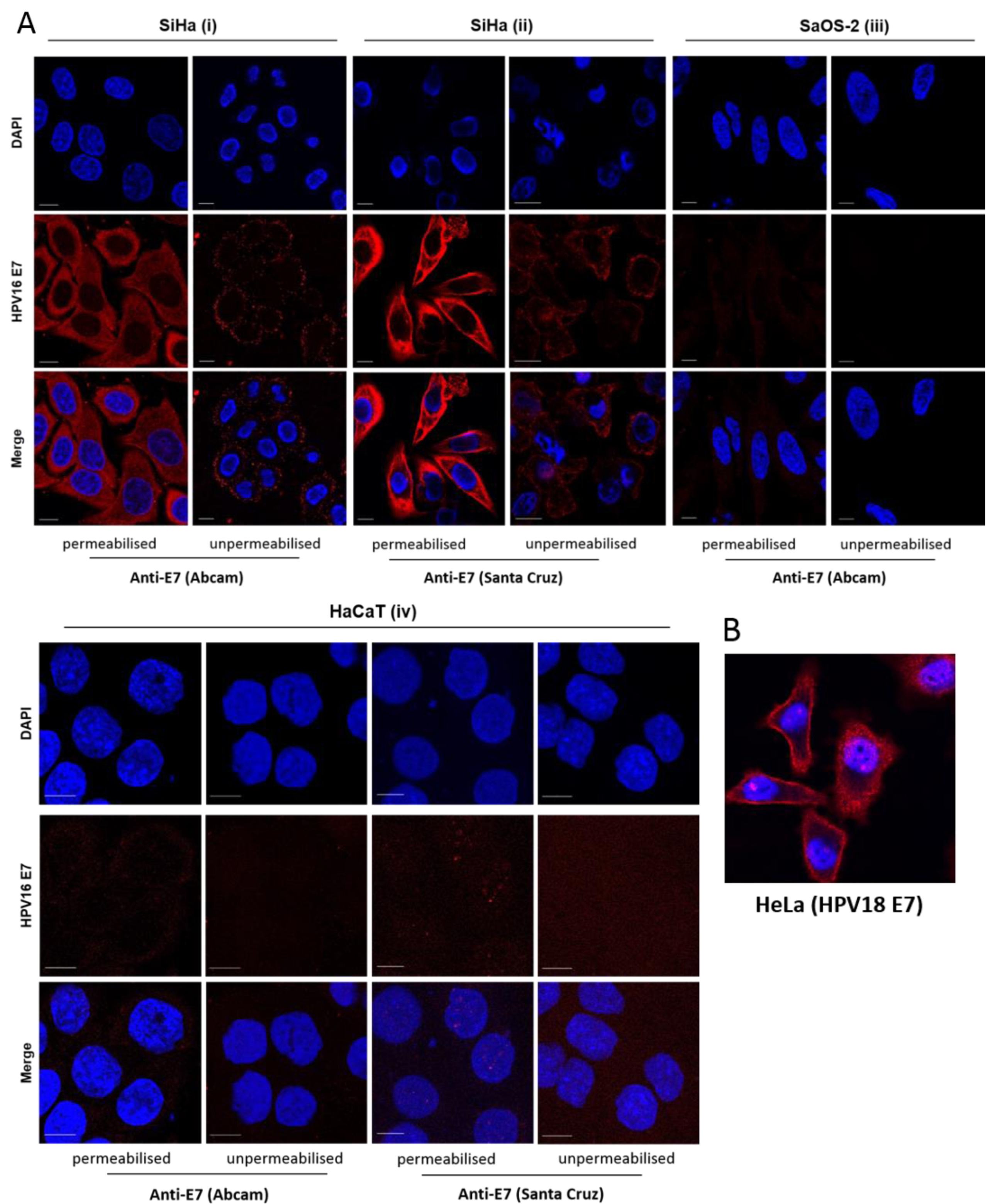
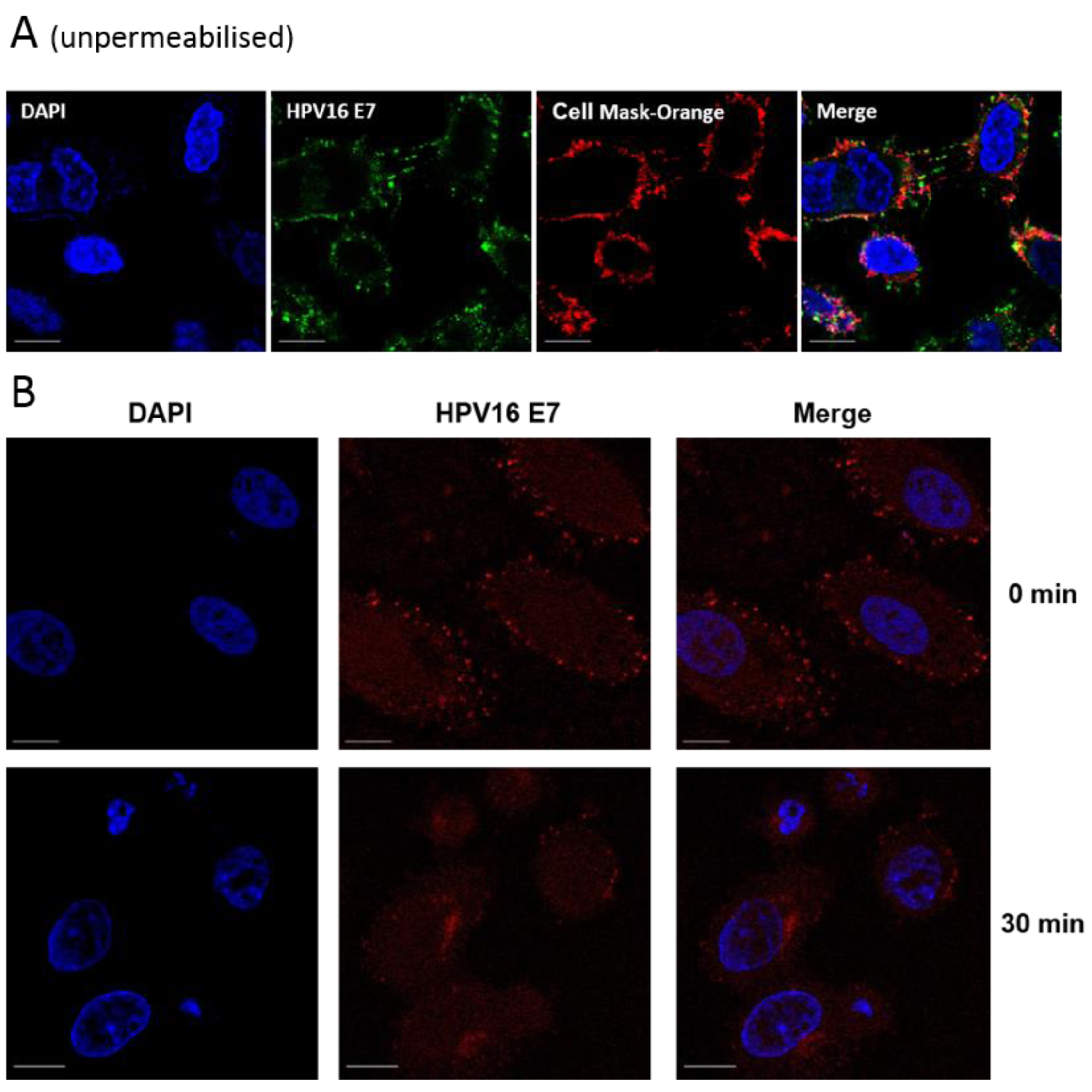
3.2. E7 Localises to the Plasma Membrane

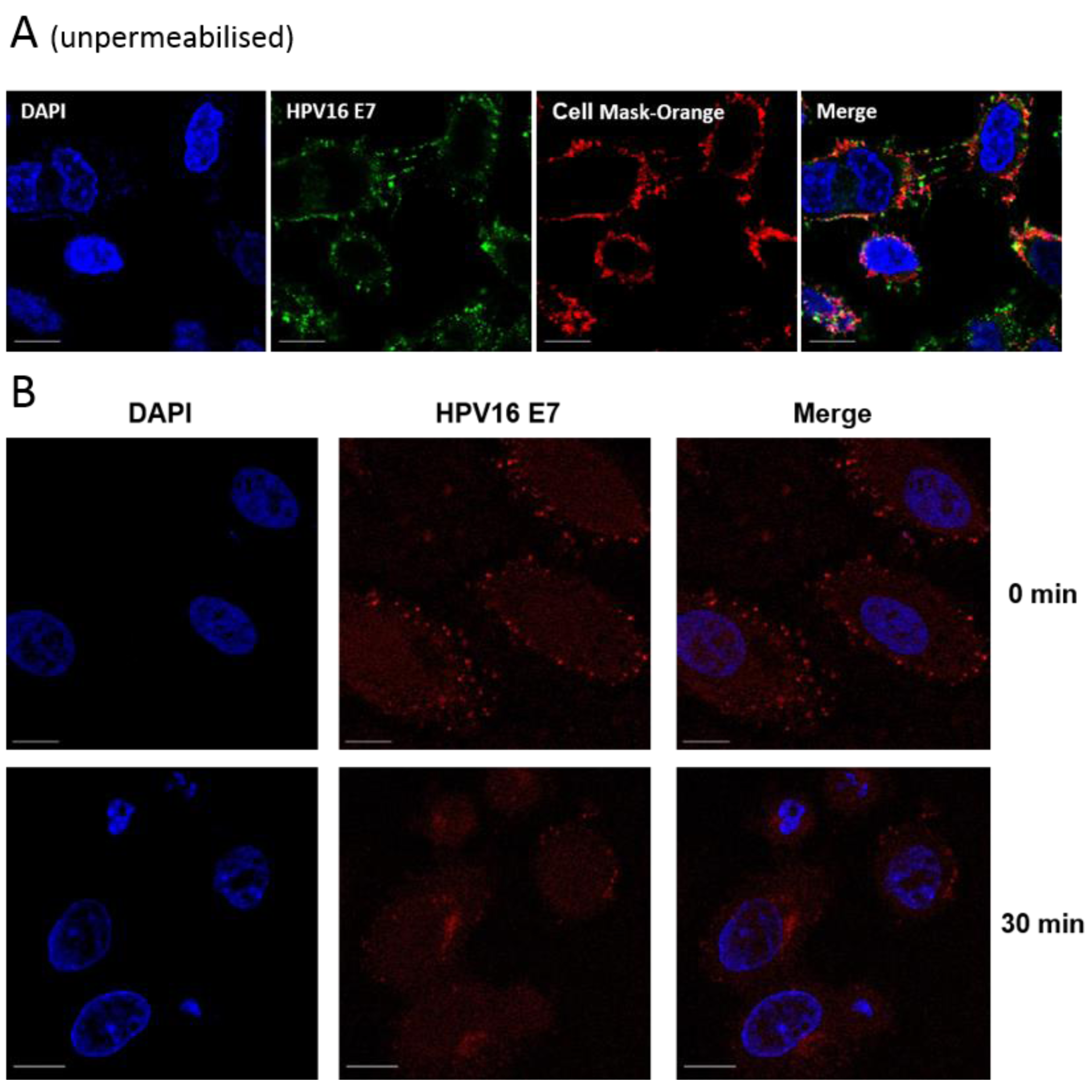
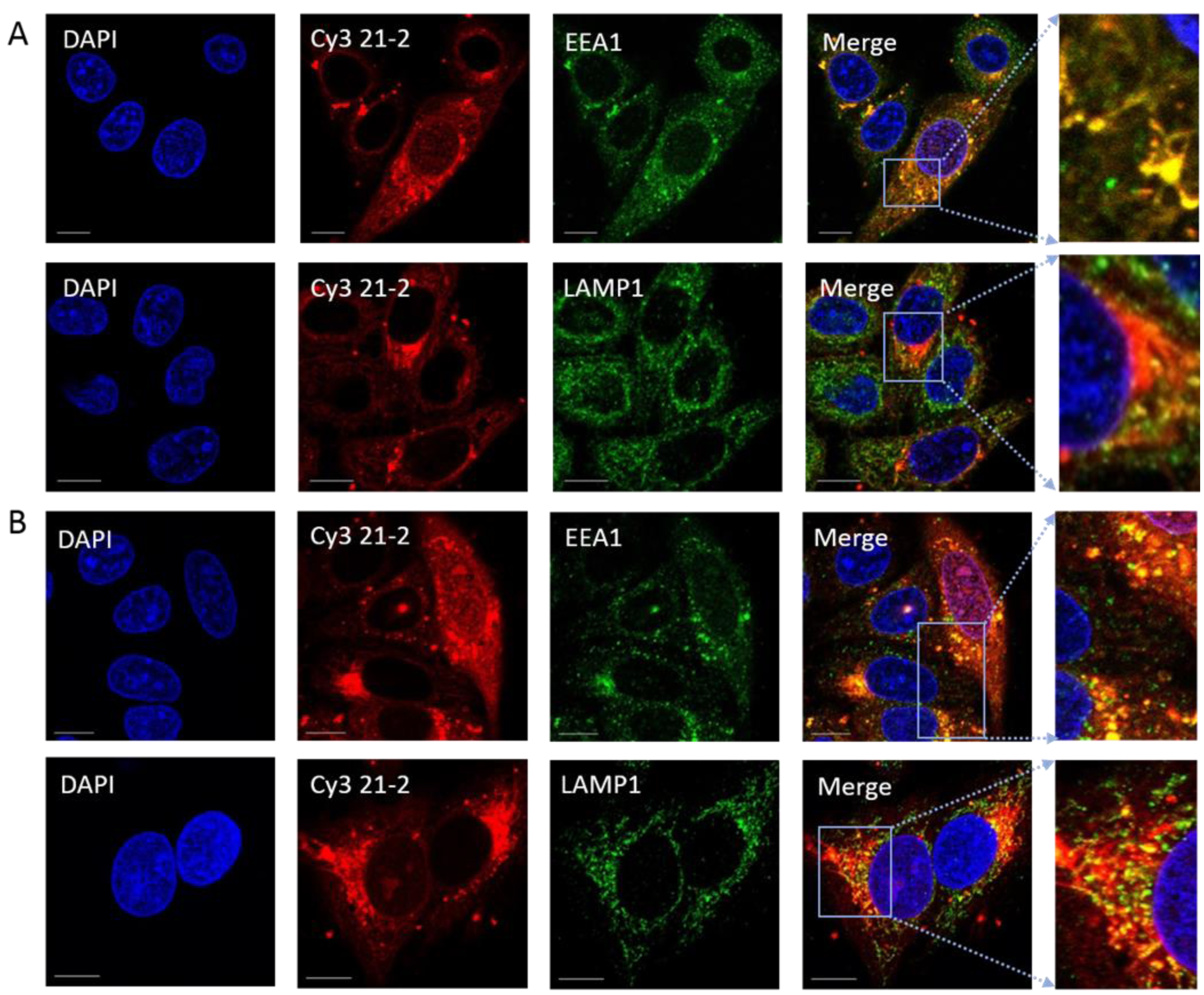
3.3. Aptamers Localise to Early/Late Endosomes upon Transfection


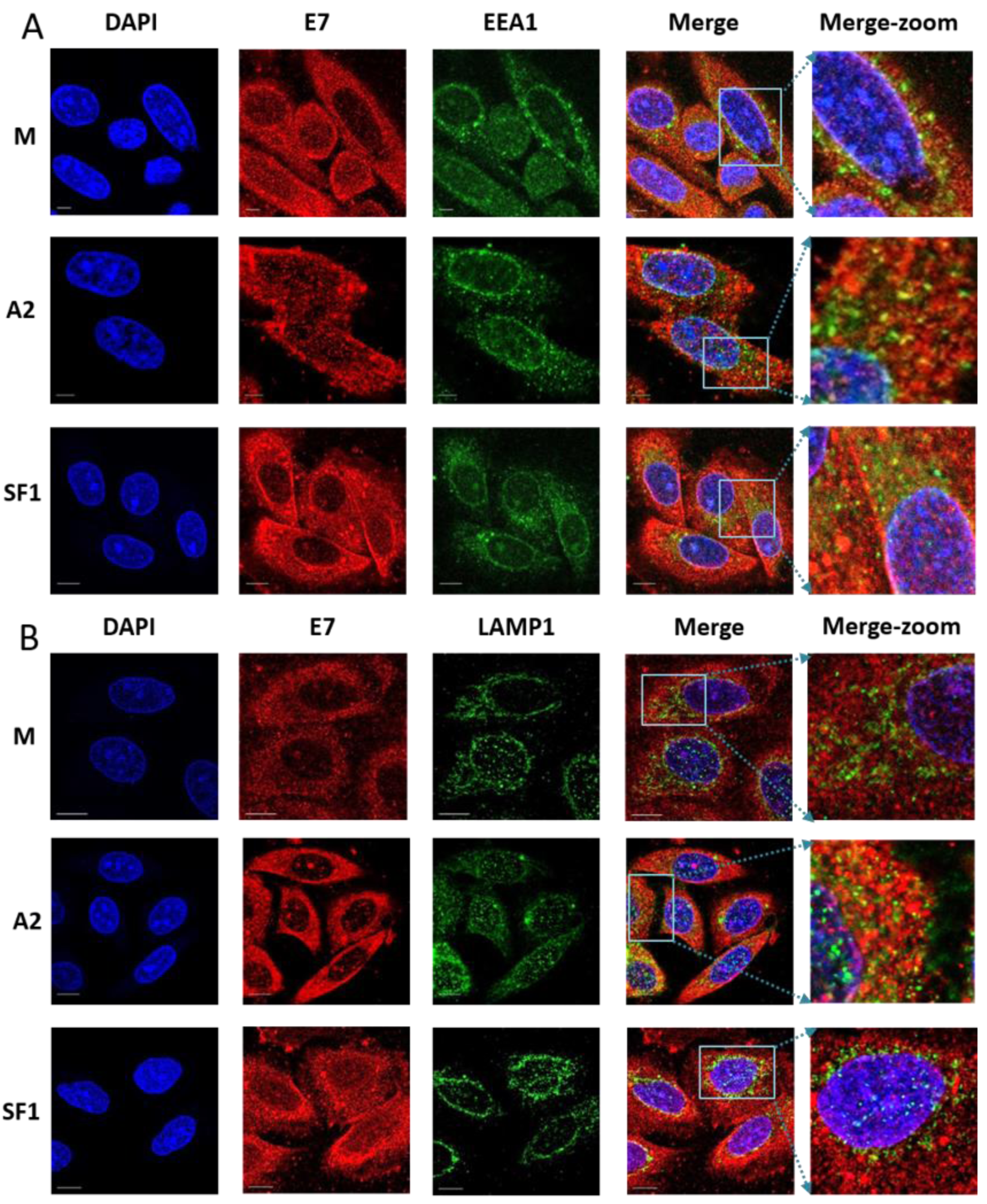
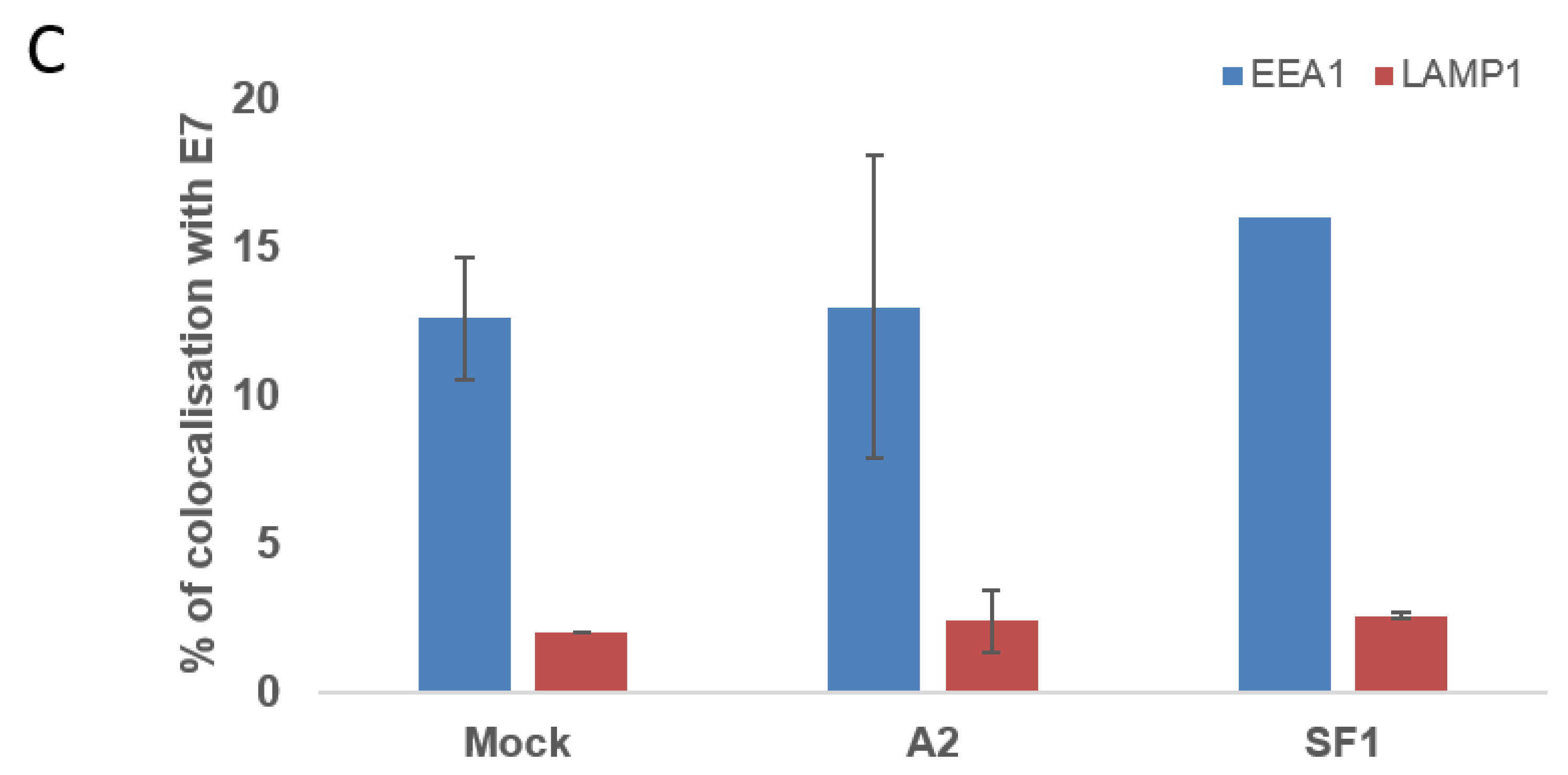
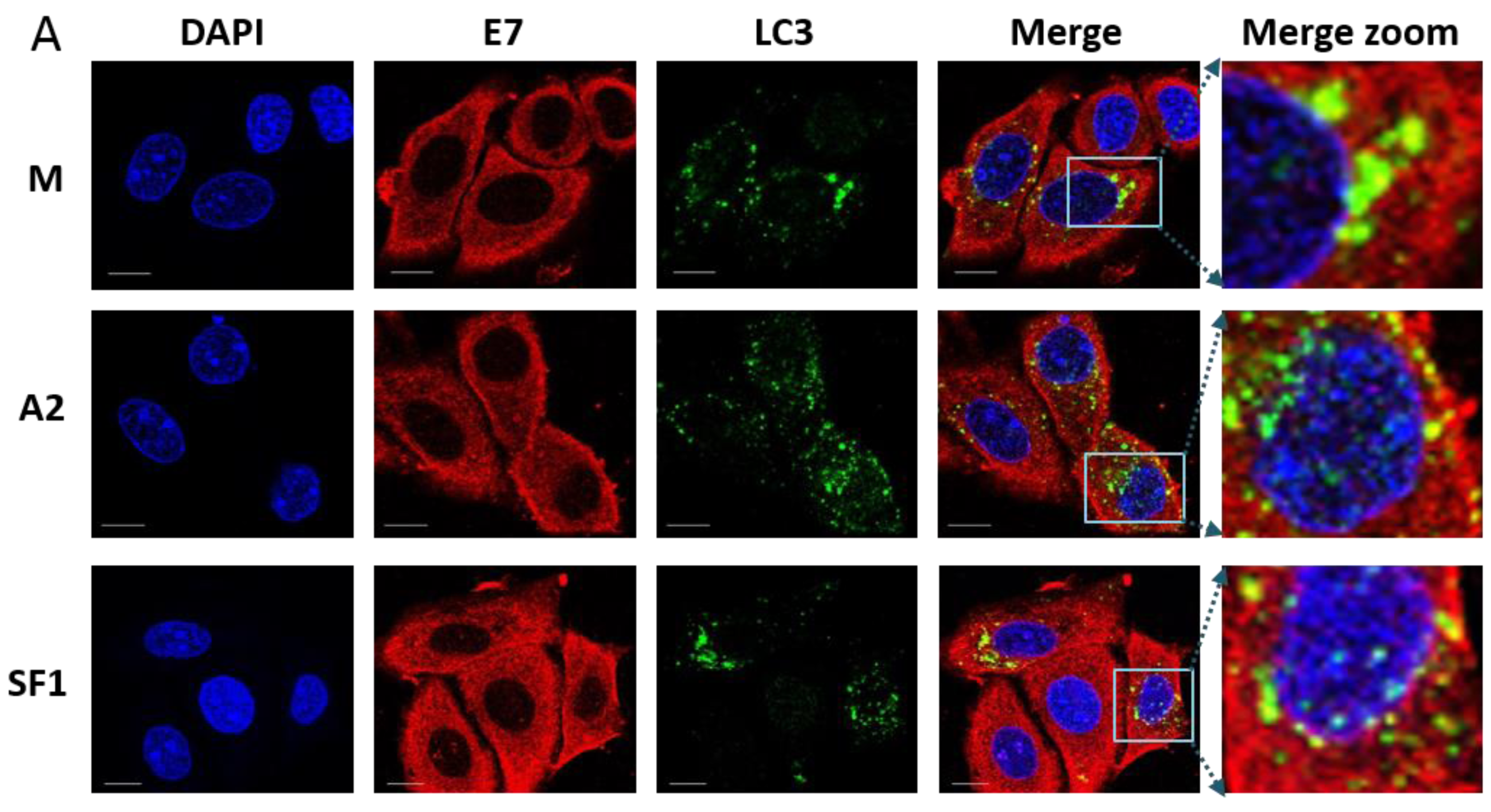
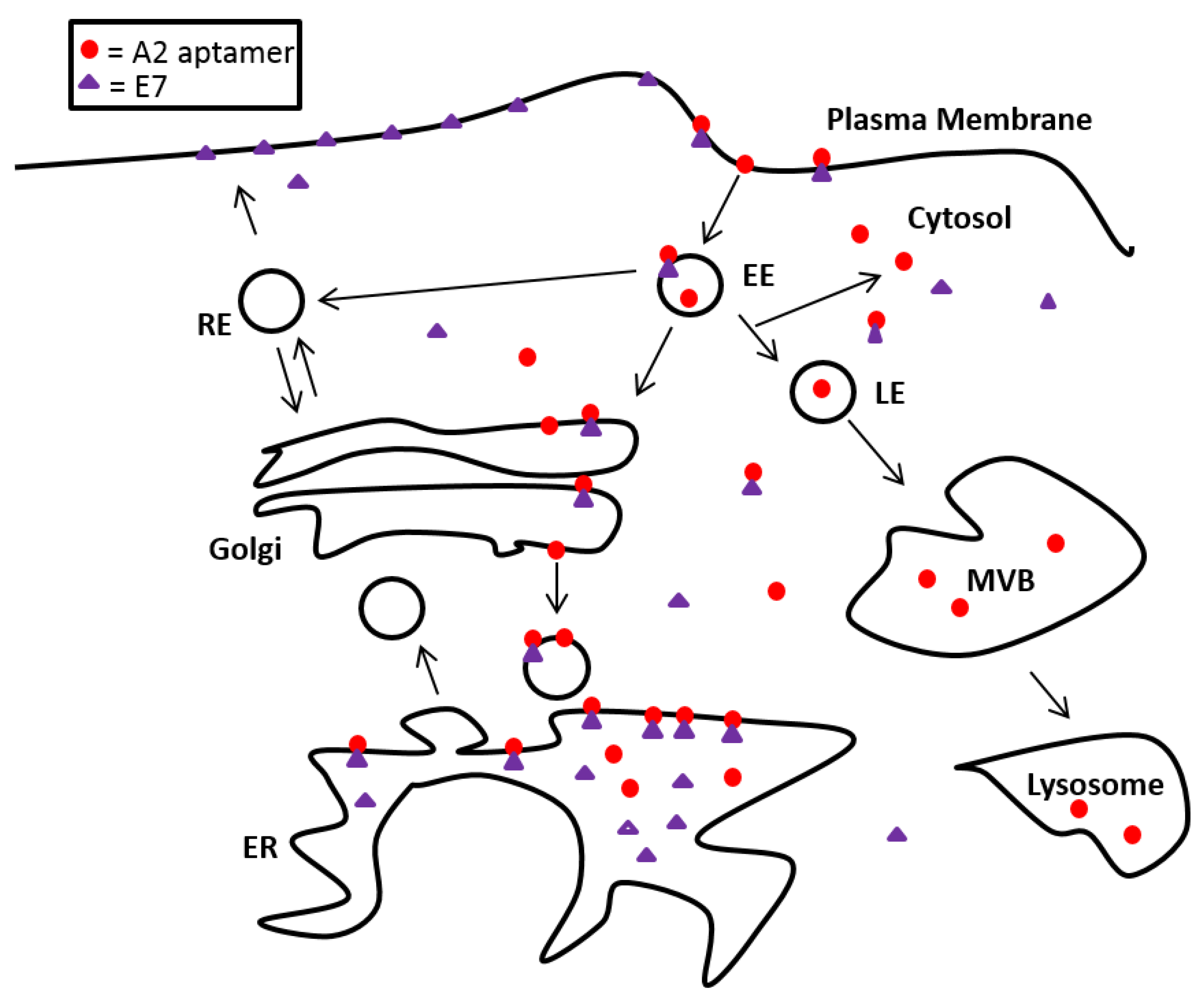
3.4. Localisation of E7 in Cellular Compartments upon Aptamer Transfection




4. Discussion

5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- PaVE: Papilloma Virus Genome Database. Available online: http://pave.niaid.nih.gov/#home (accessed on 23 June 2015).
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Naucler, P.; Mabota da Costa, F.; da Costa, J.L.; Ljungberg, O.; Bugalho, A.; Dillner, J. Human papillomavirus type-specific risk of cervical cancer in a population with high human immunodeficiency virus prevalence: Case-control study. J. Gen. Virol. 2011, 92, 2784–2791. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.X.; Lorincz, A.; Muñoz, N.; Meijer, C.J.; Shah, K.V. The causal relation between human papillomavirus and cervical cancer. J. Clin. Pathol. 2002, 55, 244–265. [Google Scholar] [CrossRef] [PubMed]
- Human papillomavirus vaccines: WHO position paper, October 2014-Recommendations. Vaccine. [CrossRef]
- McLaughlin-Drubin, M.E.; Munger, K. The human papillomavirus E7 oncoprotein. Virology 2009, 384, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Clements, A.; Zhao, K.; Marmorstein, R. Structure of the human Papillomavirus E7 oncoprotein and its mechanism for inactivation of the retinoblastoma tumor suppressor. J. Biol. Chem. 2006, 281, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Ohlenschlager, O.; Seiboth, T.; Zengerling, H.; Briese, L.; Marchanka, A.; Ramachandran, R.; Baum, M.; Korbas, M.; Meyer-Klaucke, W.; Dürst, M.; et al. Solution structure of the partially folded high-risk human papilloma virus 45 oncoprotein E7. Oncogene 2006, 25, 5953–5959. [Google Scholar] [CrossRef]
- McIntyre, M.C.; Frattini, M.G.; Grossman, S.R.; Laimins, L.A. Human papillomavirus type 18 E7 protein requires intact Cys-X-X-Cys motifs for zinc binding, dimerization, and transformation but not for Rb binding. J. Virol. 1993, 67, 3142–3150. [Google Scholar] [PubMed]
- Clements, A.; Johnston, K.; Mazzarelli, J.M.; Ricciardi, R.P.; Marmorstein, R. Oligomerization properties of the viral oncoproteins adenovirus E1A and human papillomavirus E7 and their complexes with the retinoblastoma protein. Biochemistry 2000, 39, 16033–16045. [Google Scholar] [CrossRef] [PubMed]
- Alonso, L.G.; García-Alai, M.M.; Smal, C.; Centeno, J.M.; Iacono, R.; Castaño, E.; Gualfetti, P.; de Prat-Gay, G. The HPV16 E7 viral oncoprotein self-assembles into defined spherical oligomers. Biochemistry 2004, 43, 3310–3317. [Google Scholar] [CrossRef] [PubMed]
- Smotkin, D.; Wettstein, F.O. The major human papillomavirus protein in cervical cancers is a cytoplasmic phosphoprotein. J. Virol. 1987, 61, 1686–1689. [Google Scholar] [PubMed]
- Smith-McCune, K.; Kalman, D.; Robbins, C.; Shivakumar, S.; Yuschenkoff, L.; Bishop, J.M. Intranuclear localization of human papillomavirus 16 E7 during transformation and preferential binding of E7 to the Rb family member p130. Proc. Natl. Acad. Sci. USA 1999, 96, 6999–7004. [Google Scholar] [CrossRef] [PubMed]
- Zatsepina, O.; Braspenning, J.; Robberson, D.; Hajibagheri, M.A.; Blight, K.J.; Ely, S.; Hibma, M.; Spitkovsky, D.; Trendelenburg, M.; Crawford, L.; et al. The human papillomavirus type 16 E7 protein is associated with the nucleolus in mammalian and yeast cells. Oncogene 1997, 14, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Dantur, K.; Alonso, L.; Castaño, E.; Morelli, L.; Centeno-Crowley, J.M.; Vighi, S.; de Prat-Gay, G. Cytosolic accumulation of HPV16 E7 oligomers supports different transformation routes for the prototypic viral oncoprotein: The amyloid-cancer connection. Int. J. Cancer 2009, 125, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- Knapp, A.A.; McManus, P.M.; Bockstall, K.; Moroianu, J. Identification of the nuclear localization and export signals of high risk HPV16 E7 oncoprotein. Virology 2009, 383, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Laurson, J.; Raj, K. Localisation of human papillomavirus 16 E7 oncoprotein changes with cell confluence. PLoS ONE 2011, 6, e21501. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Cload, S.T. SELEX with modified nucleotides. Curr. Opin. Chem. Biol. 2008, 12, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Blind, M.; Blank, M. Aptamer Selection Technology and Recent Advances. Mol. Ther. Nucleic Acids 2015, 4, e223. [Google Scholar] [CrossRef]
- Belyaeva, T.A.; Nicol, C.; Cesur, O.; Travé, G.; Blair, G.E.; Stonehouse, N.J. An RNA Aptamer Targets the PDZ-Binding Motif of the HPV16 E6 Oncoprotein. Cancers 2014, 6, 1553–1569. [Google Scholar] [CrossRef] [PubMed]
- Nicol, C.; Bunka, D.H.; Blair, G.E.; Stonehouse, N.J. Effects of single nucleotide changes on the binding and activity of RNA aptamers to human papillomavirus 16 E7 oncoprotein. Biochem. Biophys. Res. Commun. 2011, 405, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Nicol, C.; Cesur, Ö.; Forrest, S.; Belyaeva, T.A.; Bunka, D.H.; Blair, G.E.; Stonehouse, N.J. An RNA aptamer provides a novel approach for the induction of apoptosis by targeting the HPV16 E7 oncoprotein. PLoS ONE 2013, 8, e64781. [Google Scholar] [CrossRef] [PubMed]
- Ellingham, M.; Bunka, D.H.; Rowlands, D.J.; Stonehouse, N.J. Selection and characterization of RNA aptamers to the RNA-dependent RNA polymerase from foot-and-mouth disease virus. RNA 2006, 12, 1970–1979. [Google Scholar] [CrossRef] [PubMed]
- Forrest, S.; Lear, Z.; Herod, M.R.; Ryan, M.; Rowlands, D.J.; Stonehouse, N.J. Inhibition of the foot-and-mouth disease virus subgenomic replicon by RNA aptamers. J. Gen. Virol. 2014, 95, 2649–2657. [Google Scholar] [CrossRef] [PubMed]
- Bellecave, P.; Cazenave, C.; Rumi, J.; Staedel, C.; Cosnefroy, O.; Andreola, M.L.; Ventura, M.; Tarrago-Litvak, L.; Astier-Gin, T. Inhibition of hepatitis C virus (HCV) RNA polymerase by DNA aptamers: Mechanism of inhibition of in vitro RNA synthesis and effect on HCV-infected cells. Antimicrob. Agents Chemother. 2008, 52, 2097–2110. [Google Scholar] [CrossRef] [PubMed]
- Biroccio, A.; Hamm, J.; Incitti, I.; De Francesco, R.; Tomei, L. Selection of RNA aptamers that are specific and high-affinity ligands of the hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 2002, 76, 3688–3696. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.J.; Kahlon, J.B.; Thomas, S.D.; Trent, J.O.; Miller, D.M. Antiproliferative activity of G-rich oligonucleotides correlates with protein binding. J. Biol. Chem. 1999, 274, 26369–26377. [Google Scholar] [CrossRef] [PubMed]
- Sayyed, S.G.; Hägele, H.; Kulkarni, O.P.; Endlich, K.; Segerer, S.; Eulberg, D.; Klussmann, S.; Anders, H.J. Podocytes produce homeostatic chemokine stromal cell-derived factor-1/CXCL12, which contributes to glomerulosclerosis, podocyte loss and albuminuria in a mouse model of type 2 diabetes. Diabetologia 2009, 52, 2445–2454. [Google Scholar] [CrossRef] [PubMed]
- Doble, R.; McDermott, M.F.; Cesur, O.; Stonehouse, N.J.; Wittmann, M. IL-17A RNA aptamer: Possible therapeutic potential in some cells, more than we bargained for in others? J. Investig. Dermatol. 2014, 134, 852–855. [Google Scholar] [CrossRef] [PubMed]
- Nafissi, N.; Alqawlaq, S.; Lee, E.A.; Foldvari, M.; Spagnuolo, P.A.; Slavcev, R.A. DNA ministrings: Highly safe and effective gene delivery vectors. Mol. Ther. Nucleic Acids 2014, 3, e165. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F.; La Frazia, S.; Chiappa, L.; Ciucci, A.; Santoro, M.G. Thiazolides, a new class of anti-influenza molecules targeting viral hemagglutinin at the post-translational level. J. Biol. Chem. 2009, 284, 29798–29808. [Google Scholar] [CrossRef] [PubMed]
- Mankouri, J.; Taneja, T.K.; Smith, A.J.; Ponnambalam, S.; Sivaprasadarao, A. Kir6.2 mutations causing neonatal diabetes prevent endocytosis of ATP-sensitive potassium channels. EMBO J. 2006, 25, 4142–4151. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, I.; Nickerson, J.; Penman, S.; Stanley, M. Human papillomavirus 16 E7 protein is associated with the nuclear matrix. Proc. Natl. Acad. Sci. USA 1991, 88, 11217–11221. [Google Scholar] [CrossRef] [PubMed]
- Reinstein, E.; Scheffner, M.; Oren, M.; Ciechanover, A.; Schwartz, A. Degradation of the E7 human papillomavirus oncoprotein by the ubiquitin-proteasome system: Targeting via ubiquitination of the N-terminal residue. Oncogene 2000, 19, 5944–5950. [Google Scholar] [CrossRef] [PubMed]
- Dominska, M.; Dykxhoorn, D.M. Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 2010, 123, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Valdovinos-Torres, H.; Orozco-Morales, M.; Pedroza-Saavedra, A.; Padilla-Noriega, L.; Esquivel-Guadarrama, F.; Gutierrez-Xicotencatl, L. Different Isoforms of HPV-16 E7 Protein are Present in Cytoplasm and Nucleus. Open Virol. J. 2008, 2, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, R.; Stern, P.; Diehn, M.; Shamu, C.; Osario, M.; Zúñiga, M.; Brown, P.O.; Ploegh, H. Degradation of proteins from the ER of S. cerevisiae requires an intact unfolded protein response pathway. Mol. Cell 2000, 5, 729–735. [Google Scholar] [CrossRef]
- Liu, C.Y.; Kaufman, R.J. The unfolded protein response. J. Cell Sci. 2003, 116, 1861–1862. [Google Scholar] [CrossRef] [PubMed]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cesur, Ö.; Nicol, C.; Groves, H.; Mankouri, J.; Blair, G.E.; Stonehouse, N.J. The Subcellular Localisation of the Human Papillomavirus (HPV) 16 E7 Protein in Cervical Cancer Cells and Its Perturbation by RNA Aptamers. Viruses 2015, 7, 3443-3461. https://doi.org/10.3390/v7072780
Cesur Ö, Nicol C, Groves H, Mankouri J, Blair GE, Stonehouse NJ. The Subcellular Localisation of the Human Papillomavirus (HPV) 16 E7 Protein in Cervical Cancer Cells and Its Perturbation by RNA Aptamers. Viruses. 2015; 7(7):3443-3461. https://doi.org/10.3390/v7072780
Chicago/Turabian StyleCesur, Özlem, Clare Nicol, Helen Groves, Jamel Mankouri, George Eric Blair, and Nicola J. Stonehouse. 2015. "The Subcellular Localisation of the Human Papillomavirus (HPV) 16 E7 Protein in Cervical Cancer Cells and Its Perturbation by RNA Aptamers" Viruses 7, no. 7: 3443-3461. https://doi.org/10.3390/v7072780
APA StyleCesur, Ö., Nicol, C., Groves, H., Mankouri, J., Blair, G. E., & Stonehouse, N. J. (2015). The Subcellular Localisation of the Human Papillomavirus (HPV) 16 E7 Protein in Cervical Cancer Cells and Its Perturbation by RNA Aptamers. Viruses, 7(7), 3443-3461. https://doi.org/10.3390/v7072780