
A Glimpse of Nucleo-Cytoplasmic Large DNA Virus Biodiversity through the Eukaryotic Genomics Window


Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. NCLDV Protein Markers in Eukaryotes
3.2. Phylogeny of Eukaryotic NCLDV-Like Proteins
3.3. Genomic Context around NCLDV-Like Genes
3.4. Hints on Viral Functions
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Iyer, L.M.; Balaji, S.; Koonin, E.V.; Aravind, L. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 2006, 117, 156–184. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Yutin, N. Nucleo-cytoplasmic Large DNA Viruses (NCLDV) of Eukaryotes. eLS 2012. [Google Scholar]
- Philippe, N.; Legendre, M.; Doutre, G.; Couté, Y.; Poirot, O.; Lescot, M.; Arslan, D.; Seltzer, V.; Bertaux, L.; Bruley, C.; et al. Pandoraviruses: Amoeba Viruses with Genomes Up to 2.5 Mb Reaching That of Parasitic Eukaryotes. Science 2013, 341, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.G. Giant viruses come of age. Curr. Opin. Microbiol. 2016, 31, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Wommack, K.E.; Nasko, D.J.; Chopyk, J.; Sakowski, E.G. Counts and sequences, observations that continue to change our understanding of viruses in nature. J. Microbiol. Seoul Korea 2015, 53, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Hingamp, P.; Grimsley, N.; Acinas, S.G.; Clerissi, C.; Subirana, L.; Poulain, J.; Ferrera, I.; Sarmento, H.; Villar, E.; Lima-Mendez, G.; et al. Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes. ISME J. 2013, 7, 1678–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihara, T.; Nishimura, Y.; Shimizu, Y.; Nishiyama, H.; Yoshikawa, G.; Uehara, H.; Hingamp, P.; Goto, S.; Ogata, H. Linking Virus Genomes with Host Taxonomy. Viruses 2016, 8, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanc, G.; Gallot-Lavallée, L.; Maumus, F. Provirophages in the Bigelowiella genome bear testimony to past encounters with giant viruses. Proc. Natl. Acad. Sci. USA 2015, 112, E5318–E5326. [Google Scholar] [CrossRef] [PubMed]
- Maumus, F.; Epert, A.; Nogué, F.; Blanc, G. Plant genomes enclose footprints of past infections by giant virus relatives. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Colson, P.; Giorgi, R.; Pontarotti, P.; Raoult, D. DNA-Dependent RNA Polymerase Detects Hidden Giant Viruses in Published Databanks. Genome Biol. Evol. 2014, 6, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, S.; Liu, T.; Sun, J.; Chi, S.; Liu, C.; Li, X.; Yin, J.; Wang, X.; Yu, J. Endogenous viral elements in algal genomes. Acta Oceanol. Sin. 2014, 33, 102–107. [Google Scholar] [CrossRef]
- Delaroque, N.; Boland, W. The genome of the brown alga Ectocarpus siliculosus contains a series of viral DNA pieces, suggesting an ancient association with large dsDNA viruses. BMC Evol. Biol. 2008, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Filée, J. Multiple occurrences of giant virus core genes acquired by eukaryotic genomes: The visible part of the iceberg? Virology 2014, 466–467, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Meints, R.H.; Ivey, R.G.; Lee, A.M.; Choi, T.-J. Identification of Two Virus Integration Sites in the Brown Alga Feldmannia Chromosome. J. Virol. 2008, 82, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Delaroque, N.; Maier, I.; Knippers, R.; Müller, D.G. Persistent virus integration into the genome of its algal host, Ectocarpus siliculosus (Phaeophyceae). J. Gen. Virol. 1999, 80, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Maumus, F.; Blanc, G. Study of gene trafficking between Acanthamoeba and giant viruses suggests an undiscovered family of amoeba-infecting viruses. Genome Biol. Evol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Wolf, Y.I.; Raoult, D.; Koonin, E.V. Eukaryotic large nucleo-cytoplasmic DNA viruses: Clusters of orthologous genes and reconstruction of viral genome evolution. Virol. J. 2009, 6, 223. [Google Scholar] [CrossRef] [PubMed]
- Available online: ftp://ftp.ncbi.nih.gov/pub/wolf/COGs/NCVOG/ (accessed on 30 November 2016).
- Available online: https://www.bioinfodata.org/Blast4OneKP (accessed on 30 November 2016).
- Available online: ftp://ftp.imicrobe.us/projects/104/CAM_P_0001000.pep.fa.gz (accessed on 30 November 2016).
- Available online: ftp://ftp.imicrobe.us/projects/104/ Callum_FINAL_biosample_ids.xls (accessed on 30 November 2016).
- Available online: http://www.onekp.com/samples/list.php (accessed on 30 November 2016).
- Yutin, N.; Raoult, D.; Koonin, E.V. Virophages, polintons, and transpovirons: a complex evolutionary network of diverse selfish genetic elements with different reproduction strategies. Virol. J. 2013, 10, 158. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Audic, S.; Claverie, J.-M.; Blanc, G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol. Biol. 2010, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J.; Burki, F.; Wilcox, H.M.; Allam, B.; Allen, E.E.; Amaral-Zettler, L.A.; Armbrust, E.V.; Archibald, J.M.; Bharti, A.K.; Bell, C.J.; et al. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): Illuminating the Functional Diversity of Eukaryotic Life in the Oceans through Transcriptome Sequencing. PLoS Biol 2014, 12, e1001889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matasci, N.; Hung, L.-H.; Yan, Z.; Carpenter, E.J.; Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Ayyampalayam, S.; Barker, M.; et al. Data access for the 1,000 Plants (1KP) project. GigaScience 2014, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Ishida, K.-I.; Hirakawa, Y. Diurnal transcriptional regulation of endosymbiotically derived genes in the chlorarachniophyte Bigelowiella natans. Genome Biol. Evol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Cho, B.C.; Simpson, A.G.B. Halocafeteria seosinensis gen. et sp. nov. (Bicosoecida), a halophilic bacterivorous nanoflagellate isolated from a solar saltern. Extrem. Life Extreme Cond. 2006, 10, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.G.; Allen, M.J.; Wilson, W.H.; Suttle, C.A. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc. Natl. Acad. Sci. USA 2010, 107, 19508–19513. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.G.; Suttle, C.A. A virophage at the origin of large DNA transposons. Science 2011, 332, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L.; Dunigan, D.D. Chloroviruses: not your everyday plant virus. Trends Plant Sci. 2012, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Clerissi, C.; Grimsley, N.; Ogata, H.; Hingamp, P.; Poulain, J.; Desdevises, Y. Unveiling of the Diversity of Prasinoviruses (Phycodnaviridae) in Marine Samples by Using High-Throughput Sequencing Analyses of PCR-Amplified DNA Polymerase and Major Capsid Protein Genes. Appl. Environ. Microbiol. 2014, 80, 3150–3160. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Van Etten, J.L.; Allen, M.J. The Phycodnaviridae: The Story of How Tiny Giants Rule the World. Curr. Top. Microbiol. Immunol. 2009, 328, 1–42. [Google Scholar] [PubMed]
- Hanson, L.A.; Rudis, M.R.; Vasquez-Lee, M.; Montgomery, R.D. A broadly applicable method to characterize large DNA viruses and adenoviruses based on the DNA polymerase gene. Virol. J. 2006, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Suttle, C.A. Evolutionary relationships among large double-stranded DNA viruses that infect microalgae and other organisms as inferred from DNA polymerase genes. Virology 1996, 219, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Takemura, M.; Yokobori, S.; Ogata, H. Evolution of Eukaryotic DNA Polymerases via Interaction Between Cells and Large DNA Viruses. J. Mol. Evol. 2015, 81, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Koonin, E.V. Hidden evolutionary complexity of Nucleo-Cytoplasmic Large DNA viruses of eukaryotes. Virol. J. 2012, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Colson, P.; Raoult, D.; Koonin, E.V. Mimiviridae: clusters of orthologous genes, reconstruction of gene repertoire evolution and proposed expansion of the giant virus family. Virol. J. 2013, 10, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier Academic Press: San Diego, CA, USA, 2011. [Google Scholar]
- Colbourne, J.K.; Pfrender, M.E.; Gilbert, D.; Thomas, W.K.; Tucker, A.; Oakley, T.H.; Tokishita, S.; Aerts, A.; Arnold, G.J.; Basu, M.K.; et al. The ecoresponsive genome of Daphnia pulex. Science 2011, 331, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Baumgarten, S.; Simakov, O.; Esherick, L.Y.; Liew, Y.J.; Lehnert, E.M.; Michell, C.T.; Li, Y.; Hambleton, E.A.; Guse, A.; Oates, M.E.; et al. The genome of Aiptasia, a sea anemone model for coral symbiosis. Proc. Natl. Acad. Sci. USA 2015, 112, 11893–11898. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Maruyama, F.; Fujisawa, T.; Togashi, T.; Yamamoto, N.; Seo, M.; Sato, S.; Yamada, T.; Mori, H.; Tajima, N.; et al. Klebsormidium flaccidum genome reveals primary factors for plant terrestrial adaptation. Nat. Commun. 2014, 5, 3978. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Zhang, X.; Miao, M.; Fan, X.; Zheng, Y.; Xu, D.; Wang, J.; Zhou, L.; Wang, D.; Gao, Y.; et al. Saccharina genomes provide novel insight into kelp biology. Nat. Commun. 2015, 6, 6986. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Wang, S.; Sekimoto, S.; Aerts, A.L.; Choi, C.; Clum, A.; LaButti, K.M.; Lindquist, E.A.; Yee Ngan, C.; Ohm, R.A.; et al. Phylogenomic Analyses Indicate that Early Fungi Evolved Digesting Cell Walls of Algal Ancestors of Land Plants. Genome Biol. Evol. 2015, 7, 1590–1601. [Google Scholar] [CrossRef] [PubMed]
- Filée, J.; Chandler, M. Gene Exchange and the Origin of Giant Viruses. Intervirology 2010, 53, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Stram, Y.; Kuzntzova, L. Inhibition of viruses by RNA interference. Virus Genes 2006, 32, 299–306. [Google Scholar] [CrossRef] [PubMed]
- McQueen-Mason, S.; Durachko, D.M.; Cosgrove, D.J. Two endogenous proteins that induce cell wall extension in plants. Plant Cell 1992, 4, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Yennawar, N.H.; Li, L.-C.; Dudzinski, D.M.; Tabuchi, A.; Cosgrove, D.J. Crystal structure and activities of EXPB1 (Zea m 1), a beta-expansin and group-1 pollen allergen from maize. Proc. Natl. Acad. Sci. USA 2006, 103, 14664–14671. [Google Scholar] [CrossRef] [PubMed]
- Nikolaidis, N.; Doran, N.; Cosgrove, D.J. Plant expansins in bacteria and fungi: evolution by horizontal gene transfer and independent domain fusion. Mol. Biol. Evol. 2014, 31, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.M. Cyclin’ on the viral path to destruction. Nat. Cell Biol. 2000, 2, E203–E204. [Google Scholar] [CrossRef] [PubMed]
- Bagga, S.; Bouchard, M.J. Cell cycle regulation during viral infection. Methods Mol. Biol. Clifton NJ 2014, 1170, 165–227. [Google Scholar]
- Bowman, S.M.; Free, S.J. The structure and synthesis of the fungal cell wall. BioEssays 2006, 28, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Onimatsu, H.; Van Etten, J.L. Chlorella viruses. Adv. Virus Res. 2006, 66, 293–336. [Google Scholar] [PubMed]
- Klose, T.; Rossmann, M.G. Structure of large dsDNA viruses. Biol. Chem. 2014, 395, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Bartoli, J.; Shmakova, L.; Jeudy, S.; Labadie, K.; Adrait, A.; Lescot, M.; Poirot, O.; Bertaux, L.; Bruley, C.; et al. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc. Natl. Acad. Sci. USA 2014, 111, 4274–4279. [Google Scholar] [CrossRef] [PubMed]
- Short, S.M. The ecology of viruses that infect eukaryotic algae. Environ. Microbiol. 2012, 14, 2253–2271. [Google Scholar] [CrossRef] [PubMed]
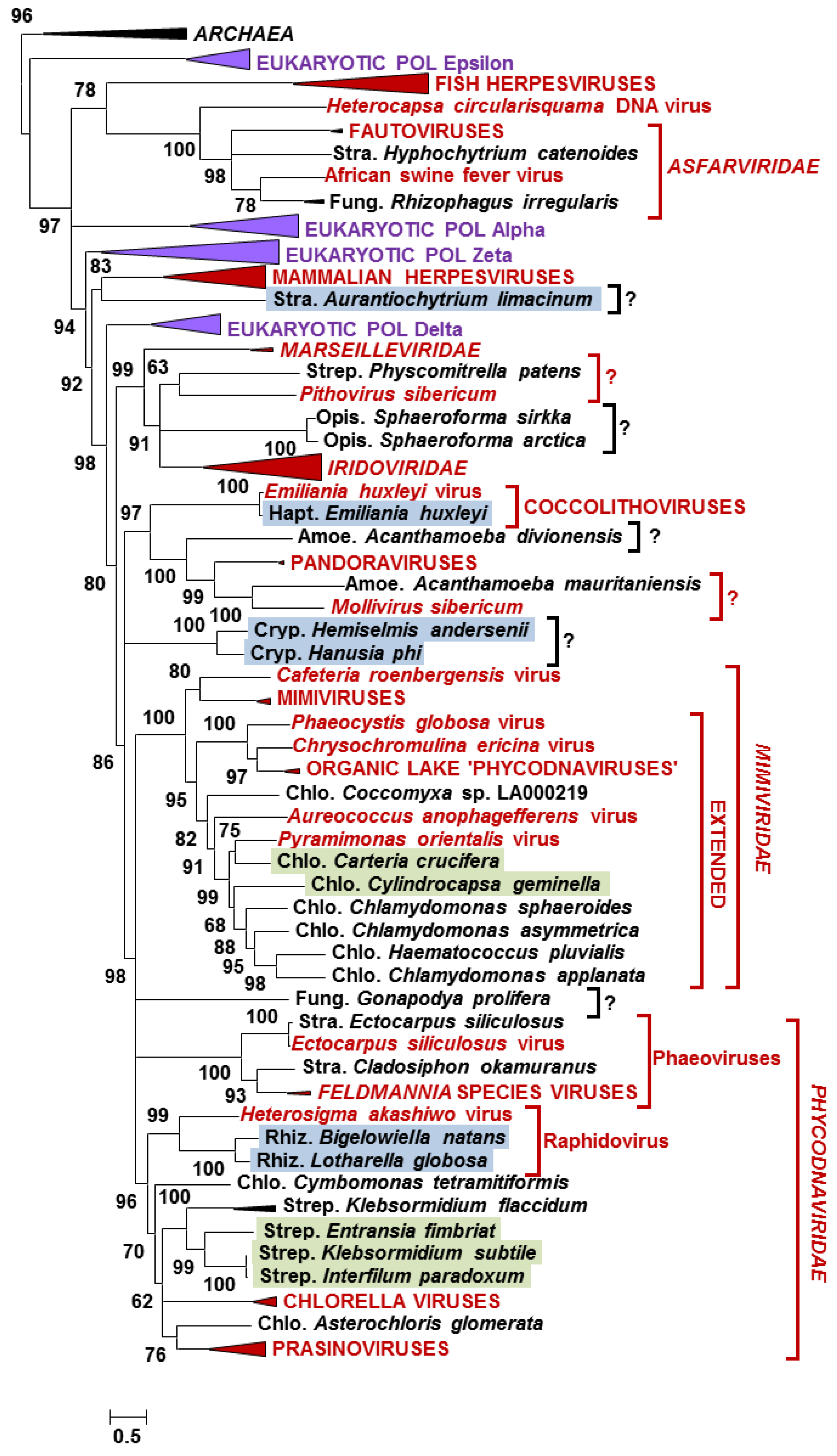

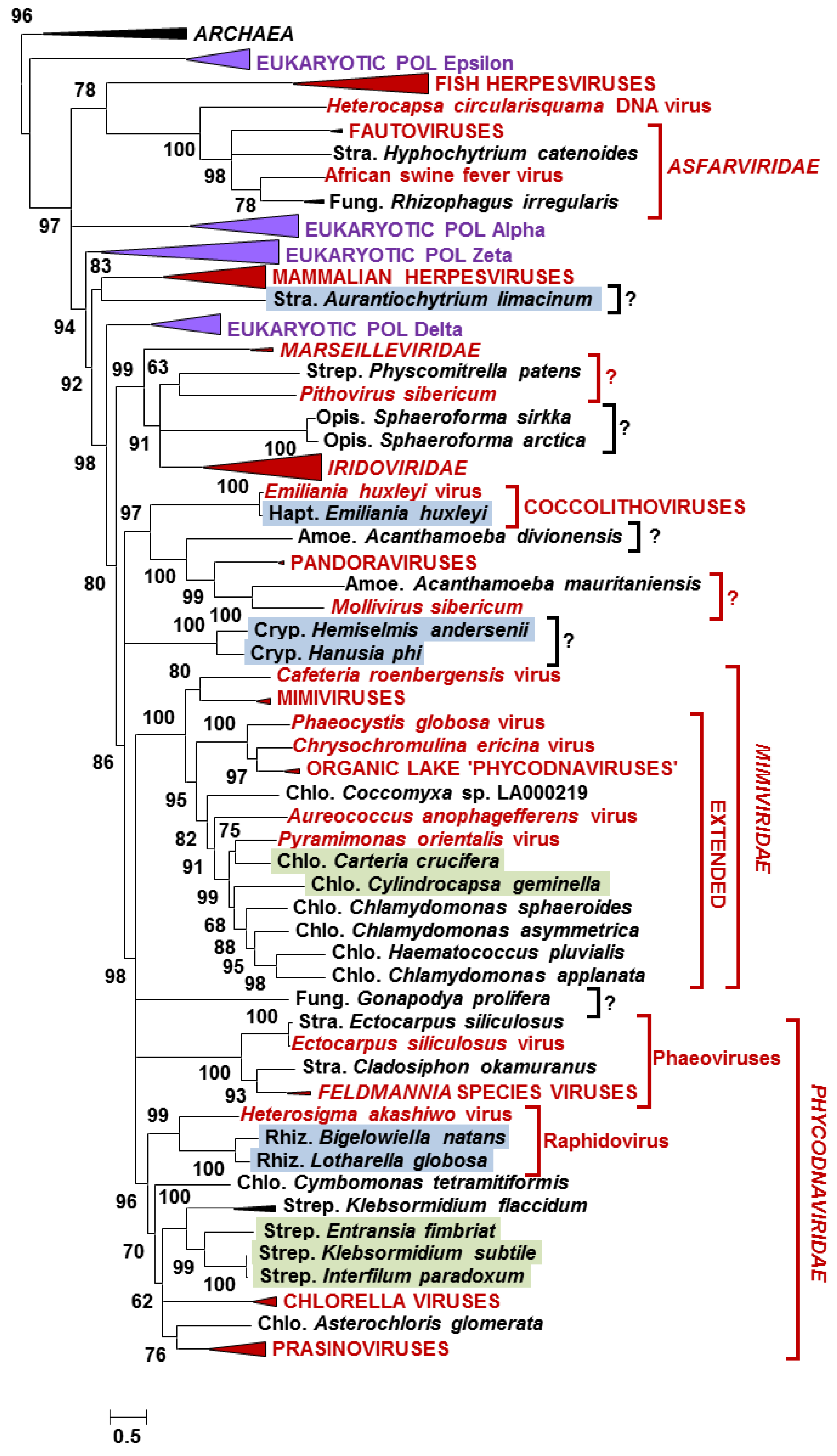{kind=link}
| Eukaryotic Clade | Species | Habitat | Database | DNAP * | MCP * | ATPase * | D5 * | VLTF3 * |
|---|---|---|---|---|---|---|---|---|
| Genomic datasets | ||||||||
| Amoebozoa (Discosea) | Acanthamoeba astronyxis | terrestrial and aquatic | Assembly | √ | √ | √ | ||
| Acanthamoeba castellanii | RefSeq | √ | √ | √ | √ | |||
| Acanthamoeba divionensis | Assembly | √ | √ | √ | ||||
| Acanthamoeba healyi | Assembly | √ | ||||||
| Acanthamoeba lenticulata | Assembly | √ | √ | √ | √ | |||
| Acanthamoeba lugdunensis | Assembly | √ | √ | |||||
| Acanthamoeba mauritaniens | Assembly | √ | √ | √ | √ | |||
| Acanthamoeba pearcei | Assembly | √ | √ | √ | ||||
| Acanthamoeba polyphaga | Assembly | √ | √ | √ | ||||
| Acanthamoeba quina | Assembly | √ | √ | √ | √ | |||
| Acanthamoeba rhysodes | Assembly | √ | ||||||
| Cryptophyta (Pyrenomonadales) | Guillardia theta | sea | RefSeq | MI | ||||
| Euglenozoa | Euglena gracilis | freshwater | Assembly | √ | ||||
| Fungi (Chytridiomycota) | Gonapodya prolifera | freshwater | RefSeq | Phy | √ | √ | Phy | √ |
| Fungi (Glomeromycota) | Rhizophagus irregularis | terrestrial | Assembly | Asf | ||||
| Fungi (Blastocladiomycota) | Allomyces macrogynus | freshwater | RefSeq | Phy | ||||
| Metazoa (Arthropoda) | Daphnia pulex | freshwater | RefSeq | √ | ||||
| Metazoa (Cnidaria) | Exaiptasia pallida | sea | RefSeq | Asf | ||||
| Hydra vulgaris | freshwater | RefSeq | Mi | √ | Mi | √ | ||
| Opisthokonta (Ichthyosporea) | Sphaeroforma arctica | sea | RefSeq | Irma | Irma | |||
| Sphaeroforma sirkka | sea | Assembly | Irma | Irma | Irma | √ | Irma | |
| Rhizaria (Cercozoa) | Bigelowiella natans | sea | RefSeq | Phy + √ | √ | √ | Phy | √ |
| Stramenopiles (Bicosoecida) | Halocafeteria seosinensis | saltern pond | Assembly | √ | ||||
| Stramenopiles (Eustigmatophyceae) | Nannochloropsis limnetica | freshwater | Assembly | Pha | Pha | |||
| Stramenopiles (Hyphochytriomycetes) | Hyphochytrium catenoides | terrestrial | Assembly | Asf | Asf | Asf | Asf | Asf |
| Stramenopiles (Oomycetes) | Phytophthora sp. totara | soilborne plant pathogen | Assembly | Asf | ||||
| Phytophthora agathidicida | Assembly | Asf | ||||||
| Phytophthora alni | Assembly | Asf | ||||||
| Phytophthora cambivora | Assembly | Asf | ||||||
| Phytophthora cryptogea | Assembly | Asf | ||||||
| Phytophthora nicotianae | Assembly | Asf | Asf | |||||
| Phytophthora parasitica | RefSeq | Asf | Asf | |||||
| Pythium irregulare | Assembly | Asf | ||||||
| Pythium oligadrum | Assembly | Asf | ||||||
| Pythium ultimum | Assembly | Asf | Asf | |||||
| Stramenopiles (Phaeophyceae) | Cladosiphon okamuranus | sea | Assembly | Pha | Pha | Pha | Pha | Pha |
| Ectocarpus siliculosus | RefSeq | Pha | Pha | Pha | Pha | Pha | ||
| Saccharina japonica | RefSeq | Pha | ||||||
| Viridiplantae (Chlorophyta) | Asterochloris glomerata | lichen photobiont | Assembly | Phy | Phy | Phy | Phy | |
| Chlamydomonas applanata | terrestrial | Assembly | Mi | |||||
| Chlamydomonas asymmetrica | freshwater | Assembly | Mi | Mi | Mi | Mi + Phy | Mi | |
| Chlamydomonas sphaeroides | freshwater | Assembly | Mi | Mi | Mi | Mi + Phy | Mi + √ | |
| Chlorella vulgaris | freshwater | Assembly | √ | |||||
| Coccomyxa sp. LA000219 | unknown | Assembly | Mi | Mi | Mi | Mi | Mi | |
| Cymbomonas tetramitiformis | sea | Assembly | Phy | Phy | Phy | Phy | Phy | |
| Haematococcus pluvialis | freshwater | Assembly | Mi | Mi | Mi | Mi + Phy | Mi | |
| Viridiplantae (Streptophyta) | Klebsormidium flaccidum | terrestrial | RefSeq | Phy | Phy | Phy | √ | Phy |
| Viridiplantae (Streptophyta) | Physcomitrella patens | terrestrial | RefSeq | Pitho | Pitho | |||
| Transcriptomic datasets | ||||||||
| Cryptophyta (Cryptomonadales) | Hemiselmis andersenii | sea | MMETSP | √ | ||||
| Cryptophyta (Pyrenomonadales) | Hanusia phi | sea | MMETSP | √ | ||||
| Haptophyceae (Coccolithales) | Pleurochrysis carterae | sea | MMETSP | √ | √ | √ | ||
| Haptophyceae (Isochrysidales) | Chrysochromulina polylepis | sea | MMETSP | √ | ||||
| Isochrysis galbana | sea | MMETSP | √ | |||||
| Haptophyceae (Phaeocystales) | Phaeocystis antarctica | sea | MMETSP | √ | ||||
| Haptophyceae (Prymnesiales) | Emiliania huxleyi | sea | MMETSP | Coc | Coc | Coc | ||
| Rhizaria (Cercozoa) | Lotharella globosa | sea | MMETSP | Phy | ||||
| Stramenopiles (Labyrinthulomycetes) | Aurantiochytrium limacinum | sea | MMETSP | Pha | ||||
| Schizochytrium aggregatum | sea | MMETSP | √ | √ | ||||
| Thraustochytrium sp. | sea | MMETSP | √ | √ | ||||
| Undescribed Strain | CCMP2135 | sea | MMETSP | √ | ||||
| Undescribed Strain | CCMP2436 | sea | MMETSP | √ | ||||
| Viridiplantae (Chlorophyta) | Carteria crucifera | freshwater | 1KP | Mi | Mi | |||
| Cylindrocapsa geminella | freshwater | 1KP | Mi | |||||
| Viridiplantae (Streptophyta) | Entransia fimbriat | freshwater | 1KP | Phy | ||||
| Interfilum paradoxum | terrestrial | 1KP | Phy | |||||
| Klebsormidium subtile | terrestrial | 1KP | Phy | |||||
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallot-Lavallée, L.; Blanc, G. A Glimpse of Nucleo-Cytoplasmic Large DNA Virus Biodiversity through the Eukaryotic Genomics Window. Viruses 2017, 9, 17. https://doi.org/10.3390/v9010017
Gallot-Lavallée L, Blanc G. A Glimpse of Nucleo-Cytoplasmic Large DNA Virus Biodiversity through the Eukaryotic Genomics Window. Viruses. 2017; 9(1):17. https://doi.org/10.3390/v9010017
Chicago/Turabian StyleGallot-Lavallée, Lucie, and Guillaume Blanc. 2017. "A Glimpse of Nucleo-Cytoplasmic Large DNA Virus Biodiversity through the Eukaryotic Genomics Window" Viruses 9, no. 1: 17. https://doi.org/10.3390/v9010017