
The E3 Ubiquitin Ligase TMEM129 Is a Tri-Spanning Transmembrane Protein


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Lentiviral Infection
2.2. Antibodies
2.3. Plasmids
2.4. Flow Cytometry
2.5. Immunoblotting
2.6. Immunoprecipitation
2.7. Deglycosylation Studies
3. Results
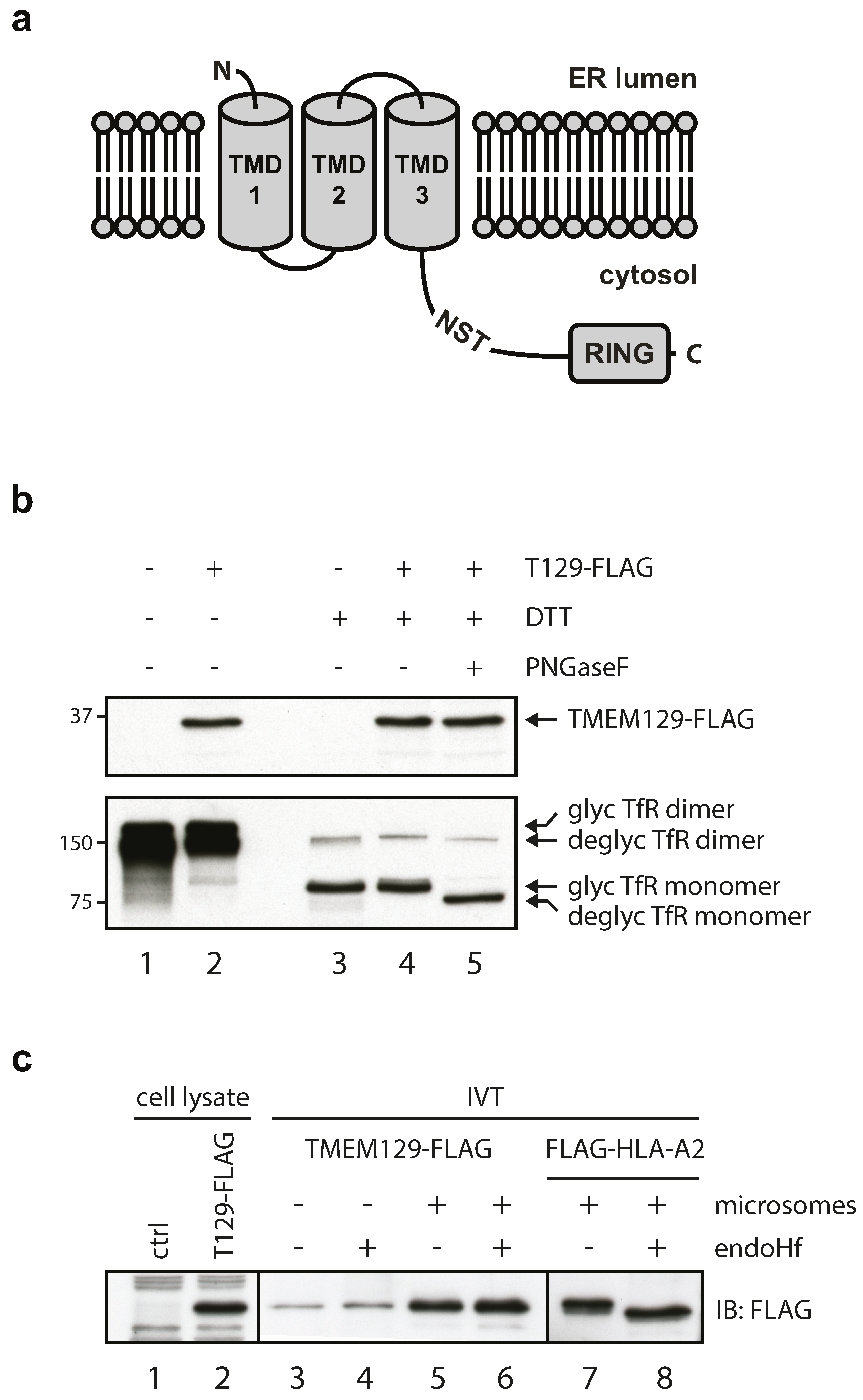
3.1. TMEM129 Is a Non-Glycosylated Protein Lacking a Cleavable Signal Sequence and Disulphide Bonds
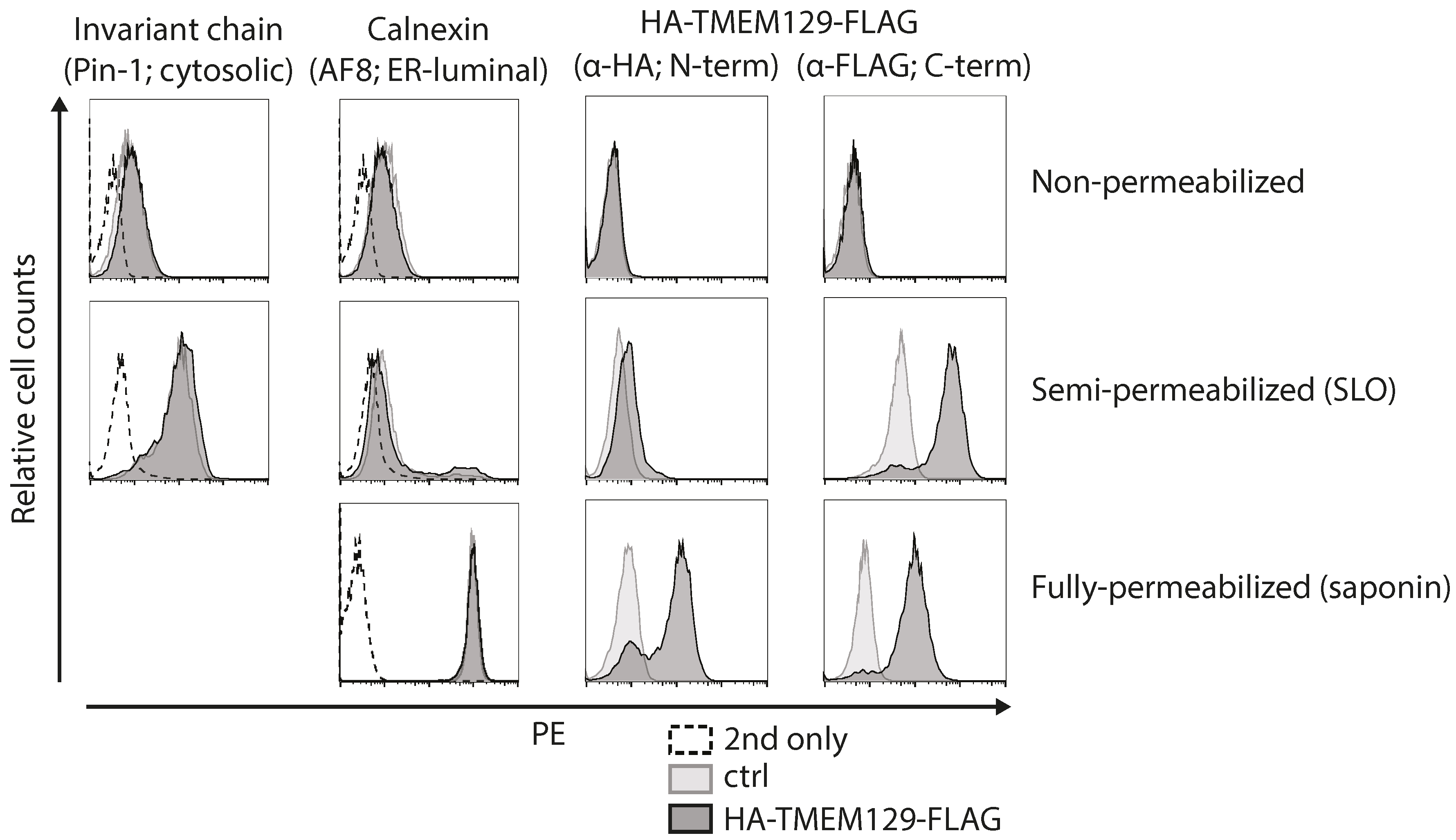
3.2. TMEM129 Is Localized in the Endoplasmic Reticulum Membrane in an Nexo–Ccyto Orientation
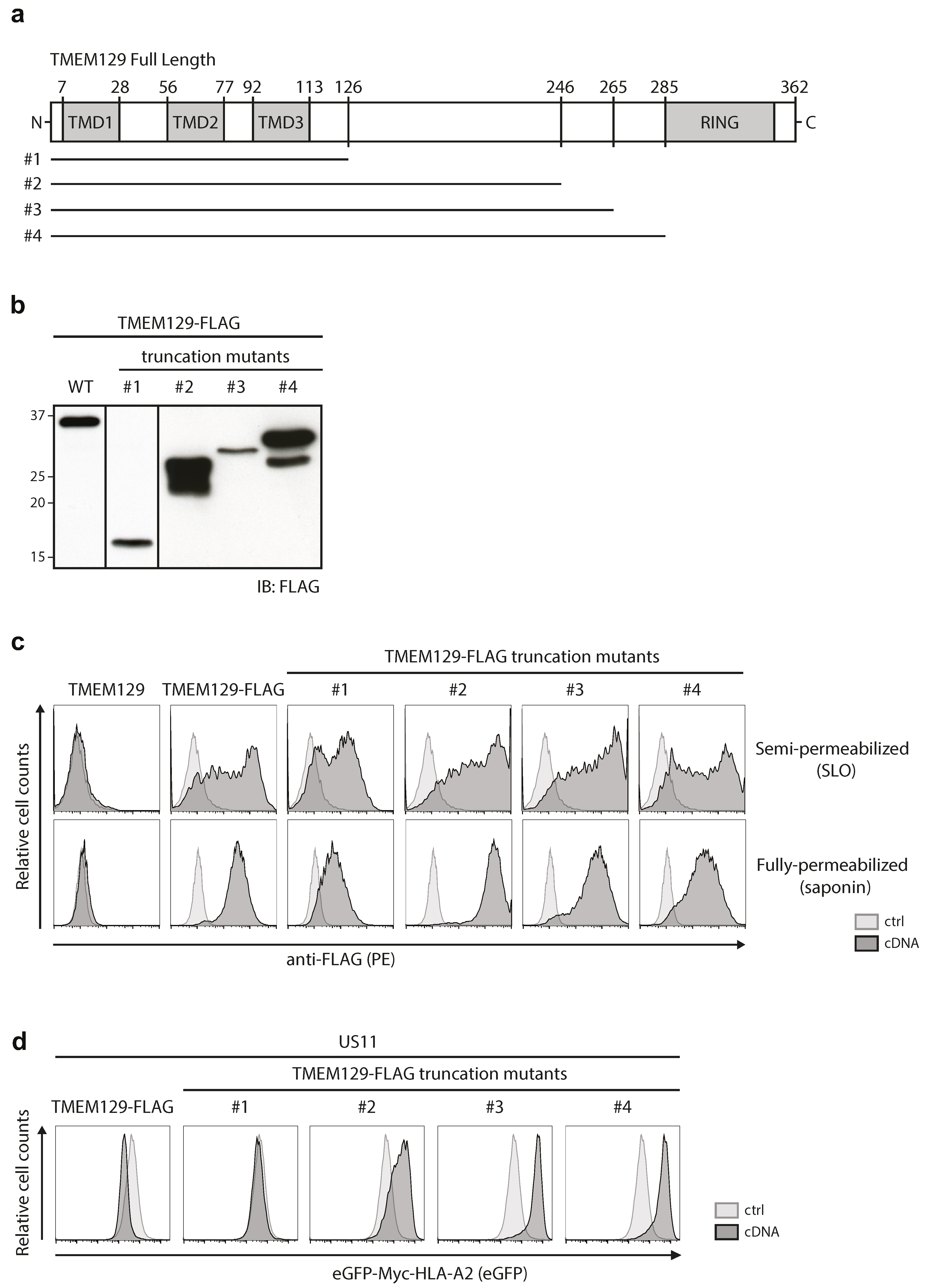
3.3. The Cytosolic Tail of TMEM129 Is Essential for Activity
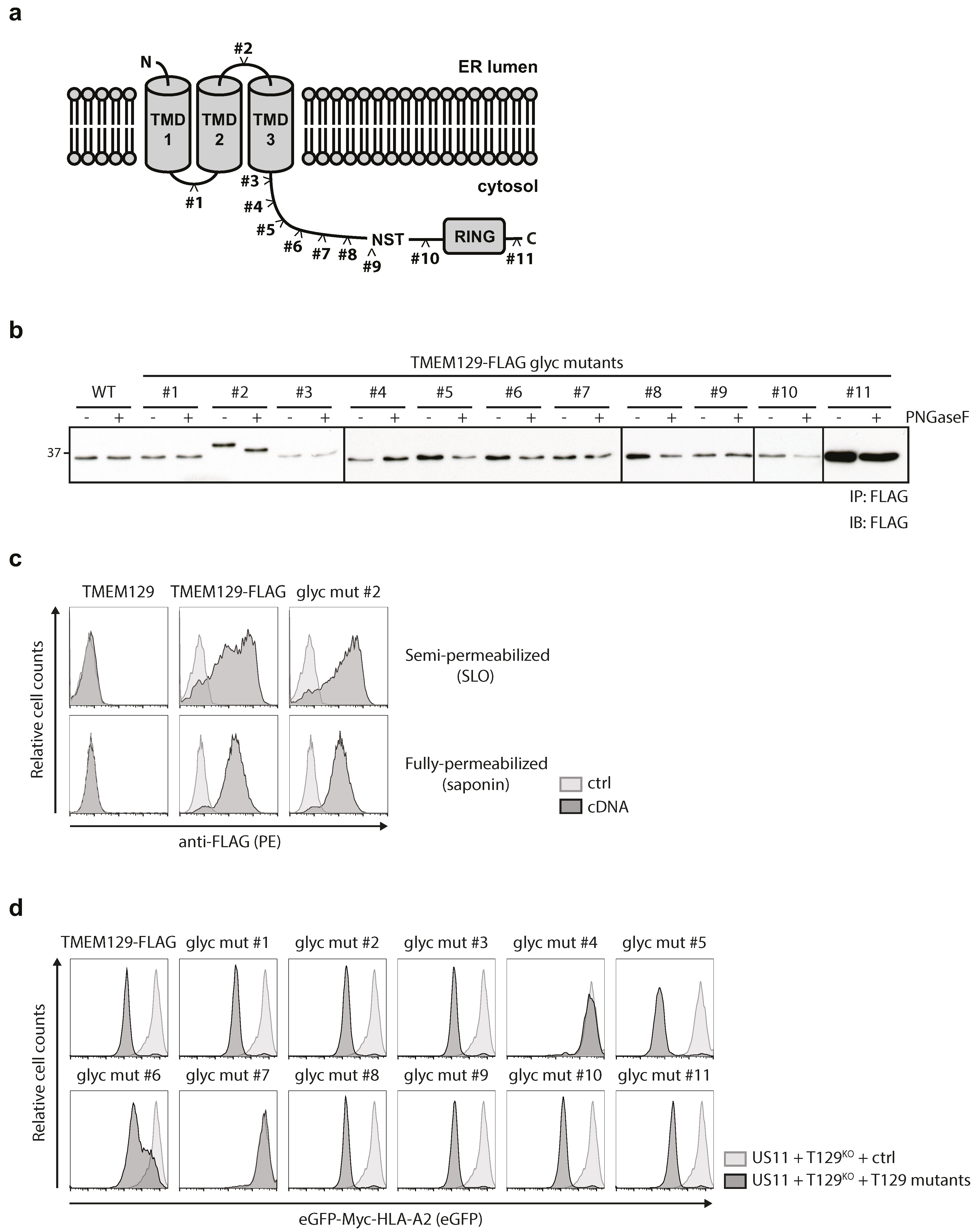
3.4. TMEM129 Contains Three Transmembrane Domains
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Hampton, R.Y. ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 2002, 14, 476–482. [Google Scholar] [CrossRef]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system. Biochim. Biophys. Acta 2014, 1843, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Needham, P.G.; Brodsky, J.L. How early studies on secreted and membrane protein quality control gave rise to the ER associated degradation (ERAD) pathway: The early history of ERAD. Biochim. Biophys. Acta 2013, 1833, 2447–2457. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Kopito, R.R.; Christianson, J.C. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Christianson, J.C.; Olzmann, J.A.; Shaler, T.A.; Sowa, M.E.; Bennett, E.J.; Richter, C.M.; Tyler, R.E.; Greenblatt, E.J.; Harper, J.W.; Kopito, R.R. Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 2012, 14, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Denic, V.; Quan, E.M.; Weissman, J.S. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell 2006, 126, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Gauss, R.; Sommer, T.; Jarosch, E. The Hrd1p ligase complex forms a linchpin between ER-lumenal substrate selection and Cdc48p recruitment. EMBO J. 2006, 25, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.; Goder, V.; Rapoport, T.A. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 2006, 126, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.; Stanley, A.M.; Rapoport, T.A. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 2010, 143, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Bays, N.W.; Gardner, R.G.; Seelig, L.P.; Joazeiro, C.A.; Hampton, R.Y. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat. Cell Biol. 2001, 3, 24–29. [Google Scholar] [PubMed]
- Swanson, R.; Locher, M.; Hochstrasser, M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes. Dev. 2001, 15, 2660–2674. [Google Scholar] [CrossRef] [PubMed]
- Foresti, O.; Rodriguez-Vaello, V.; Funaya, C.; Carvalho, P. Quality control of inner nuclear membrane proteins by the Asi complex. Science 2014, 346, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Khmelinskii, A.; Blaszczak, E.; Pantazopoulou, M.; Fischer, B.; Omnus, D.J.; Le Dez, G.; Brossard, A.; Gunnarsson, A.; Barry, J.D.; Meurer, M.; et al. Protein quality control at the inner nuclear membrane. Nature 2014, 516, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Claessen, J.H.; Kundrat, L.; Ploegh, H.L. Protein quality control in the ER: Balancing the ubiquitin checkbook. Trends. Cell Biol. 2012, 22, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Kikkert, M.; Doolman, R.; Dai, M.; Avner, R.; Hassink, G.; van Voorden, S.; Thanedar, S.; Roitelman, J.; Chau, V.; Wiertz, E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 3525–3534. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Ferrone, M.; Yang, C.; Jensen, J.P.; Tiwari, S.; Weissman, A.M. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2001, 98, 14422–14427. [Google Scholar] [CrossRef] [PubMed]
- Bartee, E.; Mansouri, M.; Hovey Nerenberg, B.T.; Gouveia, K.; Fruh, K. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J. Virol. 2004, 78, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Hassink, G.; Kikkert, M.; van Voorden, S.; Lee, S.J.; Spaapen, R.; van Laar, T.; Coleman, C.S.; Bartee, E.; Fruh, K.; Chau, V.; et al. TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem. J. 2005, 388, 647–655. [Google Scholar] [CrossRef] [PubMed]
- van de Weijer, M.L.; Bassik, M.C.; Luteijn, R.D.; Voorburg, C.M.; Lohuis, M.A.; Kremmer, E.; Hoeben, R.C.; LeProust, E.M.; Chen, S.; Hoelen, H.; et al. A high-coverage shRNA screen identifies TMEM129 as an E3 ligase involved in ER-associated protein degradation. Nat. Commun. 2014, 5, 3832. [Google Scholar] [CrossRef] [PubMed]
- van den Boomen, D.J.; Timms, R.T.; Grice, G.L.; Stagg, H.R.; Skodt, K.; Dougan, G.; Nathan, J.A.; Lehner, P.J. TMEM129 is a Derlin-1 associated ERAD E3 ligase essential for virus-induced degradation of MHC-I. Proc. Natl. Acad. Sci. USA 2014, 111, 11425–11430. [Google Scholar] [CrossRef] [PubMed]
- Stagg, H.R.; Thomas, M.; van den Boomen, D.; Wiertz, E.J.; Drabkin, H.A.; Gemmill, R.M.; Lehner, P.J. The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J. Cell Biol. 2009, 186, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.H.; Lan, W.M.; Chau, L.Y. TRC8 suppresses tumorigenesis through targeting heme oxygenase-1 for ubiquitination and degradation. Oncogene 2013, 32, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Younger, J.M.; Chen, L.; Ren, H.Y.; Rosser, M.F.; Turnbull, E.L.; Fan, C.Y.; Patterson, C.; Cyr, D.M. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 2006, 126, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Morito, D.; Hirao, K.; Oda, Y.; Hosokawa, N.; Tokunaga, F.; Cyr, D.M.; Tanaka, K.; Iwai, K.; Nagata, K. Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRDeltaF508. Mol. Biol. Cell 2008, 19, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Y.; Yamada, M.; Takahashi, K.; Yamada, M. Ubiquitin ligase Kf-1 is involved in the endoplasmic reticulum-associated degradation pathway. Biochem. Biophys. Res. Commun. 2008, 374, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.P.; Wang, Y.; Sliter, D.A.; Pearce, M.M.; Wojcikiewicz, R.J. RNF170 protein, an endoplasmic reticulum membrane ubiquitin ligase, mediates inositol 1,4,5-trisphosphate receptor ubiquitination and degradation. J. Biol. Chem. 2011, 286, 24426–24433. [Google Scholar] [CrossRef] [PubMed]
- Lerner, M.; Corcoran, M.; Cepeda, D.; Nielsen, M.L.; Zubarev, R.; Ponten, F.; Uhlen, M.; Hober, S.; Grander, D.; Sangfelt, O. The RBCC gene RFP2 (Leu5) encodes a novel transmembrane E3 ubiquitin ligase involved in ERAD. Mol. Biol. Cell 2007, 18, 1670–1682. [Google Scholar] [CrossRef] [PubMed]
- Altier, C.; Garcia-Caballero, A.; Simms, B.; You, H.; Chen, L.; Walcher, J.; Tedford, H.W.; Hermosilla, T.; Zamponi, G.W. The Cavbeta subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat. Neurosci. 2011, 14, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Neutzner, A.; Neutzner, M.; Benischke, A.S.; Ryu, S.W.; Frank, S.; Youle, R.J.; Karbowski, M. A systematic search for endoplasmic reticulum (ER) membrane-associated RING finger proteins identifies Nixin/ZNRF4 as a regulator of calnexin stability and ER homeostasis. J. Biol. Chem. 2011, 286, 8633–8643. [Google Scholar] [CrossRef] [PubMed]
- Wiertz, E.J.; Jones, T.R.; Sun, L.; Bogyo, M.; Geuze, H.J.; Ploegh, H.L. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996, 84, 769–779. [Google Scholar] [CrossRef]
- Wiertz, E.J.; Tortorella, D.; Bogyo, M.; Yu, J.; Mothes, W.; Jones, T.R.; Rapoport, T.A.; Ploegh, H.L. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 1996, 384, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Van de Weijer, M.L.; Luteijn, R.D.; Wiertz, E.J. Viral immune evasion: Lessons in MHC class I antigen presentation. Semin. Immunol. 2015, 27, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Kall, L.; Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef] [PubMed]
- Hitt, R.; Wolf, D.H. Der1p, a protein required for degradation of malfolded soluble proteins of the endoplasmic reticulum: Topology and Der1-like proteins. FEMS Yeast Res. 2004, 4, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Lilley, B.N.; Ploegh, H.L. A membrane protein required for dislocation of misfolded proteins from the ER. Nature 2004, 429, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Shibata, Y.; Yun, C.; Ron, D.; Rapoport, T.A. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 2004, 429, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, E.J.; Olzmann, J.A.; Kopito, R.R. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant alpha-1 antitrypsin from the endoplasmic reticulum. Nat. Struct. Mol. Biol. 2011, 18, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Fairbank, M.; St-Pierre, P.; Nabi, I.R. The complex biology of autocrine motility factor/phosphoglucose isomerase (AMF/PGI) and its receptor, the gp78/AMFR E3 ubiquitin ligase. Mol. BioSyst. 2009, 5, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Landolt-Marticorena, C.; Reithmeier, R.A. Asparagine-linked oligosaccharides are localized to single extracytosolic segments in multi-span membrane glycoproteins. Biochem. J. 1994, 302, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Popov, M.; Tam, L.Y.; Li, J.; Reithmeier, R.A. Mapping the ends of transmembrane segments in a polytopic membrane protein. Scanning N-glycosylation mutagenesis of extracytosolic loops in the anion exchanger, band 3. J. Biol. Chem. 1997, 272, 18325–18332. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, I.M.; von Heijne, G. Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J. Biol. Chem. 1993, 268, 5798–5801. [Google Scholar] [PubMed]
- Christianson, J.C.; Ye, Y. Cleaning up in the endoplasmic reticulum: Ubiquitin in charge. Nat. Struct. Mol. Biol. 2014, 21, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Plemper, R.K.; Bohmler, S.; Bordallo, J.; Sommer, T.; Wolf, D.H. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature 1997, 388, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Pilon, M.; Schekman, R.; Romisch, K. Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J. 1997, 16, 4540–4548. [Google Scholar] [CrossRef] [PubMed]
- Plemper, R.K.; Bordallo, J.; Deak, P.M.; Taxis, C.; Hitt, R.; Wolf, D.H. Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J. Cell Sci. 1999, 112, 4123–4134. [Google Scholar] [PubMed]
- Zhou, M.; Schekman, R. The engagement of Sec61p in the ER dislocation process. Mol. Cell 1999, 4, 925–934. [Google Scholar] [CrossRef]
- Gillece, P.; Pilon, M.; Romisch, K. The protein translocation channel mediates glycopeptide export across the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2000, 97, 4609–4614. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, B.M.; Tyson, J.R.; Reid, P.J.; Stirling, C.J. Distinct domains within yeast Sec61p involved in post-translational translocation and protein dislocation. J. Biol. Chem. 2000, 275, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, A.; Herrgen, H.; Winkeler, A.; Herzog, V. Cholera toxin is exported from microsomes by the Sec61p complex. J. Cell Biol. 2000, 148, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.C.; Schekman, R. Role of Sec61p in the ER-associated degradation of short-lived transmembrane proteins. J. Cell Biol. 2008, 181, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Willer, M.; Forte, G.M.; Stirling, C.J. Sec61p is required for ERAD-L: Genetic dissection of the translocation and ERAD-L functions of Sec61P using novel derivatives of CPY. J. Biol. Chem. 2008, 283, 33883–33888. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.; Wolf, D.H. Sec61p is part of the endoplasmic reticulum-associated degradation machinery. EMBO J. 2009, 28, 2874–2884. [Google Scholar] [CrossRef] [PubMed]
- Wahlman, J.; DeMartino, G.N.; Skach, W.R.; Bulleid, N.J.; Brodsky, J.L.; Johnson, A.E. Real-time fluorescence detection of ERAD substrate retrotranslocation in a mammalian in vitro system. Cell 2007, 129, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Hsiao, H.T.; Chu, Y.R.; Ye, Y.; Chen, X. Derlin2 protein facilitates HRD1-mediated retro-translocation of sonic hedgehog at the endoplasmic reticulum. J. Biol. Chem. 2013, 288, 25330–25339. [Google Scholar] [CrossRef] [PubMed]
- Hoelen, H.; Zaldumbide, A.; van Leeuwen, W.F.; Torfs, E.C.; Engelse, M.A.; Hassan, C.; Lebbink, R.J.; de Koning, E.J.; Resssing, M.E.; de Ru, A.H.; et al. Proteasomal Degradation of Proinsulin Requires Derlin-2, HRD1 and p97. PLoS ONE 2015, 10, e0128206. [Google Scholar] [CrossRef] [PubMed]
- Deak, P.M.; Wolf, D.H. Membrane topology and function of Der3/Hrd1p as a ubiquitin-protein ligase (E3) involved in endoplasmic reticulum degradation. J. Biol. Chem. 2001, 276, 10663–10669. [Google Scholar] [CrossRef] [PubMed]
- Nadav, E.; Shmueli, A.; Barr, H.; Gonen, H.; Ciechanover, A.; Reiss, Y. A novel mammalian endoplasmic reticulum ubiquitin ligase homologous to the yeast Hrd1. Biochem. Biophys. Res. Commun. 2003, 303, 91–97. [Google Scholar] [CrossRef]
- Burr, M.L.; Cano, F.; Svobodova, S.; Boyle, L.H.; Boname, J.M.; Lehner, P.J. HRD1 and UBE2J1 target misfolded MHC class I heavy chains for endoplasmic reticulum-associated degradation. Proc. Natl. Acad. Sci. USA 2011, 108, 2034–2039. [Google Scholar] [CrossRef] [PubMed]
- Burr, M.L.; van den Boomen, D.J.; Bye, H.; Antrobus, R.; Wiertz, E.J.; Lehner, P.J. MHC class I molecules are preferentially ubiquitinated on endoplasmic reticulum luminal residues during HRD1 ubiquitin E3 ligase-mediated dislocation. Proc. Natl. Acad. Sci. USA 2013, 110, 14290–14295. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Ruggiano, A.; Carvalho, P.; Rapoport, T.A. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell 2014, 158, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, R.D.; Rapoport, T.A. Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell 2016, 166, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Kreft, S.G.; Wang, L.; Hochstrasser, M. Membrane topology of the yeast endoplasmic reticulum-localized ubiquitin ligase Doa10 and comparison with its human ortholog TEB4 (MARCH-VI). J. Biol. Chem. 2006, 281, 4646–4653. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glyc Mutant | Forward or Reverse | Sequence 5′–3′ |
|---|---|---|
| Backbone | Forward | TGAGCTAGCAGTATTAATTAACCAC |
| Reverse | ATGACTAAGCTAGTACCGGTTAG | |
| #1 Ser43 NAT | Forward | GTCGGGCTGGCTGGGCAGCAACGCAACAGAGGACGCCGCCTTCGTG |
| Reverse | CACGAAGGCGGCGTCCTCTGTTGCGTTGCTGCCCAGCCAGCCCGAC | |
| #2 Ala85 GAAGGAANATEGAAEGAAGG | Forward | ACCGCCAGCTGCACCTCCTGCCGCGCCCTCTGTTGCGTTAGCTGCACCTCCTGCCGCGCCGGCGTGGAGCCGCTTTTCTG |
| Reverse | GGCGCGGCAGGAGGTGCAGCTAACGCAACAGAGGGCGCGGCAGAAGGTGCAGCTGGCGGTCTCAGCCAGGCCCCTGAG | |
| #3 Asp117 NATE | Forward | CTACTACTGGTCCCGTGACAACGCAACAGAGCGGTGGGCCTGCCACCC |
| Reverse | GGGTGGCAGGCCCACCGCTCTGTTGCGTTGTCACGGGACCAGTAGTAG | |
| #4 Ala125 NATE | Forward | GCCTGCCACCCACTGGCGAACGCAACAGAGCGCACCCTGGCCCTCTACG |
| Reverse | CGTAGAGGGCCAGGGTGCGCTCTGTTGCGTTCGCCAGTGGGTGGCAGGC | |
| #5 Ala140 NATE | Forward | CACAGTCTGGCTGGCAGGCTAACGCAACAGAGGTTGCCTCCTCTGTCAACAC |
| Reverse | GTGTTGACAGAGGAGGCAACCTCTGTTGCGTTAGCCTGCCAGCCAGACTGTG | |
| #6 Ile152 NAT | Forward | CACTGAGTTCCGGCGGATTAACGCAACAGAGGACAAGTTTGCCACCGGTG |
| Reverse | CACCGGTGGCAAACTTGTCCTCTGTTGCGTTAATCCGCCGGAACTCAGTG | |
| #7 Trp170 NATE | Forward | GTGATTGTGACAGACACGTGGAACGCAACAGAGGTGATGAAGGTAACCACCTAC |
| Reverse | GTAGGTGGTTACCTTCATCACCTCTGTTGCGTTCCACGTGTCTGTCACAATCAC | |
| #8 Val191 NA | Forward | GGACGTGCACCTGACTGTGAACGCAACGGAGTCTCGGCAGCATG |
| Reverse | CATGCTGCCGAGACTCCGTTGCGTTCACAGTCAGGTGCACGTCC | |
| #9 Ser230 NA | Forward | CTTTGACATCTGGCTGAACTCCAACGCAACTGAGTACGGGGAGCTCTG |
| Reverse | CAGAGCTCCCCGTACTCAGTTGCGTTGGAGTTCAGCCAGATGTCAAAG | |
| #10 Ser275 NATE | Forward | GAGGTCAACCCGGCCTACTCAAACGCAACAGAGGTGCCCAGCAGCCAGGAG |
| Reverse | CTCCTGGCTGCTGGGCACCTCTGTTGCGTTTGAGTAGGCCGGGTTGACCTC | |
| #11 Phe363 NATE | Reverse | ATGACTAAGCTAGTACCGGTTAGGATGCATTCACTTGTCGTCATCGTCTTTGTAGTCTTCCTCTGTTGCGTTGAAGCGCACGGTGCACAC |
| Truncation Mutant | Forward or Reverse | Sequence 5′–3′ |
|---|---|---|
| #1 | Reverse | ATGACTAAGCTAGTACCGGTTAGGATGCATTCACTTGTCGTCATCGTCTTTGTAGTCTTCGAAGGCAAATGTCTCCAGG |
| #2 | Reverse | ATGACTAAGCTAGTACCGGTTAGGATGCATTCACTTGTCGTCATCGTCTTTGTAGTCTTCGAACCTGCGGATGGGTGCCCG |
| #3 | Reverse | ATGACTAAGCTAGTACCGGTTAGGATGCATTCACTTGTCGTCATCGTCTTTGTAGTCTTCGAATGGCGAGAGCTCATGCTGC |
| #4 | Reverse | ATGACTAAGCTAGTACCGGTTAGGATGCATTCACTTGTCGTCATCGTCTTTGTAGTCTTCGAAGCGCGCCAGTGGGTGGC |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van de Weijer, M.L.; Van Muijlwijk, G.H.; Visser, L.J.; Costa, A.I.; Wiertz, E.J.H.J.; Lebbink, R.J. The E3 Ubiquitin Ligase TMEM129 Is a Tri-Spanning Transmembrane Protein. Viruses 2016, 8, 309. https://doi.org/10.3390/v8110309
Van de Weijer ML, Van Muijlwijk GH, Visser LJ, Costa AI, Wiertz EJHJ, Lebbink RJ. The E3 Ubiquitin Ligase TMEM129 Is a Tri-Spanning Transmembrane Protein. Viruses. 2016; 8(11):309. https://doi.org/10.3390/v8110309
Chicago/Turabian StyleVan de Weijer, Michael L., Guus H. Van Muijlwijk, Linda J. Visser, Ana I. Costa, Emmanuel J. H. J. Wiertz, and Robert Jan Lebbink. 2016. "The E3 Ubiquitin Ligase TMEM129 Is a Tri-Spanning Transmembrane Protein" Viruses 8, no. 11: 309. https://doi.org/10.3390/v8110309