Characterization of Nucleoside Reverse Transcriptase Inhibitor-Associated Mutations in the RNase H Region of HIV-1 Subtype C Infected Individuals

 , , and
, , and {kind=link}
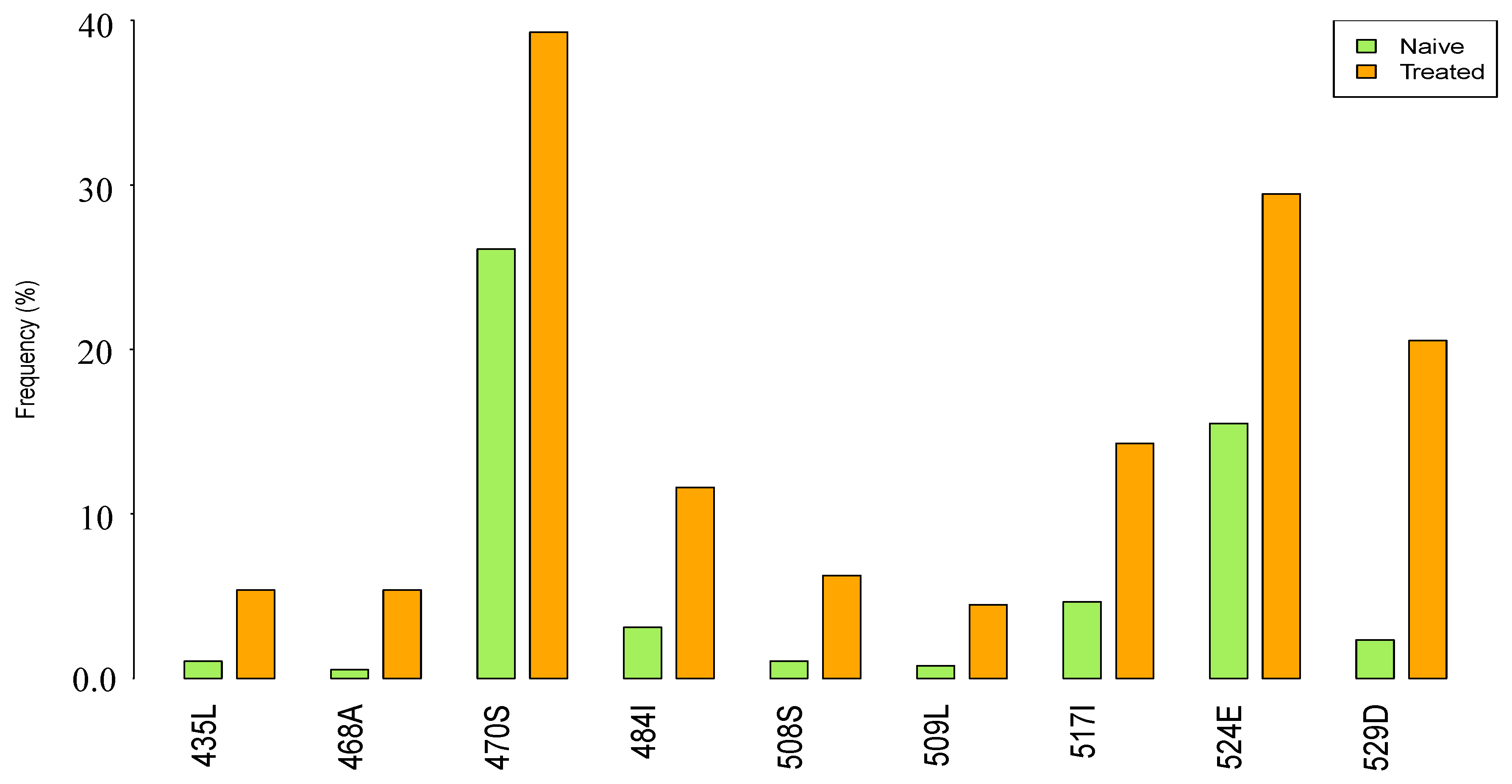{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. Amplification, Sequencing and Phylogenetic Analyses of the RNase H Domain
2.3. Identification of Treatment-Related Mutations in RNase H of HIV-1 Subtype C
2.4. Bayesian Network Construction
3. Results
3.1. Prevalence of RNase H Mutations in ART-Naive Subtype C Sequences
3.2. Prevalence of RNase H Mutations in NRTI-Treatment-Experienced Compared to ART-Naive Subtype C Sequences
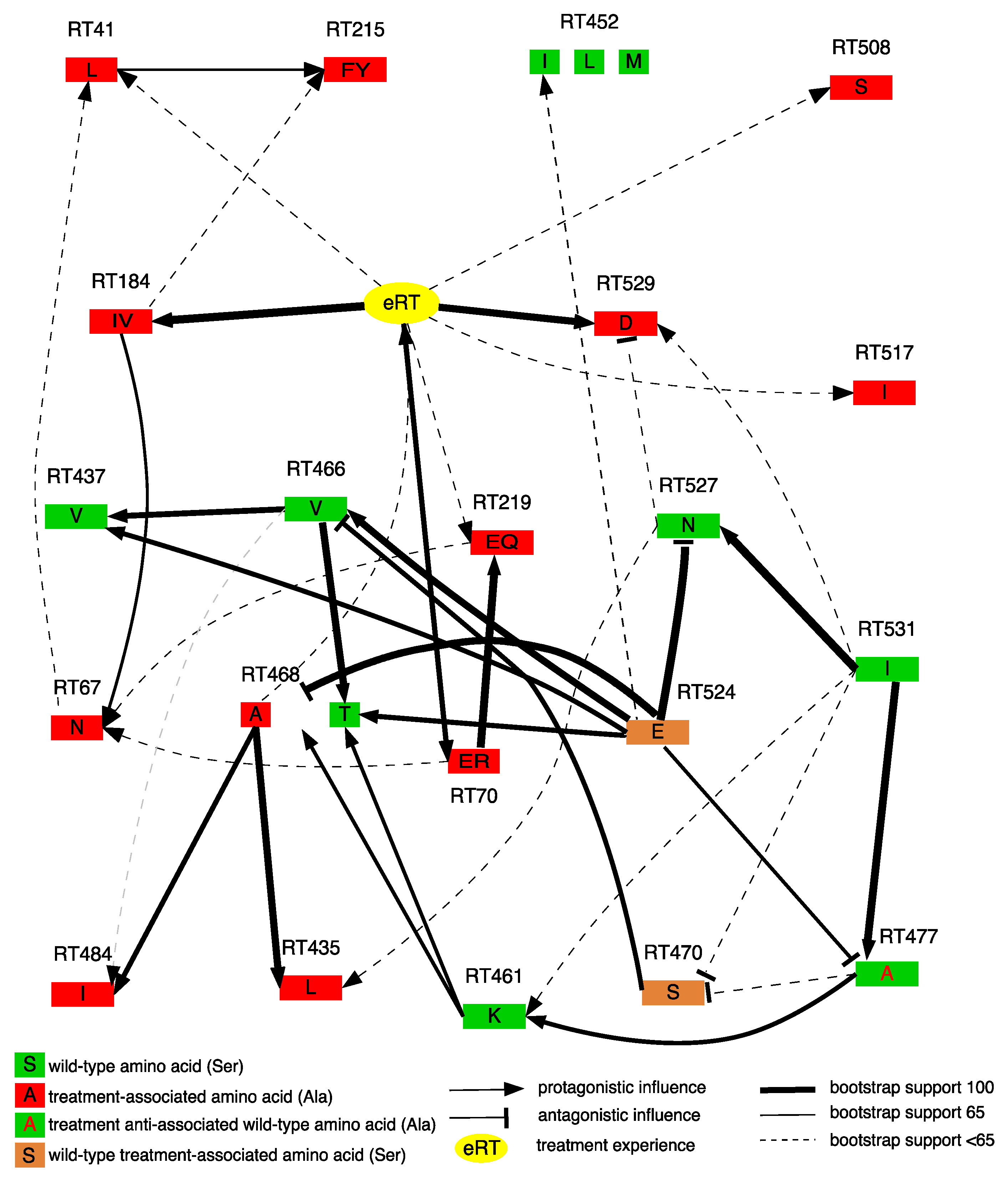
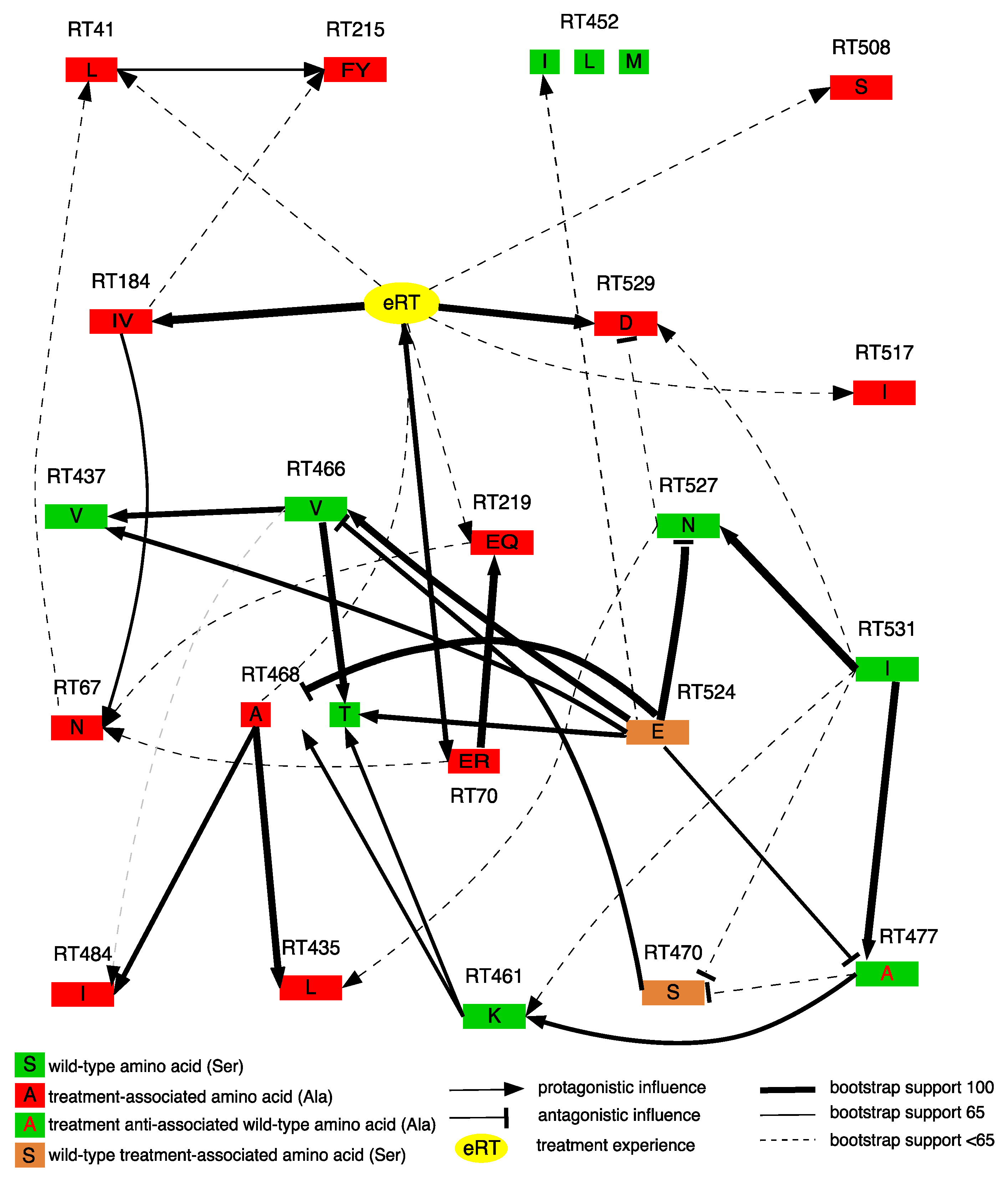
3.3. Dependencies between RNase H domain Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Authors Contributions
Conflicts of Interest
References
- UNAIDS. Global AIDS Update. 2016. Available online: http://www.unaids.org/en/resources/documents/2016/Global-AIDS-update-2016 (accessed on 7 June 2017).
- Department of Health National Comprehensive HIV and AIDS plan statistics; South Africa. 2008. Available online: http://www.health.gov.za/edp.php (accessed on 7 June 2017).
- Meintjes, G.; Moorhouse, M.A.; Carmona, S.; Davies, N.; Dlamini, S.; van Vuuren, C.; Manzini, T.; Mathe, M.; Moosa, Y.; Nash, J.; et al. Adult antiretroviral therapy guidelines 2017. S. Afr. J. HIV Med. 2017, 18, 1–24. [Google Scholar] [CrossRef]
- Hemelaar, J.; Gouws, E.; Ghys, P.D.; Osmanov, S. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. AIDS 2006, 20, W13–W23. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Sobieszczyk, M.E.; McCutchan, F.E.; Hammer, S.M. The challenge of HIV-1 subtype diversity. N. Engl. J. Med. 2008, 358, 1590–1602. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cajas, J.L.; Pai, N.P.; Klein, M.B.; Wainberg, M.A. Differences in resistance mutations among HIV-1 non-subtype B infections: A systematic review of evidence (1996–2008). J. Int. AIDS Soc. 2009, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.F.; Lengruber, R.B.; Soares, E.A.; Jere, A.; Sprinz, E.; Martinez, A.M.; Silveira, J.; Sion, F.S.; Pathak, V.K.; Soares, M.A. Conservation patterns of HIV-1 RT connection and RNase H domains: identification of new mutations in NRTI-treated patients. PLoS ONE 2008, 3, e1781. [Google Scholar] [CrossRef] [PubMed]
- Barral, M.F.M.; Sousa, A.K.P.; Santos, A.F.; Abreu, C.M.; Tanuri, A.; Soares, M.A. Identification of novel resistance-related polymorphisms in HIV-1 subtype C RT connection and RNase H domains from patients under virological failure in Brazil. AIDS Res. Hum. Retrovir. 2017, 33, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Delviks-Frankenberry, K.A.; Nikolenko, G.N.; Barr, R.; Pathak, V.K. Mutations in human immunodeficiency virus type 1 RNase H primer grip enhance 3′-azido-3′-deoxythymidine resistance. J. Virol. 2007, 81, 6837–6845. [Google Scholar] [CrossRef] [PubMed]
- Nikolenko, G.N.; Palmer, S.; Maldarelli, F.; Mellors, J.W.; Coffin, J.M.; Pathak, V.K. Mechanism for nucleoside analog-mediated abrogation of HIV-1 replication: Balance between RNase H activity and nucleotide excision. Proc. Natl. Acad. Sci. USA 2005, 102, 2093–2098. [Google Scholar] [CrossRef] [PubMed]
- Ntemgwa, M.; Wainberg, M.A.; Oliveira, M.; Moisi, D.; Lalonde, R.; Micheli, V.; Brenner, B.G. Variations in reverse transcriptase and RNase H domain mutations in human immunodeficiency virus type 1 clinical isolates are associated with divergent phenotypic resistance to zidovudine. Antimicrob. Agents Chemother. 2007, 51, 3861–3869. [Google Scholar] [CrossRef] [PubMed]
- Roquebert, B.; Wirden, M.; Simon, A.; Deval, J.; Katlama, C.; Calvez, V.; Marcelin, A.G. Relationship between mutations in HIV-1 RNase H domain and nucleoside reverse transcriptase inhibitors resistance mutations in naive and pre-treated HIV infected patients. J. Med. Virol. 2007, 79, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Das, K.; Tantillo, C.; Clark, A.D., Jr.; Ding, J.; Whitcomb, J.M.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA: DNA. EMBO J. 2001, 20, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Marconi, V.C.; Sunpath, H.; Lu, Z.; Gordon, M.; Koranteng-Apeagyei, K.; Hampton, J.; Carpenter, S.; Giddy, J.; Ross, D.; Holst, H.; et al. Prevalence of HIV-1 drug resistance after failure of a first highly active antiretroviral therapy regimen in KwaZulu Natal, South Africa. Clin. Infect. Dis. 2008, 46, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Green, T.N.; Archary, M.; Gordon, M.L.; Padayachi, N.; Lie, Y.; Anton, E.D.; Reeves, J.D.; Grobler, A.; Bobat, R.; Coovadia, H.; et al. Drug resistance and coreceptor usage in HIV type 1 subtype C-infected children initiating or failing highly active antiretroviral therapy in South Africa. AIDS Res. Hum. Retrovir. 2012, 28, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Hahn, B.H.; Shaw, G.M.; Arya, S.K.; Popovic, M.; Gallo, R.C.; Wong-Staal, F. Molecular cloning and characterization of the HTLV-III virus associated with AIDS. Nature 1984, 312, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Alcantara, L.C.; Cassol, S.; Libin, P.; Deforche, K.; Pybus, O.G.; van Ranst, M.; Galvao-Castro, B.; Vandamme, A.M.; de Oliveira, T. A standardized framework for accurate, high-throughput genotyping of recombinant and non-recombinant viral sequences. Nucleic Acids Res. 2009, 37, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Pineda-Pena, A.C.; Faria, N.R.; Imbrechts, S.; Libin, P.; Abecasis, A.B.; Deforche, K.; Gomez-Lopez, A.; Camacho, R.J.; de Oliveira, T.; Vandamme, A.M. Automated subtyping of HIV-1 genetic sequences for clinical and surveillance purposes: Performance evaluation of the new REGA version 3 and seven other tools. Infect. Genet. Evol. 2013, 19, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Kantor, R.; Katzenstein, D.A.; Camacho, R.; Morris, L.; Sirivichayakul, S.; Jorgensen, L.; Brigido, L.F.; Schapiro, J.M.; Shafer, R.W. HIV-1 pol mutation frequency by subtype and treatment experience: Extension of the HIVseq program to seven non-B subtypes. AIDS 2006, 20, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Columb, M.O.; Sagadai, S. Multiple comparisons. Curr. Anaesth. Crit. Care 2006, 17, 233–236. [Google Scholar] [CrossRef]
- Brehm, J.H.; Koontz, D.; Meteer, J.D.; Pathak, V.; Sluis-Cremer, N.; Mellors, J.W. Selection of mutations in the connection and RNase H domains of human immunodeficiency virus type 1 reverse transcriptase that increase resistance to 3′-azido-3′-dideoxythymidine. J. Virol. 2007, 81, 7852–7859. [Google Scholar] [CrossRef] [PubMed]
- Hachiya, A.; Shimane, K.; Sarafianos, S.G.; Kodama, E.N.; Sakagami, Y.; Negishi, F.; Koizumi, H.; Gatanaga, H.; Matsuoka, M.; Takiguchi, M.; et al. Clinical relevance of substitutions in the connection subdomain and RNase H domain of HIV-1 reverse transcriptase from a cohort of antiretroviral treatment-naive patients. Antivir. Res. 2009, 82, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Nikolenko, G.N.; Delviks-Frankenberry, K.A.; Palmer, S.; Maldarelli, F.; Fivash, M.J., Jr.; Coffin, J.M.; Pathak, V.K. Mutations in the connection domain of HIV-1 reverse transcriptase increase 3′-azido-3′-deoxythymidine resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.; Schulze, T.; Larder, B.A.; Moelling, K. Mutations within the RNase H domain of human immunodeficiency virus type 1 reverse transcriptase abolish virus infectivity. J. Gen. Virol. 1991, 72 Pt 1, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, S.; Wohrl, B.M.; Tisdale, M.; Moelling, K. Enzymatic analysis of two HIV-1 reverse transcriptase mutants with mutations in carboxyl-terminal amino acid residues conserved among retroviral ribonucleases H. J. Biol. Chem. 1993, 268, 2674–2683. [Google Scholar] [PubMed]
- Wohrl, B.M.; Volkmann, S.; Moelling, K. Mutations of a conserved residue within HIV-1 ribonuclease H affect its exo- and endonuclease activities. J. Mol. Biol. 1991, 220, 801–818. [Google Scholar] [CrossRef]
- Lengruber, R.B.; Delviks-Frankenberry, K.A.; Nikolenko, G.N.; Baumann, J.; Santos, A.F.; Pathak, V.K.; Soares, M.A. Phenotypic characterization of drug resistance-associated mutations in HIV-1 RT connection and RNase H domains and their correlation with thymidine analogue mutations. J. Antimicrob. Chemother. 2011, 66, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Waters, J.M.; O’Neal, W.; White, K.L.; Wakeford, C.; Lansdon, E.B.; Harris, J.; Svarovskaia, E.S.; Miller, M.D.; Borroto-Esoda, K. Mutations in the thumb-connection and RNase H domain of HIV type-1 reverse transcriptase of antiretroviral treatment-experienced patients. Antivir. Ther. 2009, 14, 231–239. [Google Scholar] [PubMed]
- Deforche, K.; Camacho, R.; Grossman, Z.; Silander, T.; Soares, M.A.; Moreau, Y.; Shafer, R.W.; van Laethem, K.; Carvalho, A.P.; Wynhoven, B.; et al. Bayesian network analysis of resistance pathways against HIV-1 protease inhibitors. Infect. Genet. Evol. 2007, 7, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Theys, K.; Deforche, K.; Libin, P.; Camacho, R.J.; van Laethem, K.; Vandamme, A.M. Resistance pathways of human immunodeficiency virus type 1 against the combination of zidovudine and lamivudine. J. Gen. Virol. 2010, 91 Pt 8, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Cuypers, L.; Libin, P.; Schrooten, Y.; Theys, K.; di Maio, V.C.; Cento, V.; Lunar, M.M.; Nevens, F.; Poljak, M.; Ceccherini-Silberstein, F.; et al. Exploring resistance pathways for first-generation NS3/4A protease inhibitors boceprevir and telaprevir using Bayesian network learning. Infect. Genet. Evol. 2017, 53, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Arion, D.; Sluis-Cremer, N.; Min, K.L.; Abram, M.E.; Fletcher, R.S.; Parniak, M.A. Mutational analysis of Tyr-501 of HIV-1 reverse transcriptase. Effects on ribonuclease H activity and inhibition of this activity by N-acylhydrazones. J. Biol. Chem. 2002, 277, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- Julias, J.G.; McWilliams, M.J.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. Mutations in the RNase H domain of HIV-1 reverse transcriptase affect the initiation of DNA synthesis and the specificity of RNase H cleavage in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 9515–9520. [Google Scholar] [CrossRef] [PubMed]
- Abram, M.E.; Parniak, M.A. Virion instability of human immunodeficiency virus type 1 reverse transcriptase (RT) mutated in the protease cleavage site between RT p51 and the RT RNase H domain. J. Virol. 2005, 79, 11952–11961. [Google Scholar] [CrossRef] [PubMed]
- Iordanskiy, S.; Waltke, M.; Feng, Y.; Wood, C. Subtype-associated differences in HIV-1 reverse transcription affect the viral replication. Retrovirology 2010, 7, 85. [Google Scholar] [CrossRef] [PubMed]

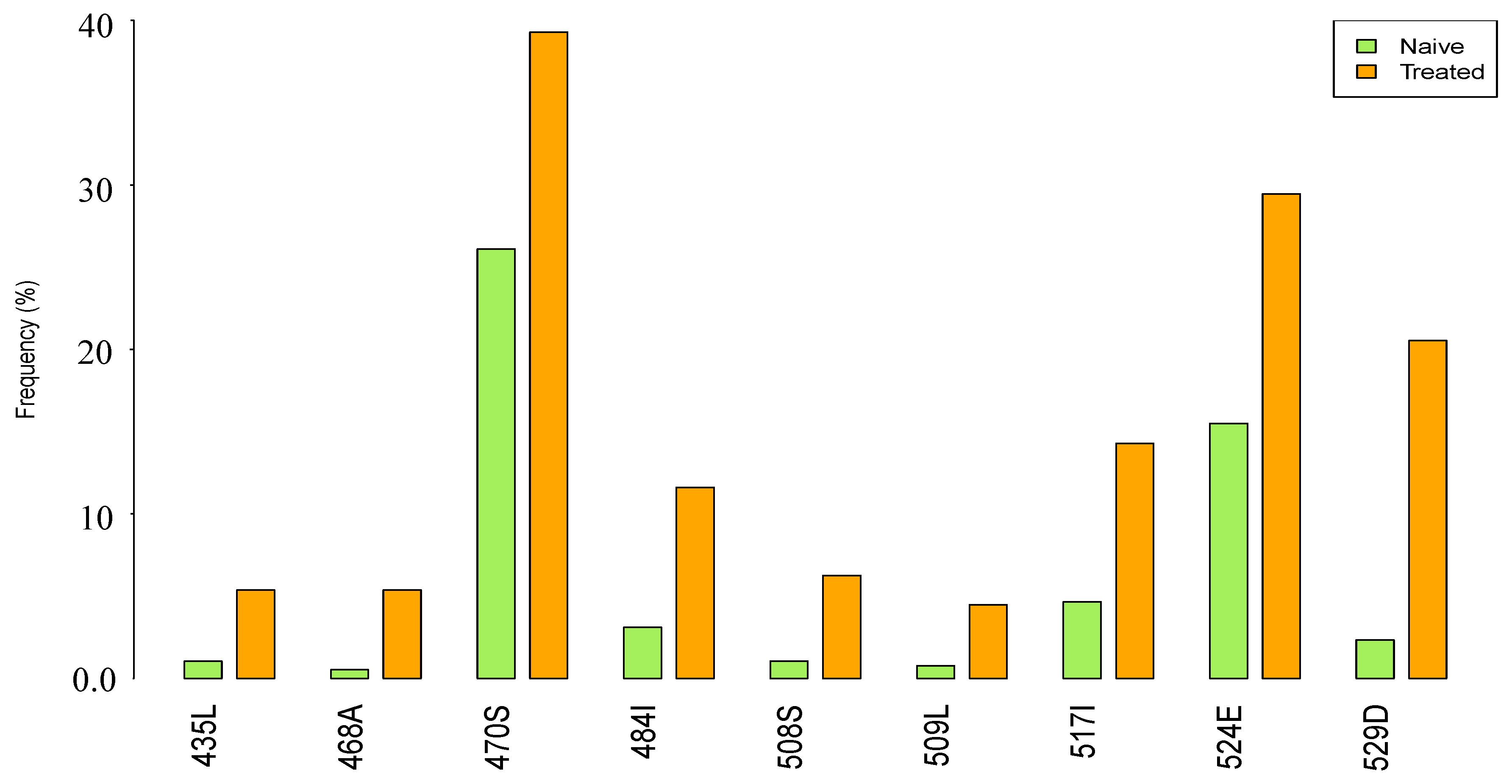
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngcapu, S.; Theys, K.; Libin, P.; Marconi, V.C.; Sunpath, H.; Ndung’u, T.; Gordon, M.L. Characterization of Nucleoside Reverse Transcriptase Inhibitor-Associated Mutations in the RNase H Region of HIV-1 Subtype C Infected Individuals. Viruses 2017, 9, 330. https://doi.org/10.3390/v9110330
Ngcapu S, Theys K, Libin P, Marconi VC, Sunpath H, Ndung’u T, Gordon ML. Characterization of Nucleoside Reverse Transcriptase Inhibitor-Associated Mutations in the RNase H Region of HIV-1 Subtype C Infected Individuals. Viruses. 2017; 9(11):330. https://doi.org/10.3390/v9110330
Chicago/Turabian StyleNgcapu, Sinaye, Kristof Theys, Pieter Libin, Vincent C. Marconi, Henry Sunpath, Thumbi Ndung’u, and Michelle L. Gordon. 2017. "Characterization of Nucleoside Reverse Transcriptase Inhibitor-Associated Mutations in the RNase H Region of HIV-1 Subtype C Infected Individuals" Viruses 9, no. 11: 330. https://doi.org/10.3390/v9110330