
Metaviromics of Namib Desert Salt Pans: A Novel Lineage of Haloarchaeal Salterproviruses and a Rich Source of ssDNA Viruses



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing

2.2. Library Construction and Sequencing
2.3. Raw Read Processing and Assembly
2.4. In Silico Analyses
3. Results and Discussion
3.1. The Hosabes and Eisfeld Playas
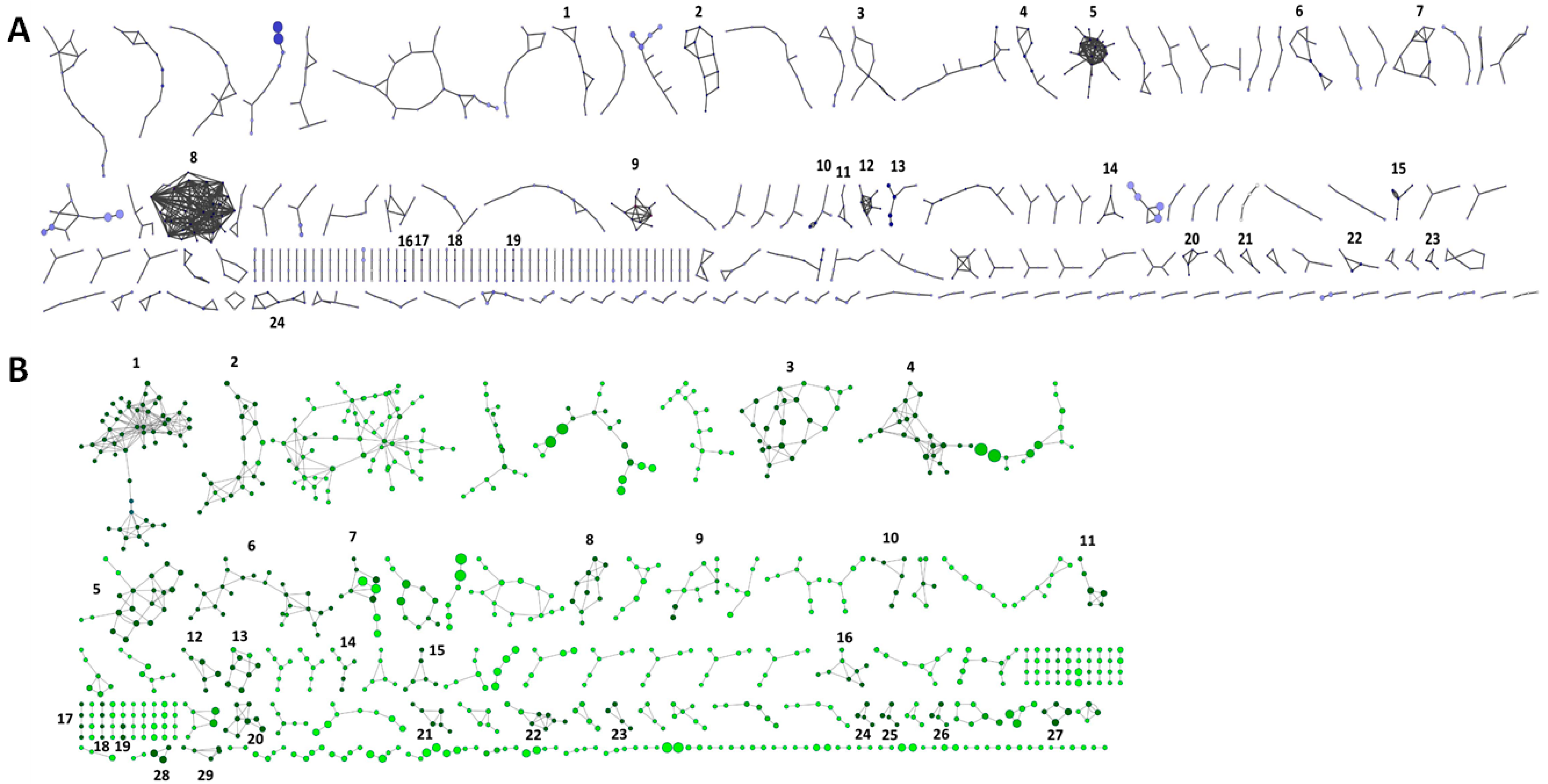
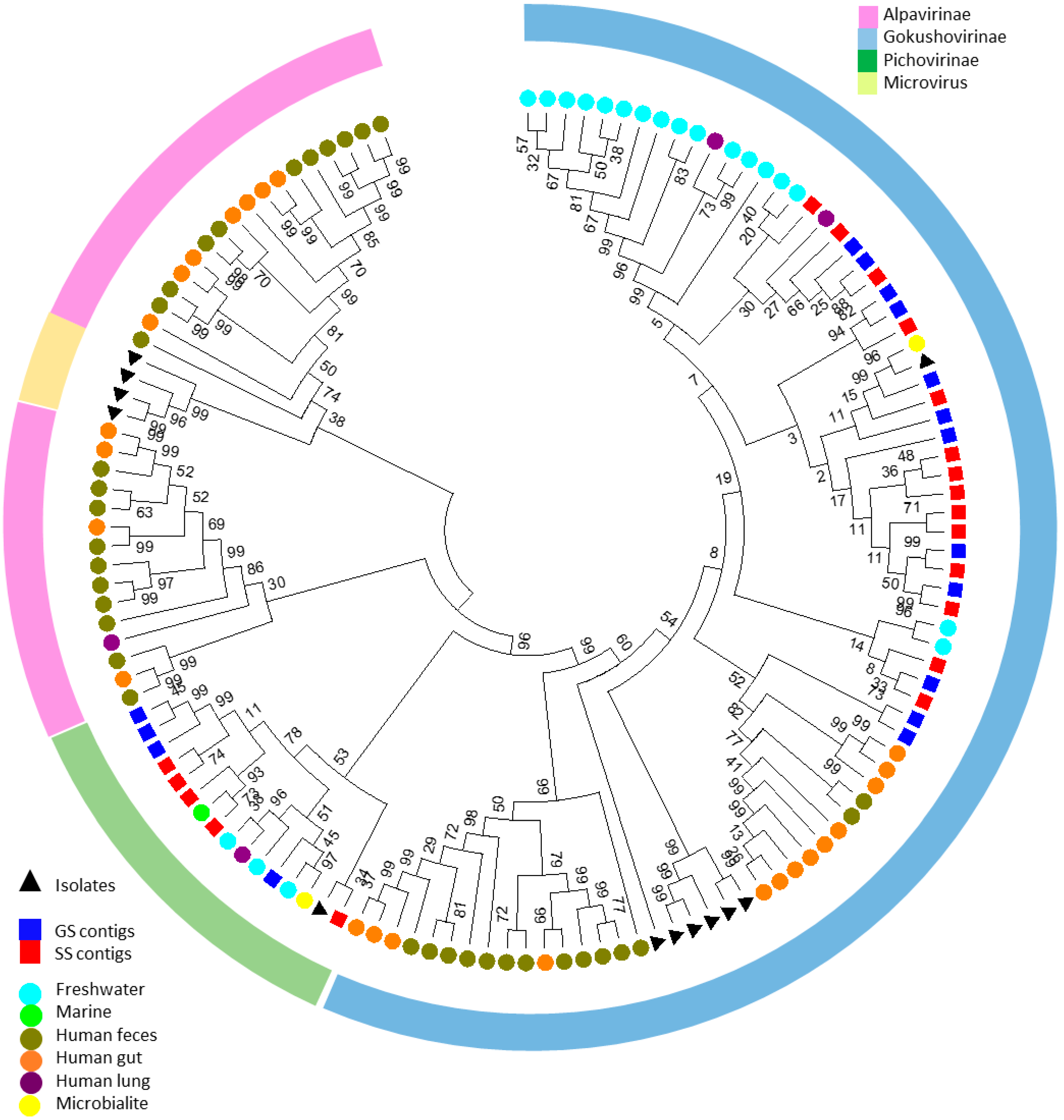
3.2. Identification of Unknown Contigs and Reads Shows an Extended Range of ssDNA Viruses with a High Prevalence of Members of the Microviridae Family

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxonomic Breakdown 1 | Gobabeb Saline (GS) | Swakopmund Saline (SS) |
|---|---|---|
| Unknown reads (MetaVir) | 95% | 92% |
| Unknown contigs (MetaVir) | 80% | 79% |
| ORFans (VIROME) | 47% | 48% |
| Viral metagenomic ORFs (VIROME) | 7% | 9% |
| Microbial metagenomic ORFs (VIROME) | 3% | 5% |
| ORFs designated functional proteins (VIROME) | 31% | 30% |
| ORFs designated unassigned proteins (VIROME) | 11% | 8% |

| Network 1 | Number of Contigs 2 | Maximum Contig Coverage | Average Network Coverage | Putative Taxonomic Assignment of Network | Marker Genes Present |
|---|---|---|---|---|---|
| 1 | 5 | 1204× | 739× | unknown | - |
| 2 | 6 | 5193× | 2849× | Inoviridae | Assembly protein |
| 3 | 6 | 20,648× | 4037× | ssDNA viruses | Rep protein, capsid protein |
| 4 | 5 | 1247× | 646× | unknown | - |
| 5 | 12 | 77,280× | 24,754× | ssDNA viruses | - |
| 6 | 5 | 1083× | 430× | unicellular algae | - |
| 7 | 6 | 1070× | 562× | unknown | - |
| 8 | 15 | 90,340× | 16,676× | ssDNA viruses | Coat protein, rep protein |
| 9 | 6 | 159,565× | 40,887× | Microviridae | protein D |
| 10 | 3 | 4599× | 1568× | Gokushovirinae | VP1, replication initiation protein |
| 11 | 3 | 1660× | 885× | bacteria | - |
| 12 | 5 | 1733× | 1128× | bacteria | - |
| 13 | 3 | 1600× | 898× | Myoviridae | Integrase, thioredo×in, primase |
| 14 | 3 | 4477× | 4096× | unknown | - |
| 15 | 3 | 17,074× | 6620× | unknown | Replication initiation factor |
| 16 | 2 | 9629× | 4971× | ssDNA viruses | - |
| 17 | 2 | 73,466× | 36,764× | Geminiviridae | Coat protein |
| 18 | 2 | 72,629× | 36,423× | unknown | - |
| 19 | 2 | 28,306× | 16,364× | unknown | - |
| 20 | 3 | 3887× | 2776× | unknown | - |
| 21 | 2 | 8056× | 4856× | unknown | - |
| 22 | 3 | 1011× | 522× | unknown | - |
| 23 | 2 | 10,800× | 8738× | unknown | - |
| 24 | 4 | 1909× | 992× | bacteria, fungi | - |
| Network 1 | Number of Contigs 2 | Maximum Contig Coverage | Average Network Coverage | Putative Taxonomic Assignment of Network | Marker Genes Present |
|---|---|---|---|---|---|
| 1 | 27 | 445,272× | 49,366× | ssDNA viruses | Rep protein |
| 2 | 11 | 2553× | 1327× | Gokushovirinae | Capsid protein, replication initiator, portal protein |
| 3 | 12 | 2708× | 1534× | unknown | Replication initiator domain |
| 4 | 11 | 20,653× | 6595× | ssDNA viruses | Rep protein, coat protein |
| 5 | 10 | 7461× | 3782× | bacteria | Rep protein |
| 6 | 13 | 2449× | 1261× | Gokushovirinae | Capsid protein, replication initiator |
| 7 | 5 | 1640× | 741× | Caudovirales | VirE, integrase |
| 8 | 4 | 7756× | 2660× | Circoviridae | Rep protein |
| 9 | 6 | 1055× | 412× | ssDNA viruses | Rep protein |
| 10 | 3 | 1178× | 992× | Circoviridae | Rep protein |
| 11 | 3 | 5901× | 3905× | Circoviridae | Rep protein |
| 12 | 3 | 4081× | 2463× | bacteria | - |
| 13 | 4 | 1599× | 945× | Halobacteria | Rep protein |
| 14 | 3 | 1645× | 838× | unknown | - |
| 15 | 3 | 2242× | 1207× | Circoviridae | Capsid protein |
| 16 | 4 | 1480× | 812× | unknown | - |
| 17 | 2 | 1287× | 1123× | unknown | - |
| 18 | 2 | 2230× | 1638× | unknown | - |
| 19 | 2 | 1118× | 600× | unknown | - |
| 20 | 4 | 9613× | 6416× | Inoviridae | Assembly protein |
| 21 | 3 | 1578× | 1178× | Circoviridae | Capsid protein |
| 22 | 4 | 6853× | 3898× | Circoviridae | Replication-associated protein |
| 23 | 2 | 79,472× | 49,830× | unknown | - |
| 24 | 2 | 9622× | 7527× | ssDNA viruses | Replication-associated protein |
| 25 | 2 | 17,505× | 10,399× | ssDNA viruses | Capsid protein |
| 26 | 2 | 1782× | 1208× | unknown | - |
| 27 | 2 | 5295× | 4949× | unknown | - |
| 28 | 2 | 3670× | 2825× | Cyanobacteria | - |
| 29 | 3 | 2157× | 1447× | unknown | - |
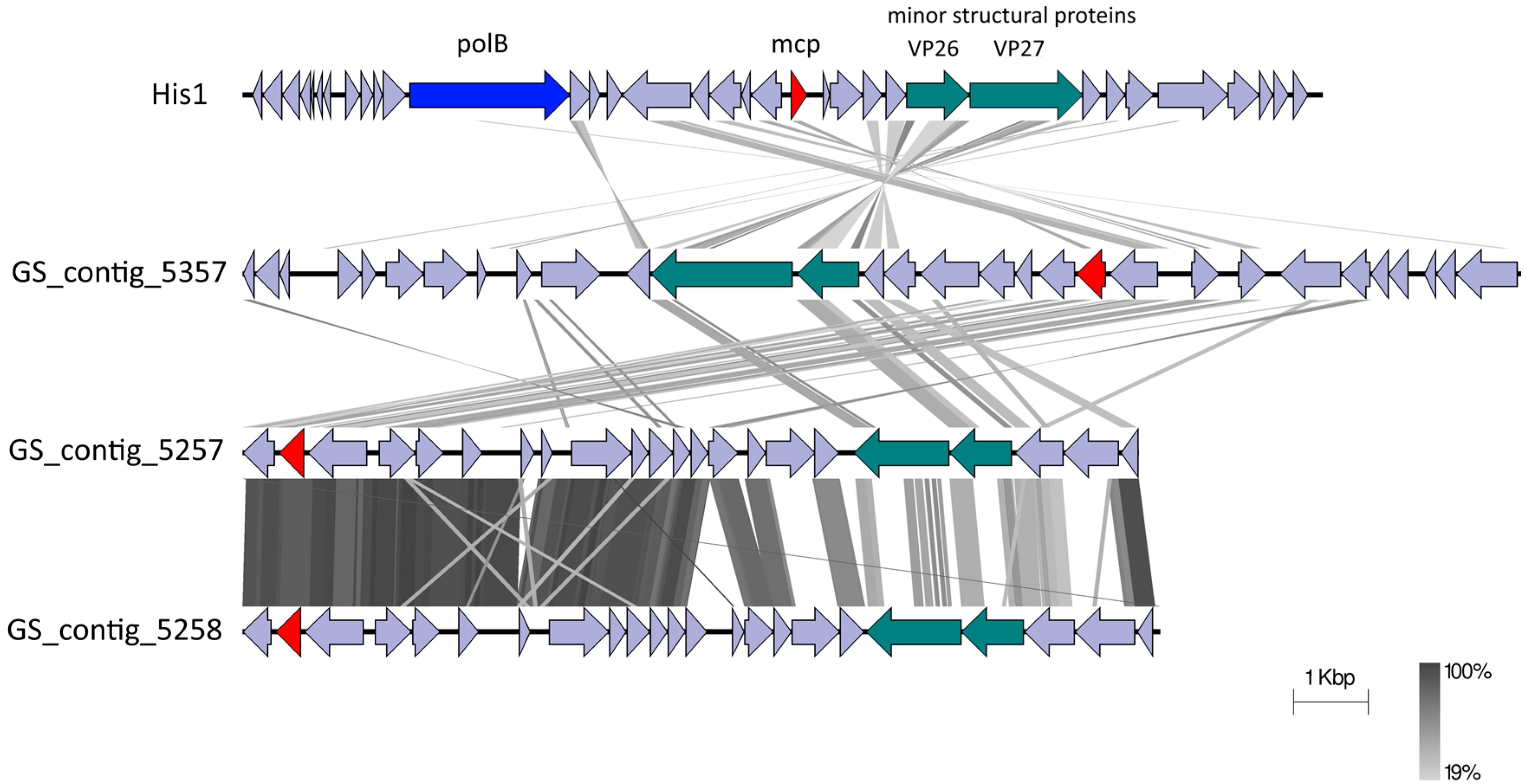
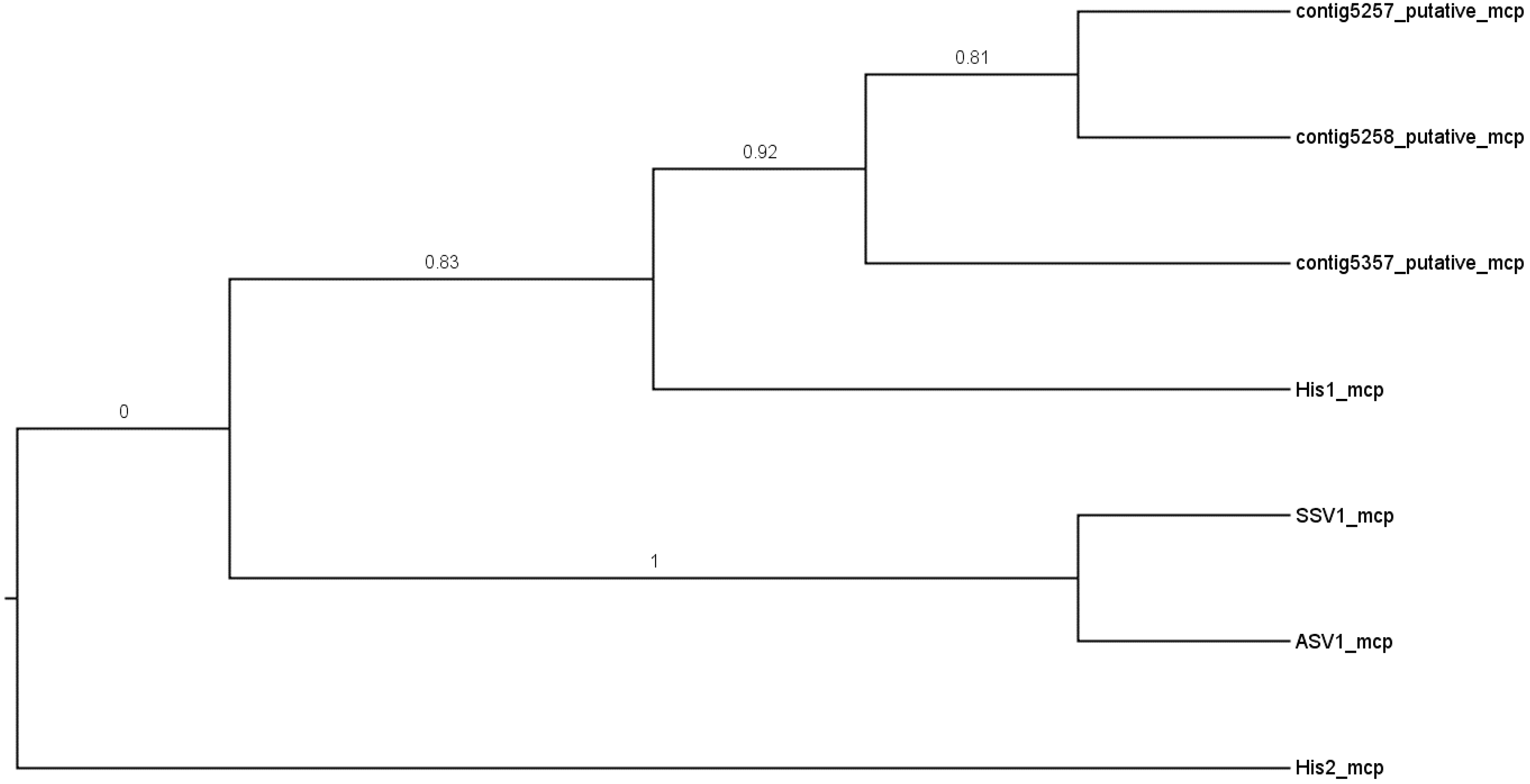
3.3. The Gobabeb Saline Site Contains Novel Haloviral Genomes Related to the Genus Salterprovirus


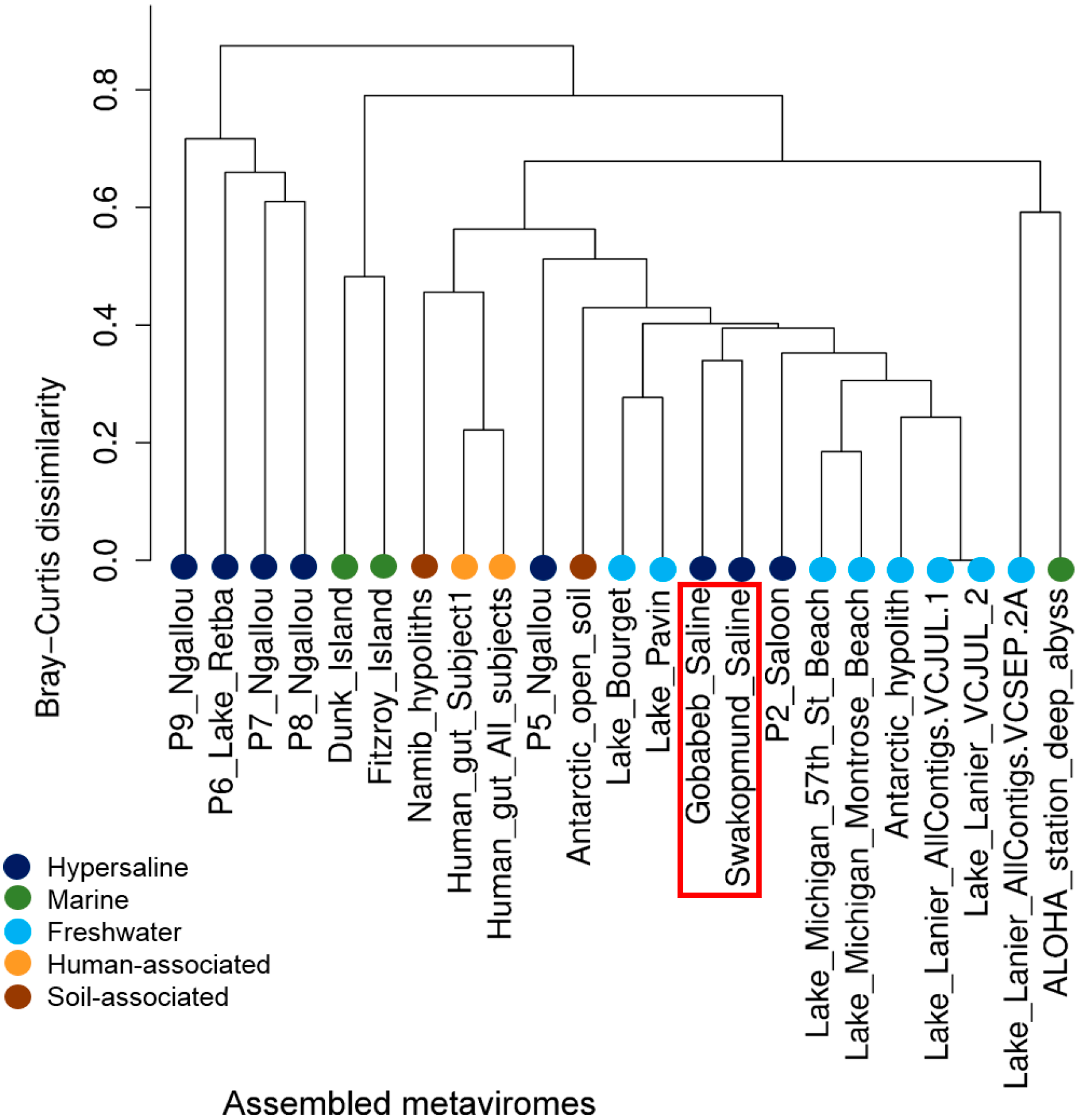
3.4. The Gobabeb and Swakopmund Saline Metaviromes are Novel and More Closely Related to Each Other Than to Other Metaviromes
3.4.1. Read Mapping

3.4.2. Comparison of the Presence/Absence of Viral Groups

4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rodriguez-Brito, B.; Li, L.; Wegley, L.; Furlan, M.; Angly, F.; Breitbart, M.; Buchanan, J.; Desnues, C.; Dinsdale, E.; Edwards, R.; et al. Viral and microbial community dynamics in four aquatic environments. ISME J. 2010, 4, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Baxter, B.K.; Mangalea, M.R.; Willcox, S.; Sabet, S.; Nagoulat, M.-N.; Griffith, J.D. Haloviruses of Great Salt Lake: A model for understanding viral diversity. In Halophiles and Hypersaline Environments; Ventosa, A., Oren, A., Ma, Y., Eds.; Springer: Berlin, Germany, 2011; pp. 173–190. [Google Scholar]
- Boujelben, I.; Yarza, P.; Almansa, C.; Villamor, J.; Maalej, S.; Antón, J.; Santos, F. Virioplankton community structure in Tunisian solar salterns. Appl. Environ. Microbiol. 2012, 78, 7429–7437. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.; Yarza, P.; Parro, V.; Briones, C.; Anton, J. The metavirome of a hypersaline environment. Environ. Microbiol. 2010, 12, 2965–2976. [Google Scholar] [CrossRef] [PubMed]
- Emerson, J.B.; Thomas, B.C.; Andrade, K.; Allen, E.E.; Heidelberg, K.B.; Banfield, J.F. Dynamic viral populations in hypersaline systems as revealed by metagenomic assembly. Appl. Environ. Microbiol. 2012, 78, 6309–6320. [Google Scholar] [CrossRef] [PubMed]
- Emerson, J.B.; Thomas, B.C.; Andrade, K.; Heidelberg, K.B.; Banfield, J.F. New approaches indicate constant viral diversity despite shifts in assemblage structure in an Australian hypersaline lake. Appl. Environ. Microbiol. 2013, 79, 6754–6764. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M. Marine Viruses: Truth or Dare. Ann. Rev. Mar. Sci. 2012, 4, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Arnold, H.P.; Zillig, W.; Ziese, U.; Holz, I.; Crosby, M.; Utterback, T.; Weidmann, J.F.; Kristjanson, J.K.; Klenk, H.P.; Nelson, K.E.; et al. A novel lipothrixvirus, SIFV, of the extremely thermophilic crenarchaeon Sulfolobus. Virology 2000, 267, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wei, D.; Wang, Y.; Zhang, X. The role of interactions between bacterial chaperone, aspartate aminotransferase, and viral protein during virus infection in high temperature environment: The interactions between bacterium and virus proteins. BMC Microbiol. 2013, 13, 48. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Ye, T.; Zhang, X. Roles of bacteriophage GVE2 endolysin in host lysis at high temperatures. Microbiology 2013, 159, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.J.; DiFrancesco, R.A.; Gowda, K.; Klingele, A.J.; Sugar, D.R.; Stocki, S.; Mead, D.A.; Schoenfeld, T.W. Thermostable DNA polymerase from a viral metagenome is a potent RT-PCR enzyme. PLoS ONE 2012, 7, e38371. [Google Scholar] [CrossRef] [PubMed]
- Plotka, M.; Kaczorowska, A.K.; Stefanska, A.; Morzywolek, A.; Fridjonsson, O.H.; Dunin-Horkawicz, S.; Kozlowski, L.; Hreggvidsson, G.O.; Kristjansson, J.K.; Dabrowski, S.; et al. Novel highly thermostable endolysin from Thermus scotoductus MAT2119 bacteriophage Ph2119 with amino acid sequence similarity to eukaryotic peptidoglycan recognition proteins. Appl. Environ. Microbiol. 2014, 80, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Sevostyanova, A.; Djordjevic, M.; Kuznedelov, K.; Naryshkina, T.; Gelfand, M.S.; Severinov, K.; Minakhin, L. Temporal regulation of viral transcription during development of Thermus thermophilus bacteriophage φYS40. J. Mol. Biol. 2007, 366, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, X. Deep-sea thermophilic Geobacillus bacteriophage GVE2 transcriptional profile and proteomic characterization of virions. Appl. Microbiol. Biotechnol. 2008, 80, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Payet, J.; Suttle, C.A. To kill or not to kill: The balance between lytic and lysogenic viral infection is driven by trophic status. Limnol. Oceanogr. 2013, 58, 465–474. [Google Scholar]
- Rice, G.; Stedman, K.; Snyder, J.; Wiedenheft, B.; Willits, D.; Brumfield, S.; McDermott, T.; Young, M.J. Viruses from extreme thermal environments. Proc. Natl. Acad. Sci. USA 2001, 98, 13341–13345. [Google Scholar] [CrossRef] [PubMed]
- Roine, E.; Oksanen, H.M. Viruses from the hypersaline environment. In Halophiles and Hypersaline Environments; Ventosa, A., Al, E., Eds.; Springer: Berlin, Germany, 2011; pp. 153–172. [Google Scholar]
- Sime-Ngando, T.; Lucas, S.; Robin, A.; Tucker, K.P.; Colombet, J.; Bettarel, Y.; Desmond, E.; Gribaldo, S.; Forterre, P.; Breitbart, M.; et al. Diversity of virus-host systems in hypersaline Lake Retba, Senegal. Environ. Microbiol. 2011, 13, 1956–1972. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, N.S.; Roine, E.; Oren, A.; Bamford, D.H.; Oksanen, H.M. Global network of specific virus-host interactions in hypersaline environments. Environ. Microbiol. 2012, 14, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Steward, G.F. Morphological characterization of viruses in the stratified water column of alkaline, hypersaline Mono Lake. Microb. Ecol. 2010, 60, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Senčilo, A.; Roine, E. A glimpse of the genomic diversity of haloarchaeal tailed viruses. Front. Microbiol. 2014, 5, 1–6. [Google Scholar]
- Pietilä, M.K.; Roine, E.; Paulin, L.; Kalkkinen, N.; Bamford, D.H. An ssDNA virus infecting archaea: A new lineage of viruses with a membrane envelope. Mol. Microbiol. 2009, 72, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Roine, E.; Kukkaro, P.; Paulin, L.; Laurinavicius, S.; Domanska, A.; Somerharju, P.; Bamford, D.H. New, closely related haloarchaeal viral elements with different nucleic acid types. J. Virol. 2010, 84, 3682–3689. [Google Scholar] [CrossRef] [PubMed]
- Senčilo, A.; Paulin, L.; Kellner, S.; Helm, M.; Roine, E. Related haloarchaeal pleomorphic viruses contain different genome types. Nucleic Acids Res. 2012, 40, 5523–5534. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.; Meyerdierks, A.; Peña, A.; Rosselló-Mora, R.; Amann, R.; Antón, J. Metagenomic approach to the study of halophages: The environmental halophage 1. Environ. Microbiol. 2007, 9, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Heredia, I.; Martin-Cuadrado, A.B.; Mojica, F.J.M.; Santos, F.; Mira, A.; Antón, J.; Rodriguez-Valera, F. Reconstructing viral genomes from the environment using fosmid clones: The case of haloviruses. PLoS ONE 2012, 7, e33802. [Google Scholar] [CrossRef] [PubMed]
- Prestel, E.; Regeard, C.; Andrews, J.; Oger, P.; DuBow, M.S. A novel bacteriophage morphotype with a ribbon-like structure at the tail extremity. Res. J. Microbiol. 2012, 7, 75–81. [Google Scholar]
- Prestel, E.; Salamitou, S.; DuBow, M.S. An examination of the bacteriophages and bacteria of the Namib Desert. J. Microbiol. 2008, 46, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; van Zyl, L.; de Maayer, P.; Rubagotti, E.; Rybicki, E.; Tuffin, M.; Cowan, D.A. Metagenomic analysis of the viral community in Namib Desert hypoliths. Environ. Microbiol. 2015, 17, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, F.D.; Drake, N.; Goudie, A.S.; White, K.; Viles, H. The role of playas in pedogenic gypsum crust formation in the Central Namib Desert: A theoretical model. Earth Surf. Process. Landf. 2001, 26, 1177–1193. [Google Scholar] [CrossRef]
- Day, J.A.; Seely, M.K. Physical and chemical conditions in an hypersaline spring in the Namib Desert. Hydrobiologia 1988, 160, 141–153. [Google Scholar] [CrossRef]
- Day, J.A. The major ion chemistry of some southern African saline systems. Hydrobiologia 1993, 267, 37–59. [Google Scholar] [CrossRef]
- Eckardt, F.D.; Drake, N. Introducing the Namib Desert playas. In Sabkha Ecosystems; Öztürk, M., Böer, B., Barth, H.-J., Clüsener-Godt, M., Khan, M.A., Breckle, S.-W., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 2011; Volume 46, pp. 19–25. [Google Scholar]
- Eckardt, F.D.; Soderberg, K.; Coop, L.J.; Muller, A.A.; Vickery, K.J.; Grandin, R.D.; Jack, C.; Kapalanga, T.S.; Henschel, J. The nature of moisture at Gobabeb, in the central Namib Desert. J. Arid Environ. 2013, 93, 7–19. [Google Scholar] [CrossRef]
- Capece, M.C.; Clark, E.; Saleh, J.K.; Halford, D.; Heinl, N.; Hoskins, S.; Rothschild, L.J. Polyextremophiles and the constraints for terrestrial habitability. In Polyextremophiles: Life under Multiple Forms of Stress; Seckbach, J., Oren, A., Stan-Lotter, H., Eds.; Springer Science + Business Media: Dordrecht, The Netherlands, 2013; pp. 7–59. [Google Scholar]
- Schoenfeld, T.; Liles, M.; Wommack, K.E.; Polson, S.W.; Godiska, R.; Mead, D. Functional viral metagenomics and the next generation of molecular tools. Trends Microbiol. 2010, 18, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, T.; Patterson, M.; Richardson, P.M.; Wommack, K.E.; Young, M.; Mead, D. Assembly of viral metagenomes from yellowstone hot springs. Appl. Environ. Microbiol. 2008, 74, 4164–4174. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 9 March 2015).
- Roux, S.; Faubladier, M.; Mahul, A.; Paulhe, N.; Bernard, A.; Debroas, D.; Enault, F. Metavir: A web server dedicated to virome analysis. Bioinformatics 2011, 27, 3074–3075. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Tournayre, J.; Mahul, A.; Debroas, D.; Enault, F. Metavir 2: New tools for viral metagenome comparison and assembled virome analysis. BMC Bioinform. 2014, 15, 76. [Google Scholar] [CrossRef] [PubMed]
- Wommack, K.E.; Bhavsar, J.; Polson, S.W.; Chen, J.; Dumas, M.; Srinivasiah, S.; Furman, M.; Jamindar, S.; Nasko, D.J. VIROME: A standard operating procedure for analysis of viral metagenome sequences. Stand. Genomic Sci. 2012, 6, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Albertsen, M. Cytoscapeviz. Available online: http://www.github.com/MadsAlbertsen/multi-metagenome/tree/master/cytoscapeviz (accessed on 15 August 2014).
- Albertsen, M.; Hugenholtz, P.; Skarshewski, A.; Nielsen, K.L.; Tyson, G.W.; Nielsen, P.H. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 2013, 31, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Taniguchi, T.; Itoh, T. MetaGeneAnnotator: Detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 2008, 15, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Park, J.; Takagi, T. MetaGene: Prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006, 34, 5623–5630. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010, 38, D142–D148. [Google Scholar]
- Metagenomes Online. Available online: http://www.metagenomesonline.org (accessed on 6 April 2015).
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2014, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Swan, B.K.; Ehrhardt, C.J.; Reifel, K.M.; Moreno, L.I.; Valentine, D.L. Archaeal and bacterial communities respond differently to environmental gradients in anoxic sediments of a California hypersaline lake, the Salton Sea. Appl. Environ. Microbiol. 2010, 76, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Martínez-García, M.; Santos, F.; Moreno-Paz, M.; Parro, V.; Antón, J. Unveiling viral–host interactions within the “microbial dark matter”. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pride, D.T.; Wassenaar, T.M.; Ghose, C.; Blaser, M.J. Evidence of host-virus co-evolution in tetranucleotide usage patterns of bacteriophages and eukaryotic viruses. BMC Genomics 2006, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. (Eds.) Virus Taxonomy, 9th ed.; Elsevier Inc.: London, UK, 2012.
- Labonté, J.M.; Suttle, C.A. Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 2013, 7, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Wearne, K.; Bridgeford, P. Coastal birds of the Namib-Naukluft Park. In Namib: Secrets of a Desert Uncovered; Seely, M., Pallett, J., Eds.; Venture Publications: Windhoek, Namibia, 2012; pp. 37–39. [Google Scholar]
- Roberts, J.W.; Devoret, R. Lysogenic Induction. Cold Spring Harb. Monogr. Arch. 1983, 13, 123–144. [Google Scholar]
- Kim, K.-H.; Bae, J.-W. Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl. Environ. Microbiol. 2011, 77, 7663–7668. [Google Scholar] [CrossRef] [PubMed]
- Marine, R.; McCarren, C.; Vorrasane, V.; Nasko, D.; Crowgey, E.; Polson, S.W.; Wommack, K.E. Caught in the middle with multiple displacement amplification: The myth of pooling for avoiding multiple displacement amplification bias in a metagenome. Microbiome 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Polson, S.W.; Wilhelm, S.W.; Wommack, K.E. Unraveling the viral tapestry (from inside the capsid out). ISME J. 2011, 5, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Krupovic, M.; Poulet, A.; Debroas, D.; Enault, F. Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS ONE 2012, 7, e40418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, M.; Kailasan, S.; Cohen, A.; Roux, S.; Tucker, K.P.; Shevenell, A.; Agbandje-McKenna, M.; Breitbart, M. Diversity of environmental single-stranded DNA phages revealed by PCR amplification of the partial major capsid protein. ISME J. 2014, 8, 2093–2103. [Google Scholar] [CrossRef] [PubMed]
- Reavy, B.; Swanson, M.M.; Cock, P.; Dawson, L.; Freitag, T.E.; Singh, B.K.; Torrance, L.; Mushegian, A.R.; Taliansky, M. Distinct circular single-stranded DNA viruses exist in different soil types. Appl. Environ. Microbiol. 2015, 81, 3934–3945. [Google Scholar] [CrossRef] [PubMed]
- Labonté, J.M.; Hallam, S.; Suttle, C.A. Previously unknown evolutionary groups dominate the ssDNA gokushoviruses in oxic and anoxic waters of a coastal marine environment. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Bryson, S.; Thurber, A.R.; Correa, A.M.S.; Orphan, V.J.; Thurber, R.V. A novel sister clade to the Enterobacteria microviruses (family Microviridae) identified in methane seep sediments. Environ. Microbiol. 2015, 17, 3708–3721. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Forterre, P. Microviridae goes temperate: Microvirus-related proviruses reside in the genomes of Bacteroidetes. PLoS ONE 2011, 6, e19893. [Google Scholar] [CrossRef] [PubMed]
- Bath, C.; Dyall-Smith, M.L. His1, an archaeal virus of the Fuselloviridae family that infects Haloarcula Hispanica. J. Virol. 1998, 72, 9392–9395. [Google Scholar] [PubMed]
- Bath, C.; Cukalac, T.; Porter, K.; Dyall-Smith, M.L. His1 and His2 are distantly related, spindle-shaped haloviruses belonging to the novel virus group, Salterprovirus. Virology 2006, 350, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Pietilä, M.K.; Atanasova, N.S.; Oksanen, H.M.; Bamford, D.H. Modified coat protein forms the flexible spindle-shaped virion of haloarchaeal virus His1. Environ. Microbiol. 2013, 15, 1674–1686. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Quemin, E.R.J.; Bamford, D.H.; Forterre, P.; Prangishvili, D. Unification of the globally distributed spindle-shaped viruses of the Archaea. J. Virol. 2014, 88, 2354–2358. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.J.; Allen, L.Z.; Lorenzi, H.A.; Fadrosh, D.W.; Brami, D.; Thiagarajan, M.; McCrow, J.P.; Tovchigrechko, A.; Yooseph, S.; Venter, J.C. Metagenomic exploration of viruses throughout the Indian Ocean. PLoS ONE 2012, 7, e42047. [Google Scholar] [CrossRef] [PubMed]
- Zablocki, O.; van Zyl, L.; Adriaenssens, E.M.; Rubagotti, E.; Tuffin, M.; Cary, S.C.; Cowan, D. High-level diversity of tailed phages, eukaryote-associated viruses and virophage-like elements in the metaviroms of Antarctic soils. Appl. Environ. Microbiol. 2014, 80, 6888–6897. [Google Scholar] [CrossRef] [PubMed]
- Holmfeldt, K.; Solonenko, N.; Shah, M.; Corrier, K.; Riemann, L.; Verberkmoes, N.C.; Sullivan, M.B. Twelve previously unknown phage genera are ubiquitous in global oceans. Proc. Natl. Acad. Sci. USA 2013, 110, 12798–12803. [Google Scholar] [CrossRef] [PubMed]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stachler, E.; Bibby, K. Metagenomic evaluation of the highly abundant human gut bacteriophage CrAssphage for source tracking of human fecal pollution. Environ. Sci. Technol. Lett. 2014, 1, 405–409. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adriaenssens, E.M.; Van Zyl, L.J.; Cowan, D.A.; Trindade, M.I. Metaviromics of Namib Desert Salt Pans: A Novel Lineage of Haloarchaeal Salterproviruses and a Rich Source of ssDNA Viruses. Viruses 2016, 8, 14. https://doi.org/10.3390/v8010014
Adriaenssens EM, Van Zyl LJ, Cowan DA, Trindade MI. Metaviromics of Namib Desert Salt Pans: A Novel Lineage of Haloarchaeal Salterproviruses and a Rich Source of ssDNA Viruses. Viruses. 2016; 8(1):14. https://doi.org/10.3390/v8010014
Chicago/Turabian StyleAdriaenssens, Evelien M., Leonardo Joaquim Van Zyl, Don A. Cowan, and Marla I. Trindade. 2016. "Metaviromics of Namib Desert Salt Pans: A Novel Lineage of Haloarchaeal Salterproviruses and a Rich Source of ssDNA Viruses" Viruses 8, no. 1: 14. https://doi.org/10.3390/v8010014
APA StyleAdriaenssens, E. M., Van Zyl, L. J., Cowan, D. A., & Trindade, M. I. (2016). Metaviromics of Namib Desert Salt Pans: A Novel Lineage of Haloarchaeal Salterproviruses and a Rich Source of ssDNA Viruses. Viruses, 8(1), 14. https://doi.org/10.3390/v8010014