Evidence of Drug–Nutrient Interactions with Chronic Use of Commonly Prescribed Medications: An Update

Abstract
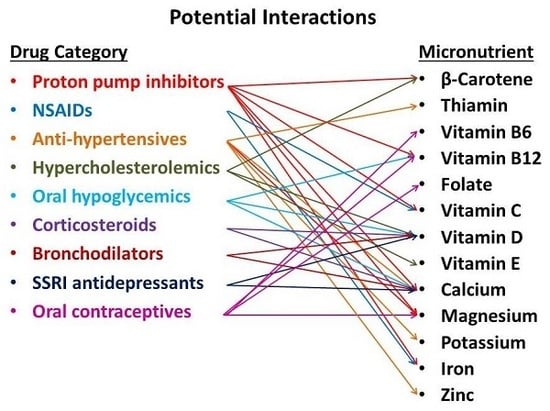
:
1. Introduction
2. Part I: Medications Most Likely to Affect Nutritional Status
2.1. Proton Pump Inhibitors (PPIs)
2.1.1. Vitamin B12
2.1.2. Vitamin C
2.1.3. Iron
2.1.4. Calcium
2.1.5. Magnesium
2.1.6. Zinc
2.1.7. Beta (β)-Carotene
2.2. NSAIDs: Aspirin
2.2.1. Vitamin C
2.2.2. Iron
2.3. Anti-Hypertensives: Diuretics
2.3.1. Calcium and Loop Diuretics
2.3.2. Calcium and Thiazides
2.3.3. Magnesium
2.3.4. Thiamin
2.3.5. Zinc
2.3.6. Potassium
2.3.7. Folate
2.4. Anti-Hypertensives: Angiotensin-Converting Enzyme (ACE) Inhibitors
2.4.1. Zinc
2.4.2. Potassium
2.5. Anti-Hypertensives: Calcium Channel Blockers (CCBs)
2.5.1. Folate
2.5.2. Potassium
2.6. Hypercholesterolemics: Statins
2.6.1. Coenzyme Q10 (CoQ10)
2.6.2. Vitamin D
2.6.3. Vitamin E and β-Carotene
2.7. Oral Hypoglycemics: Metformin
Vitamin B12
2.8. Oral Hypoglycemics: Thiazolidinediones (TZD)
Calcium and Vitamin D
2.9. Oral Corticosteroids
2.9.1. Calcium and Vitamin D
2.9.2. Sodium and Potassium
2.9.3. Chromium
2.10. Bronchodilators: Beta2-Agonists and Inhaled Corticosteroids [ICS]
Calcium and Vitamin D
2.11. Antidepressants
Calcium and Vitamin D
2.12. Oral Contraceptives (OC)
2.12.1. Vitamin B6
2.12.2. Vitamin B12
2.12.3. Folate
2.12.4. Calcium
2.12.5. Magnesium
2.12.6. Vitamin C and E
3. Part II: Medications Potentially Affected by Nutritional Status
3.1. Antidepressants and Folate
3.2. ACE Inhibitors and Iron
4. Discussion
5. Conclusions
Author Contributions
Conflicts of Interest
References
- McCabe, B.J. Prevention of food-drug interactions with special emphasis on older adults. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Samaras, D.; Samaras, N.; Lang, P.-O.; Genton, L.; Frangos, E.; Pichard, C. Effects of widely used drugs on micronutrients: A story rarely told. Nutrition 2013, 29, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Roe, D. Diet and Drug Interactions; Springer: New York, NY, USA, 1989; ISBN 978-94-011-6047-6. [Google Scholar]
- Chan, L.-N. Drug-nutrient interactions. JPEN J. Parenter. Enteral Nutr. 2013, 37, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Mason, P. Important drug-nutrient interactions. Proc. Nutr. Soc. 2010, 69, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Boullata, J.I.; Hudson, L.M. Drug-nutrient interactions: A broad view with implications for practice. J. Acad. Nutr. Diet. 2012, 112, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Hing, E.; Rui, P.; Palso, K. National Ambulatory Medical Care Survey: 2013 State and National Summary Tables. Available online: https://www.cdc.gov/nchs/data/ahcd/namcs_summary/2013_namcs_web_tables.pdf (accessed on 17 March 2018).
- National Center for Health Statistics (US). Health, United States, 2015: With Special Feature on Racial and Ethnic Health Disparities; Health, United States; National Center for Health Statistics (US): Hyattsville, MD, USA, 2016.
- Weissman, J.F.; Pratt, L.A.; Miller, E.A.; Parker, J.D. Serious Psychological Distress among Adults: United States, 2009–2013. NCHS Data Brief 2015, 203, 1–8. [Google Scholar]
- Valuck, R.J.; Ruscin, J.M. A case-control study on adverse effects: H2 blocker or proton pump inhibitor use and risk of vitamin B12 deficiency in older adults. J. Clin. Epidemiol. 2004, 57, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Dharmarajan, T.S.; Kanagala, M.R.; Murakonda, P.; Lebelt, A.S.; Norkus, E.P. Do acid-lowering agents affect vitamin B12 status in older adults? J. Am. Med. Dir. Assoc. 2008, 9, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Schenk, B.E.; Kuipers, E.J.; Klinkenberg-Knol, E.C.; Bloemena, E.C.; Sandell, M.; Nelis, G.F.; Snel, P.; Festen, H.P.; Meuwissen, S.G. Atrophic gastritis during long-term omeprazole therapy affects serum vitamin B12 levels. Aliment. Pharmacol. Ther. 1999, 13, 1343–1346. [Google Scholar] [CrossRef] [PubMed]
- Schenk, B.E.; Festen, H.P.; Kuipers, E.J.; Klinkenberg-Knol, E.C.; Meuwissen, S.G. Effect of short- and long-term treatment with omeprazole on the absorption and serum levels of cobalamin. Aliment. Pharmacol. Ther. 1996, 10, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Saltzman, J.R.; Kemp, J.A.; Golner, B.B.; Pedrosa, M.C.; Dallal, G.E.; Russell, R.M. Effect of hypochlorhydria due to omeprazole treatment or atrophic gastritis on protein-bound vitamin B12 absorption. J. Am. Coll. Nutr. 1994, 13, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Labenz, J.; Tillenburg, B.; Peitz, U.; Idstrom, J.P.; Verdu, E.F.; Stolte, M.; Borsch, G.; Blum, A.L. Helicobacter pylori augments the pH-increasing effect of omeprazole in patients with duodenal ulcer. Gastroenterology 1996, 110, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Marcuard, S.P.; Albernaz, L.; Khazanie, P.G. Omeprazole therapy causes malabsorption of cyanocobalamin (vitamin B12). Ann. Intern. Med. 1994, 120, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Sagar, M.; Janczewska, I.; Ljungdahl, A.; Bertilsson, L.; Seensalu, R. Effect of CYP2C19 polymorphism on serum levels of vitamin B12 in patients on long-term omeprazole treatment. Aliment. Pharmacol. Ther. 1999, 13, 453–458. [Google Scholar] [CrossRef] [PubMed]
- McColl, K.E.L. Effect of proton pump inhibitors on vitamins and iron. Am. J. Gastroenterol. 2009, 104 (Suppl. S2), S5–S9. [Google Scholar] [CrossRef] [PubMed]
- Mowat, C.; Williams, C.; Gillen, D.; Hossack, M.; Gilmour, D.; Carswell, A.; Wirz, A.; Preston, T.; McColl, K.E. Omeprazole, Helicobacter pylori status, and alterations in the intragastric milieu facilitating bacterial N-nitrosation. Gastroenterology 2000, 119, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Woodward, M.; Tunstall-Pedoe, H.; McColl, K. Helicobacter pylori infection reduces systemic availability of dietary vitamin C. Eur. J. Gastroenterol. Hepatol. 2001, 13, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Mowat, C.; Carswell, A.; Wirz, A.; McColl, K.E. Omeprazole and dietary nitrate independently affect levels of vitamin C and nitrite in gastric juice. Gastroenterology 1999, 116, 813–822. [Google Scholar] [CrossRef]
- Henry, E.B.; Carswell, A.; Wirz, A.; Fyffe, V.; McColl, K.E.L. Proton pump inhibitors reduce the bioavailability of dietary vitamin C. Aliment. Pharmacol. Ther. 2005, 22, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.R.; Brannon, M.A.; Carloss, E.A. Effect of omeprazole on oral iron replacement in patients with iron deficiency anemia. South. Med. J. 2004, 97, 887–889. [Google Scholar] [CrossRef] [PubMed]
- Kaye, J.A.; Jick, H. Proton pump inhibitor use and risk of hip fractures in patients without major risk factors. Pharmacotherapy 2008, 28, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Corley, D.A.; Kubo, A.; Zhao, W.; Quesenberry, C. Proton pump inhibitors and histamine-2 receptor antagonists are associated with hip fractures among at-risk patients. Gastroenterology 2010, 139, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Khalili, H.; Huang, E.S.; Jacobson, B.C.; Camargo, C.A.; Feskanich, D.; Chan, A.T. Use of proton pump inhibitors and risk of hip fracture in relation to dietary and lifestyle factors: A prospective cohort study. BMJ 2012, 344, e372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, M.B.; Madden, D.M.; Murray, A.M.; Heaney, R.P.; Kerzner, L.J. Effects of proton pump inhibitors on calcium carbonate absorption in women: A randomized crossover trial. Am. J. Med. 2005, 118, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.J.; Sullivan, R.R.; Gaffney-Stomberg, E.; Caseria, D.M.; O’Brien, K.O.; Proctor, D.D.; Simpson, C.A.; Kerstetter, J.E.; Insogna, K.L. Inhibiting gastric acid production does not affect intestinal calcium absorption in young, healthy individuals: A randomized, crossover, controlled clinical trial. J. Bone Miner. Res. 2010, 25, 2205–2211. [Google Scholar] [CrossRef] [PubMed]
- William, J.H.; Danziger, J. Proton-pump inhibitor-induced hypomagnesemia: Current research and proposed mechanisms. World J. Nephrol. 2016, 5, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Toh, J.W.T.; Ong, E.; Wilson, R. Hypomagnesaemia associated with long-term use of proton pump inhibitors. Gastroenterol. Rep. 2015, 3, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Cundy, T.; Dissanayake, A. Severe hypomagnesaemia in long-term users of proton-pump inhibitors. Clin. Endocrinol. 2008, 69, 338–341. [Google Scholar] [CrossRef] [PubMed]
- Basu, T.K. Vitamin C-aspirin interactions. Int. J. Vitam. Nutr. Res. Suppl. 1982, 23, 83–90. [Google Scholar] [PubMed]
- Loh, H.S.; Wilson, C.W.M. The Interactions of Aspirin and Ascorbic Acid in Normal Men. J. Clin. Pharmacol. 1975, 15, 36–45. [Google Scholar] [CrossRef]
- Wilson, C.W.; Greene, M. The relationship of aspirin to ascorbic acid metabolism during the common cold. J. Clin. Pharmacol. 1978, 18, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Schulz, H.U.; Schürer, M.; Krupp, S.; Dammann, H.G.; Timm, J.; Gessner, U. Effects of acetylsalicylic acid on ascorbic acid concentrations in plasma, gastric mucosa, gastric juice and urine—A double-blind study in healthy subjects. Int. J. Clin. Pharmacol. Ther. 2004, 42, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Black, D.A.; Fraser, C.M. Iron deficiency anaemia and aspirin use in old age. Br. J. Gen. Pract. 1999, 49, 729–730. [Google Scholar] [PubMed]
- Gaskell, H.; Derry, S.; Moore, R.A. Is there an association between low dose aspirin and anemia (without overt bleeding)? narrative review. BMC Geriatr. 2010, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Fleming, D.J.; Jacques, P.F.; Massaro, J.M.; D’Agostino, R.B.; Wilson, P.W.; Wood, R.J. Aspirin intake and the use of serum ferritin as a measure of iron status. Am. J. Clin. Nutr. 2001, 74, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Kaffes, A.; Cullen, J.; Mitchell, H.; Katelaris, P.H. Effect of Helicobacter pylori infection and low-dose aspirin use on iron stores in the elderly. J. Gastroenterol. Hepatol. 2003, 18, 1024–1028. [Google Scholar] [CrossRef] [PubMed]
- Coe, F.L.; Canterbury, J.M.; Firpo, J.J.; Reiss, E. Evidence for secondary hyperparathyroidism in idiopathic hypercalciuria. J. Clin. Investig. 1973, 52, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Chan, J.C.; Bartter, F.C. Effects of oral furosemide and salt loading on parathyroid function in normal subjects. Physiological basis for renal hypercalciuria. Nephron 1984, 38, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Rejnmark, L.; Vestergaard, P.; Pedersen, A.R.; Heickendorff, L.; Andreasen, F.; Mosekilde, L. Dose-effect relations of loop- and thiazide-diuretics on calcium homeostasis: A randomized, double-blinded Latin-square multiple cross-over study in postmenopausal osteopenic women. Eur. J. Clin. Investig. 2003, 33, 41–50. [Google Scholar] [CrossRef]
- Ooms, M.E.; Lips, P.; Van Lingen, A.; Valkenburg, H.A. Determinants of bone mineral density and risk factors for osteoporosis in healthy elderly women. J. Bone Miner. Res. 1993, 8, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Rejnmark, L.; Vestergaard, P.; Heickendorff, L.; Andreasen, F.; Mosekilde, L. Effects of long-term treatment with loop diuretics on bone mineral density, calcitropic hormones and bone turnover. J. Intern. Med. 2005, 257, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Rejnmark, L.; Vestergaard, P.; Heickendorff, L.; Andreasen, F.; Mosekilde, L. Loop diuretics increase bone turnover and decrease BMD in osteopenic postmenopausal women: Results from a randomized controlled study with bumetanide. J. Bone Miner. Res. 2006, 21, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Heidrich, F.E.; Stergachis, A.; Gross, K.M. Diuretic drug use and the risk for hip fracture. Ann. Intern. Med. 1991, 115, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Rejnmark, L.; Vestergaard, P.; Mosekilde, L. Fracture risk in patients treated with loop diuretics. J. Intern. Med. 2006, 259, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Tromp, A.M.; Ooms, M.E.; Popp-Snijders, C.; Roos, J.C.; Lips, P. Predictors of fractures in elderly women. Osteoporos. Int. 2000, 11, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Brickman, A.S.; Massry, S.G.; Coburn, J.W. changes in serum and urinary calcium during treatment with hydrochlorothiazide: Studies on mechanisms. J. Clin. Investig. 1972, 51, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Nguyen, T.; Sambrook, P.N.; Eisman, J.A. Thiazide diuretics and fractures: Can meta-analysis help? J. Bone Miner. Res. 1995, 10, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.; Htay, T. Thiazide diuretics and the risk of hip fracture. Cochrane Database Syst. Rev. 2011, CD005185. [Google Scholar] [CrossRef] [PubMed]
- Wermers, R.A.; Kearns, A.E.; Jenkins, G.D.; Melton, L.J. Incidence and clinical spectrum of thiazide-associated hypercalcemia. Am. J. Med. 2007, 120, 911.e9–15. [Google Scholar] [CrossRef] [PubMed]
- Chandler, P.D.; Scott, J.B.; Drake, B.F.; Ng, K.; Forman, J.P.; Chan, A.T.; Bennett, G.G.; Hollis, B.W.; Giovannucci, E.L.; Emmons, K.M.; et al. Risk of hypercalcemia in blacks taking hydrochlorothiazide and vitamin D. Am. J. Med. 2014, 127, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, A.M. The interactions of thiazide diuretics with parathyroid hormone and vitamin D. J. Clin. Investig. 1972, 51, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Sica, D.A. Diuretic-related side effects: Development and treatment. J. Clin. Hypertens. 2004, 6, 532–540. [Google Scholar] [CrossRef]
- Makam, A.N.; Boscardin, W.J.; Miao, Y.; Steinman, M.A. Risk of thiazide-induced metabolic adverse events in older adults. J. Am. Geriatr. Soc. 2014, 62, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Cumber, P.; Grimes, L.; Treby, D.; Bryant, R.; Rawlins, D.; Ising, H. The metabolic effects of thiazide therapy in the elderly: A population study. Age Ageing 1986, 15, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Brady, J.A.; Rock, C.L.; Horneffer, M.R. Thiamin status, diuretic medications, and the management of congestive heart failure. J. Am. Diet. Assoc. 1995, 95, 541–544. [Google Scholar] [CrossRef]
- Zenuk, C.; Healey, J.; Donnelly, J.; Vaillancourt, R.; Almalki, Y.; Smith, S. Thiamine deficiency in congestive heart failure patients receiving long term furosemide therapy. Can. J. Clin. Pharmacol. 2003, 10, 184–188. [Google Scholar] [PubMed]
- Seligmann, H.; Halkin, H.; Rauchfleisch, S.; Kaufmann, N.; Tal, R.; Motro, M.; Vered, Z.; Ezra, D. Thiamine deficiency in patients with congestive heart failure receiving long-term furosemide therapy: A pilot study. Am. J. Med. 1991, 91, 151–155. [Google Scholar] [CrossRef]
- Suter, P.M.; Haller, J.; Hany, A.; Vetter, W. Diuretic use: A risk for subclinical thiamine deficiency in elderly patients. J. Nutr. Health Aging 2000, 4, 69–71. [Google Scholar] [PubMed]
- McCabe-Sellers, B.J.; Sharkey, J.R.; Browne, B.A. Diuretic medication therapy use and low thiamin intake in homebound older adults. J. Nutr. Elder. 2005, 24, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.J.; Leary, W.P.; Lockett, C.J.; Alcocer, L. Diuretics and zinc. S. Afr. Med. J. 1982, 62, 373–375. [Google Scholar] [PubMed]
- Reyes, A.J.; Olhaberry, J.V.; Leary, W.P.; Lockett, C.J.; van der Byl, K. Urinary zinc excretion, diuretics, zinc deficiency and some side-effects of diuretics. S. Afr. Med. J. 1983, 64, 936–941. [Google Scholar] [PubMed]
- Wester, P.O. Urinary zinc excretion during treatment with different diuretics. Acta Med. Scand. 1980, 208, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Golik, A.; Modai, D.; Weissgarten, J.; Cohen, N.; Averbukh, Z.; Sigler, E.; Zaidenstein, R.; Shaked, U. Hydrochlorothiazide-amiloride causes excessive urinary zinc excretion. Clin. Pharmacol. Ther. 1987, 42, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Khedun, S.M.; Naicker, T.; Maharaj, B. Zinc, hydrochlorothiazide and sexual dysfunction. Cent. Afr. J. Med. 1995, 41, 312–315. [Google Scholar] [PubMed]
- Wester, P.O. Tissue zinc at autopsy--relation to medication with diuretics. Acta Med. Scand. 1980, 208, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.; Golik, A. Zinc balance and medications commonly used in the management of heart failure. Heart Fail. Rev. 2006, 11, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Dørup, I.; Skajaa, K.; Clausen, T.; Kjeldsen, K. Reduced concentrations of potassium, magnesium, and sodium-potassium pumps in human skeletal muscle during treatment with diuretics. Br. Med. J. Clin. Res. Ed. 1988, 296, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Clayton, J.A.; Rodgers, S.; Blakey, J.; Avery, A.; Hall, I.P. Thiazide diuretic prescription and electrolyte abnormalities in primary care. Br. J. Clin. Pharmacol. 2006, 61, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Peterzan, M.A.; Hardy, R.; Chaturvedi, N.; Hughes, A.D. Meta-analysis of dose-response relationships for hydrochlorothiazide, chlorthalidone, and bendroflumethiazide on blood pressure, serum potassium, and urate. Hypertens. Dallas Tex 1979 2012, 59, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.E.; Carter, B.L.; Zheng, S.; Grimm, R.H. Meta-analysis of dose-response characteristics of hydrochlorothiazide and chlorthalidone: Effects on systolic blood pressure and potassium. Am. J. Hypertens. 2010, 23, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Hulley, S.B.; Black, D.M.; Cheitlin, M.D.; Sebastian, A.; Seeley, D.G.; Hearst, N.; Fine, R. Diuretics, serum and intracellular electrolyte levels, and ventricular arrhythmias in hypertensive men. JAMA 1992, 267, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, F.L.; Bateman, J.R. Megaloblastic anemia possibly induced by triamterene in patients with alcoholic cirrhosis. Two case reports. Ann. Intern. Med. 1968, 68, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.B.; Zimmerman, J.; Otradovec, C.L.; Selhub, J.; Rosenberg, I.H. Chronic diuretic therapy with moderate doses of triamterene is not associated with folate deficiency. J. Lab. Clin. Med. 1991, 117, 365–369. [Google Scholar] [PubMed]
- O’Connor, D.T.; Strause, L.; Saltman, P.; Parmer, R.J.; Cervenka, J. Serum zinc is unaffected by effective captopril treatment of hypertension. J. Clin. Hypertens. 1987, 3, 405–408. [Google Scholar] [PubMed]
- Trasobares, E.; Corbatón, A.; González-Estecha, M.; Lopez-Colón, J.L.; Prats, P.; Olivan, P.; Sánchez, J.A.; Arroyo, M. Effects of angiotensin-converting enzyme inhibitors (ACE i) on zinc metabolism in patients with heart failure. J. Trace Elem. Med. Biol. 2007, 21 (Suppl. S1), 53–55. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.J.; Hoorntje, S.J.; Donker, A.J.M. Zinc Deficiency during Captopril Treatment. Nephron 1983, 34, 195–197. [Google Scholar] [CrossRef]
- Golik, A.; Modai, D.; Averbukh, Z.; Sheffy, M.; Shamis, A.; Cohen, N.; Shaked, U.; Dolev, E. Zinc metabolism in patients treated with captopril versus enalapril. Metabolism 1990, 39, 665–667. [Google Scholar] [CrossRef]
- Golik, A.; Zaidenstein, R.; Dishi, V.; Blatt, A.; Cohen, N.; Cotter, G.; Berman, S.; Weissgarten, J. Effects of captopril and enalapril on zinc metabolism in hypertensive patients. J. Am. Coll. Nutr. 1998, 17, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Peczkowska, M. Influence of angiotensin I converting enzyme inhibitors on selected parameters of zinc metabolism. Pol. Arch. Med. Wewn. 1996, 96, 32–38. [Google Scholar] [PubMed]
- Prasad, A.S.; Fitzgerald, J.T.; Hess, J.W.; Kaplan, J.; Pelen, F.; Dardenne, M. Zinc deficiency in elderly patients. Nutrition 1993, 9, 218–224. [Google Scholar] [PubMed]
- Abu-Hamdan, D.K.; Desai, H.; Sondheimer, J.; Felicetta, J.; Mahajan, S.; McDonald, F. Taste acuity and zinc metabolism in captopril-treated hypertensive male patients. Am. J. Hypertens. 1988, 1, 303S–308S. [Google Scholar] [CrossRef] [PubMed]
- Zumkley, H.; Bertram, H.P.; Vetter, H.; Zidek, W.; Losse, H. Zinc metabolism during captopril treatment. Horm. Metab. Res. 1985, 17, 256–258. [Google Scholar] [CrossRef] [PubMed]
- Good, C.B.; McDermott, L.; McCloskey, B. Diet and serum potassium in patients on ACE inhibitors. JAMA 1995, 274, 538. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, M.L.; Andrews, C.E. Severe Hyperkalemia during Very-Low-Calorie Diets and Angiotensin Converting Enzyme Use. JAMA 1990, 264, 2737–2738. [Google Scholar] [CrossRef] [PubMed]
- Burnakis, T.G. Captopril and Increased Serum Potassium Levels. JAMA 1984, 252, 1682–1683. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, T.S.; Freeman, D.; Mahnken, J.D.; Agraharkar, M.; Siddiqui, M.; Memon, A. Predictors of the development of hyperkalemia in patients using angiotensin-converting enzyme inhibitors. Am. J. Nephrol. 2000, 20, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Kostis, J.B.; Shelton, B.; Gosselin, G.; Goulet, C.; Hood, W.B.; Kohn, R.M.; Kubo, S.H.; Schron, E.; Weiss, M.B.; Willis, P.W.; et al. Adverse effects of enalapril in the Studies of Left Ventricular Dysfunction (SOLVD). SOLVD Investigators. Am. Heart J. 1996, 131, 350–355. [Google Scholar] [CrossRef]
- Burnakis, T.G.; Mioduch, H.J. Combined therapy with captopril and potassium supplementation. A potential for hyperkalemia. Arch. Intern. Med. 1984, 144, 2371–2372. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.S.; Sein, P.; Corio, R.; Bottomley, W.K. Nitrendipine-induced gingival hyperplasia. First case report. Oral Surg. Oral Med. Oral Pathol. 1990, 70, 593–596. [Google Scholar] [CrossRef]
- Carty, O.; Walsh, E.; Abdelsalem, A.; MaCarthy, D. Case report: Drug-induced gingival overgrowth associated with the use of a calcium channel blocker (amlodipine). J. Ir. Dent. Assoc. 2015, 61, 248–251. [Google Scholar] [PubMed]
- Joshi, S.; Bansal, S. A Rare Case Report of Amlodipine-Induced Gingival Enlargement and Review of Its Pathogenesis. Case Rep. Dent. 2013, 2013, e138248. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.B.; Weart, C.W.; Laro, J.J.; Neville, B.W. Calcium channel blocker-induced gingival hyperplasia: Case report and review of this iatrogenic disease. J. Fam. Pract. 1994, 39, 483–488. [Google Scholar] [PubMed]
- Madi, M.; Shetty, S.; Babu, S.; Achalli, S. Amlodipine-induced Gingival Hyperplasia—A Case Report and Review. West Indian Med. J. 2015, 64, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Livada, R.; Shiloah, J. Calcium channel blocker-induced gingival enlargement. J. Hum. Hypertens. 2014, 28, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.; Damm, D. Incidence of verapamil-induced gingival hyperplasia in a dental population. J. Periodontol. 1992, 63, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M. Current use of calcium channel blockers (CCBs) is associated with an increased risk of gingival hyperplasia. J. Evid.-Based Dent. Pract. 2012, 12, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Anand, V.; Nair, S. An unusual toxicity with beta blocker and calcium channel blocker. Indian J. Crit. Care Med. 2015, 19, 496–498. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, R.E. Hyperkalemia due to Salt Substitutes. JAMA 1986, 256, 1726. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Takahashi, Y.; Nakayama, T.; Asai, S. Comparative effect of angiotensin II type I receptor blockers and calcium channel blockers on laboratory parameters in hypertensive patients with type 2 diabetes. Cardiovasc. Diabetol. 2012, 11, 53. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Takahashi, Y.; Tezuka, K.; Takeuchi, S.; Nakayama, T.; Asai, S. A Comparative Effectiveness Study of Renal Parameters between Imidapril and Amlodipine in Patients with Hypertension: A Retrospective Cohort Study. Cardiol. Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.F.; Castelluccio, C.; Rice-Evans, C.; Taub, N.A.; Baum, H.; Quinn, P.J. Plasma coenzyme Q (ubiquinone) concentrations in patients treated with simvastatin. J. Clin. Pathol. 1993, 46, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Passi, S.; Stancato, A.; Aleo, E.; Dmitrieva, A.; Littarru, G.P. Statins lower plasma and lymphocyte ubiquinol/ubiquinone without affecting other antioxidants and PUFA. BioFactors 2003, 18, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.A.; Leth, A.; Agner, E.; Rohde, M. Dose-related decrease of serum coenzyme Q10 during treatment with HMG-CoA reductase inhibitors. Mol. Asp. Med. 1997, 18, 137–144. [Google Scholar] [CrossRef]
- Laaksonen, R.; Jokelainen, K.; Sahi, T.; Tikkanen, M.J.; Himberg, J.J. Decreases in serum ubiquinone concentrations do not result in reduced levels in muscle tissue during short-term simvastatin treatment in humans. Clin. Pharmacol. Ther. 1995, 57, 62–66. [Google Scholar] [CrossRef]
- Päivä, H.; Thelen, K.M.; Van Coster, R.; Smet, J.; De Paepe, B.; Mattila, K.M.; Laakso, J.; Lehtimäki, T.; von Bergmann, K.; Lütjohann, D.; et al. High-dose statins and skeletal muscle metabolism in humans: A randomized, controlled trial. Clin. Pharmacol. Ther. 2005, 78, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Asping, M.; Stride, N.; Søgaard, D.; Dohlmann, T.L.; Helge, J.W.; Dela, F.; Larsen, S. The effects of 2 weeks of statin treatment on mitochondrial respiratory capacity in middle-aged males: The LIFESTAT study. Eur. J. Clin. Pharmacol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Skarlovnik, A.; Janić, M.; Lunder, M.; Turk, M.; Šabovič, M. Coenzyme Q10 Supplementation Decreases Statin-Related Mild-to-Moderate Muscle Symptoms: A Randomized Clinical Study. Med. Sci. Monit. 2014, 20, 2183–2188. [Google Scholar] [CrossRef] [PubMed]
- Caso, G.; Kelly, P.; McNurlan, M.A.; Lawson, W.E. Effect of coenzyme Q10 on myopathic symptoms in patients treated with statins. Am. J. Cardiol. 2007, 99, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Fedacko, J.; Pella, D.; Fedackova, P.; Hänninen, O.; Tuomainen, P.; Jarcuska, P.; Lopuchovsky, T.; Jedlickova, L.; Merkovska, L.; Littarru, G.P. Coenzyme Q(10) and selenium in statin-associated myopathy treatment. Can. J. Physiol. Pharmacol. 2013, 91, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Young, J.M.; Florkowski, C.M.; Molyneux, S.L.; McEwan, R.G.; Frampton, C.M.; George, P.M.; Scott, R.S. Effect of coenzyme Q(10) supplementation on simvastatin-induced myalgia. Am. J. Cardiol. 2007, 100, 1400–1403. [Google Scholar] [CrossRef] [PubMed]
- Bookstaver, D.A.; Burkhalter, N.A.; Hatzigeorgiou, C. Effect of coenzyme Q10 supplementation on statin-induced myalgias. Am. J. Cardiol. 2012, 110, 526–529. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, B.; Ertugrul, D.T.; Cil, H.; Ata, N.; Akin, K.O.; Yalcin, A.A.; Kucukazman, M.; Dal, K.; Hokkaomeroglu, M.S.; Yavuz, B.B.; et al. Increased levels of 25 hydroxyvitamin D and 1,25-dihydroxyvitamin D after rosuvastatin treatment: A novel pleiotropic effect of statins? Cardiovasc. Drugs Ther. 2009, 23, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Ertugrul, D.T.; Yavuz, B.; Cil, H.; Ata, N.; Akin, K.O.; Kucukazman, M.; Yalcin, A.A.; Dal, K.; Yavuz, B.B.; Tutal, E. STATIN-D study: Comparison of the influences of rosuvastatin and fluvastatin treatment on the levels of 25 hydroxyvitamin D. Cardiovasc. Ther. 2011, 29, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Anagnostis, P.; Adamidou, F.; Slavakis, A.; Polyzos, S.A.; Selalmatzidou, D.; Panagiotou, A.; Athyros, V.G.; Karagiannis, A.; Kita, M. Comparative Effect of Atorvastatin and Rosuvastatin on 25-hydroxy-Vitamin D Levels in Non-diabetic Patients with Dyslipidaemia: A Prospective Randomized Open-label Pilot Study. Open Cardiovasc. Med. J. 2014, 8, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Rejnmark, L.; Vestergaard, P.; Mosekilde, L. Reduced fracture risk in users of thiazide diuretics. Calcif. Tissue Int. 2005, 76, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Ismail, F.; Corder, C.N.; Epstein, S.; Barbi, G.; Thomas, S. Effects of pravastatin and cholestyramine on circulating levels of parathyroid hormone and vitamin D metabolites. Clin. Ther. 1990, 12, 427–430. [Google Scholar] [PubMed]
- Montagnani, M.; Loré, F.; Di Cairano, G.; Gonnelli, S.; Ciuoli, C.; Montagnani, A.; Gennari, C. Effects of pravastatin treatment on vitamin D metabolites. Clin. Ther. 1994, 16, 824–829. [Google Scholar] [PubMed]
- Ott, C.; Raff, U.; Schneider, M.P.; Titze, S.I.; Schmieder, R.E. 25-hydroxyvitamin D insufficiency is associated with impaired renal endothelial function and both are improved with rosuvastatin treatment. Clin. Res. Cardiol. 2013, 102, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Sathyapalan, T.; Shepherd, J.; Arnett, C.; Coady, A.-M.; Kilpatrick, E.S.; Atkin, S.L. Atorvastatin increases 25-hydroxy vitamin D concentrations in patients with polycystic ovary syndrome. Clin. Chem. 2010, 56, 1696–1700. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Castrillón, J.L.; Abad, L.; Vega, G.; Sanz-Cantalapiedra, A.; García-Porrero, M.; Pinacho, F.; Dueñas, A. Effect of atorvastatin on bone mineral density in patients with acute coronary syndrome. Eur. Rev. Med. Pharmacol. Sci. 2008, 12, 83–88. [Google Scholar] [PubMed]
- Pérez-Castrillón, J.L.; Vega, G.; Abad, L.; Sanz, A.; Chaves, J.; Hernandez, G.; Dueñas, A. Effects of Atorvastatin on vitamin D levels in patients with acute ischemic heart disease. Am. J. Cardiol. 2007, 99, 903–905. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Khan, N.; Glueck, C.J.; Pandey, S.; Wang, P.; Goldenberg, N.; Uppal, M.; Khanal, S. Low serum 25 (OH) vitamin D levels (<32 ng/mL) are associated with reversible myositis-myalgia in statin-treated patients. Transl. Res. J. Lab. Clin. Med. 2009, 153, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Glueck, C.J.; Budhani, S.B.; Masineni, S.S.; Abuchaibe, C.; Khan, N.; Wang, P.; Goldenberg, N. Vitamin D deficiency, myositis-myalgia, and reversible statin intolerance. Curr. Med. Res. Opin. 2011, 27, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Linde, R.; Peng, L.; Desai, M.; Feldman, D. The role of vitamin D and SLCO1B1*5 gene polymorphism in statin-associated myalgias. Dermatoendocrinol. 2010, 2, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Kurnik, D.; Hochman, I.; Vesterman-Landes, J.; Kenig, T.; Katzir, I.; Lomnicky, Y.; Halkin, H.; Loebstein, R. Muscle pain and serum creatine kinase are not associated with low serum 25(OH) vitamin D levels in patients receiving statins. Clin. Endocrinol. 2012, 77, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Beulens, J.W.J.; Hart, H.E.; Kuijs, R.; Kooijman-Buiting, A.M.J.; Rutten, G.E.H.M. Influence of duration and dose of metformin on cobalamin deficiency in type 2 diabetes patients using metformin. Acta Diabetol. 2015, 52, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Iftikhar, R.; Kamran, S.M.; Qadir, A.; Iqbal, Z.; Usman, H. bin Prevalence of Vitamin B12 deficiency in patients of type 2 diabetes mellitus on metformin: A case control study from Pakistan. Pan Afr. Med. J. 2013, 16. [Google Scholar] [CrossRef] [PubMed]
- Nervo, M.; Lubini, A.; Raimundo, F.V.; Faulhaber, G.A.M.; Leite, C.; Fischer, L.M.; Furlanetto, T.W. Vitamin B12 in metformin-treated diabetic patients: A cross-sectional study in Brazil. Rev. Assoc. Médica Bras. 2011, 57, 46–49. [Google Scholar] [CrossRef]
- Damião, C.P.; Rodrigues, A.O.; Pinheiro, M.F.M.C.; da Cruz, R.A.; Cardoso, G.P.; Taboada, G.F.; Lima, G.A. Prevalence of vitamin B12 deficiency in type 2 diabetic patients using metformin: A cross-sectional study. Sao Paulo Med. J. 2016, 134, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.-H.; Ko, S.-H.; Ahn, Y.-B.; Song, K.-H.; Han, K.-D.; Park, Y.-M.; Ko, S.-H.; Kim, H.-S. Association of vitamin B12 deficiency and metformin use in patients with type 2 diabetes. J. Korean Med. Sci. 2014, 29, 965–972. [Google Scholar] [CrossRef] [PubMed]
- De Groot-Kamphuis, D.M.; van Dijk, P.R.; Groenier, K.H.; Houweling, S.T.; Bilo, H.J.G.; Kleefstra, N. Vitamin B12 deficiency and the lack of its consequences in type 2 diabetes patients using metformin. Neth. J. Med. 2013, 71, 386–390. [Google Scholar] [PubMed]
- Ting, R.Z.-W.; Szeto, C.C.; Chan, M.H.-M.; Ma, K.K.; Chow, K.M. Risk factors of vitamin B(12) deficiency in patients receiving metformin. Arch. Intern. Med. 2006, 166, 1975–1979. [Google Scholar] [CrossRef] [PubMed]
- Pflipsen, M.C.; Oh, R.C.; Saguil, A.; Seehusen, D.A.; Seaquist, D.; Topolski, R. The prevalence of vitamin B(12) deficiency in patients with type 2 diabetes: A cross-sectional study. J. Am. Board Fam. Med. 2009, 22, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, S.; Quan, H.; Li, J. Vitamin B12 Status in Metformin Treated Patients: Systematic Review. PLoS ONE 2014, 9, e100379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, S.; Li, L.; Li, Q.; Ren, K.; Sun, X.; Li, J. Metformin Treatment and Homocysteine: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Nutrients 2016, 8, 798. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, T.S.; Hadden, D.R.; Tomkin, G.H. Megaloblastic anaemia due to vitamin B12 malabsorption associated with long-term metformin treatment. Br. Med. J. 1980, 280, 1214–1215. [Google Scholar] [CrossRef] [PubMed]
- Bauman, W.A.; Shaw, S.; Jayatilleke, E.; Spungen, A.M.; Herbert, V. Increased intake of calcium reverses vitamin B12 malabsorption induced by metformin. Diabetes Care 2000, 23, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Habib, Z.A.; Havstad, S.L.; Wells, K.; Divine, G.; Pladevall, M.; Williams, L.K. Thiazolidinedione use and the longitudinal risk of fractures in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2010, 95, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.H.; Cadarette, S.M.; Choudhry, N.K.; Canning, C.; Levin, R.; Stürmer, T. A cohort study of thiazolidinediones and fractures in older adults with diabetes. J. Clin. Endocrinol. Metab. 2009, 94, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.V.; Chen, H.; Ambrosius, W.T.; Sood, A.; Josse, R.G.; Bonds, D.E.; Schnall, A.M.; Vittinghoff, E.; Bauer, D.C.; Banerji, M.A.; et al. Effects of TZD Use and Discontinuation on Fracture Rates in ACCORD Bone Study. J. Clin. Endocrinol. Metab. 2015, 100, 4059–4066. [Google Scholar] [CrossRef] [PubMed]
- Billington, E.O.; Grey, A.; Bolland, M.J. The effect of thiazolidinediones on bone mineral density and bone turnover: Systematic review and meta-analysis. Diabetologia 2015, 58, 2238–2246. [Google Scholar] [CrossRef] [PubMed]
- Loke, Y.K.; Singh, S.; Furberg, C.D. Long-term use of thiazolidinediones and fractures in type 2 diabetes: A meta-analysis. CMAJ Can. Med. Assoc. J. 2009, 180, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Briot, K.; Roux, C. Glucocorticoid-induced osteoporosis. RMD Open 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Van Staa, T.P.; Leufkens, H.G.; Cooper, C. The Epidemiology of Corticosteroid-Induced Osteoporosis: A Meta-analysis. Osteoporos. Int. 2002, 13, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Hahn, T.J.; Halstead, L.R.; Baran, D.T. Effects off short term glucocorticoid administration on intestinal calcium absorption and circulating vitamin D metabolite concentrations in man. J. Clin. Endocrinol. Metab. 1981, 52, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Buckley, L.M.; Leib, E.S.; Cartularo, K.S.; Vacek, P.M.; Cooper, S.M. Calcium and vitamin D3 supplementation prevents bone loss in the spine secondary to low-dose corticosteroids in patients with rheumatoid arthritis. A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 1996, 125, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Adachi, J.D.; Bensen, W.G.; Bianchi, F.; Cividino, A.; Pillersdorf, S.; Sebaldt, R.J.; Tugwell, P.; Gordon, M.; Steele, M.; Webber, C.; Goldsmith, C.H. Vitamin D and calcium in the prevention of corticosteroid induced osteoporosis: A 3 year followup. J. Rheumatol. 1996, 23, 995–1000. [Google Scholar] [PubMed]
- Bijlsma, J.W.; Raymakers, J.A.; Mosch, C.; Hoekstra, A.; Derksen, R.H.; Baart de la Faille, H.; Duursma, S.A. Effect of oral calcium and vitamin D on glucocorticoid-induced osteopenia. Clin. Exp. Rheumatol. 1988, 6, 113–119. [Google Scholar] [PubMed]
- Ringe, J.D.; Cöster, A.; Meng, T.; Schacht, E.; Umbach, R. Treatment of glucocorticoid-induced osteoporosis with alfacalcidol/calcium versus vitamin D/calcium. Calcif. Tissue Int. 1999, 65, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Ringe, J.D.; Faber, H.; Fahramand, P.; Schacht, E. Alfacalcidol versus plain vitamin D in the treatment of glucocorticoid/inflammation-induced osteoporosis. J. Rheumatol. Suppl. 2005, 76, 33–40. [Google Scholar] [PubMed]
- Homik, J.; Suarez-Almazor, M.E.; Shea, B.; Cranney, A.; Wells, G.; Tugwell, P. Calcium and vitamin D for corticosteroid-induced osteoporosis. Cochrane Database Syst. Rev. 2000, CD000952. [Google Scholar] [CrossRef] [PubMed]
- Richy, F.; Bousquet, J.; Ehrlich, G.E.; Meunier, P.J.; Israel, E.; Morii, H.; Devogelaer, J.-P.; Peel, N.; Haim, M.; Bruyere, O.; et al. Inhaled corticosteroids effects on bone in asthmatic and COPD patients: A quantitative systematic review. Osteoporos. Int. 2003, 14, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.; Fay, J.K.; Burr, M.; Stone, M.; Hood, K.; Roberts, G. Inhaled corticosteroid effects on bone metabolism in asthma and mild chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2002, CD003537. [Google Scholar] [CrossRef] [PubMed]
- Loke, Y.K.; Gilbert, D.; Thavarajah, M.; Blanco, P.; Wilson, A.M. Bone mineral density and fracture risk with long-term use of inhaled corticosteroids in patients with asthma: Systematic review and meta-analysis. BMJ Open 2015, 5, e008554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weatherall, M.; James, K.; Clay, J.; Perrin, K.; Masoli, M.; Wijesinghe, M.; Beasley, R. Dose-response relationship for risk of non-vertebral fracture with inhaled corticosteroids. Clin. Exp. Allergy 2008, 38, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Loke, Y.K.; Cavallazzi, R.; Singh, S. Risk of fractures with inhaled corticosteroids in COPD: Systematic review and meta-analysis of randomised controlled trials and observational studies. Thorax 2011, 66, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.J.; Lawlor, D.A.; Davey Smith, G.; Araya, R.; Timpson, N.; Day, I.N.M.; Ebrahim, S. The thermolabile variant of MTHFR is associated with depression in the British Women’s Heart and Health Study and a meta-analysis. Mol. Psychiatry 2006, 11, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Coppen, A.; Bailey, J. Enhancement of the antidepressant action of fluoxetine by folic acid: A randomised, placebo controlled trial. J. Affect. Disord. 2000, 60, 121–130. [Google Scholar] [CrossRef]
- Alpert, M.; Silva, R.R.; Pouget, E.R. Prediction of treatment response in geriatric depression from baseline folate level: Interaction with an SSRI or a tricyclic antidepressant. J. Clin. Psychopharmacol. 2003, 23, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Papakostas, G.I.; Petersen, T.; Mischoulon, D.; Ryan, J.L.; Nierenberg, A.A.; Bottiglieri, T.; Rosenbaum, J.F.; Alpert, J.E.; Fava, M. Serum folate, vitamin B12, and homocysteine in major depressive disorder, Part 1: Predictors of clinical response in fluoxetine-resistant depression. J. Clin. Psychiatry 2004, 65, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Papakostas, G.I.; Petersen, T.; Lebowitz, B.D.; Mischoulon, D.; Ryan, J.L.; Nierenberg, A.A.; Bottiglieri, T.; Alpert, J.E.; Rosenbaum, J.F.; Fava, M. The relationship between serum folate, vitamin B12, and homocysteine levels in major depressive disorder and the timing of improvement with fluoxetine. Int. J. Neuropsychopharmacol. 2005, 8, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Papakostas, G.I.; Shelton, R.C.; Zajecka, J.M.; Etemad, B.; Rickels, K.; Clain, A.; Baer, L.; Dalton, E.D.; Sacco, G.R.; Schoenfeld, D.; et al. l-methylfolate as adjunctive therapy for SSRI-resistant major depression: Results of two randomized, double-blind, parallel-sequential trials. Am. J. Psychiatry 2012, 169, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, L.D.; Oubre, A.Y.; Daoud, Y.A. l-methylfolate Plus SSRI or SNRI from Treatment Initiation Compared to SSRI or SNRI Monotherapy in a Major Depressive Episode. Innov. Clin. Neurosci. 2011, 8, 19–28. [Google Scholar] [PubMed]
- Shelton, R.C.; Sloan Manning, J.; Barrentine, L.W.; Tipa, E.V. Assessing Effects of l-Methylfolate in Depression Management: Results of a Real-World Patient Experience Trial. Prim. Care Companion CNS Disord. 2013, 15. [Google Scholar] [CrossRef]
- Rizzoli, R.; Cooper, C.; Reginster, J.-Y.; Abrahamsen, B.; Adachi, J.D.; Brandi, M.L.; Bruyère, O.; Compston, J.; Ducy, P.; Ferrari, S.; et al. Antidepressant medications and osteoporosis. Bone 2012, 51, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Eom, C.-S.; Lee, H.-K.; Ye, S.; Park, S.M.; Cho, K.-H. Use of selective serotonin reuptake inhibitors and risk of fracture: A systematic review and meta-analysis. J. Bone Miner. Res. 2012, 27, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Panday, K.; Gona, A.; Humphrey, M.B. Medication-induced osteoporosis: Screening and treatment strategies. Ther. Adv. Musculoskelet. Dis. 2014, 6, 185–202. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.M.; Chan, B.K.S.; Diem, S.J.; Ensrud, K.E.; Cauley, J.A.; Barrett-Connor, E.; Orwoll, E.; Bliziotes, M.M.; Osteoporotic Fractures in Men Study Group. Association of low bone mineral density with selective serotonin reuptake inhibitor use by older men. Arch. Intern. Med. 2007, 167, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.M.C.; Bivins, B.N.; Russell, K.A.; Bailey, L.B. Oral contraceptive use: Impact on folate, vitamin B6, and vitamin B12; status. Nutr. Rev. 2011, 69, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Wertalik, L.F.; Metz, E.N.; LoBuglio, A.F.; Balcerzak, S.P. Decreased serum B12 levels with oral contraceptive use. JAMA 1972, 221, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Sütterlin, M.W.; Bussen, S.S.; Rieger, L.; Dietl, J.; Steck, T. Serum folate and Vitamin B12 levels in women using modern oral contraceptives (OC) containing 20 microg ethinyl estradiol. Eur. J. Obstet. Gynecol. Reprod. Biol. 2003, 107, 57–61. [Google Scholar] [CrossRef]
- Veninga, K.S. Effects of oral contraceptives on vitamins B6, B12, C, and folacin. J. Nurse. Midwifery 1984, 29, 386–390. [Google Scholar] [PubMed]
- Berenson, A.B.; Rahman, M. Effect of hormonal contraceptives on vitamin B12 level and the association of the latter with bone mineral density. Contraception 2012, 86, 481–487. [Google Scholar] [CrossRef] [PubMed]
- McArthur, J.O.; Tang, H.; Petocz, P.; Samman, S. Biological variability and impact of oral contraceptives on vitamins B(6), B(12) and folate status in women of reproductive age. Nutrients 2013, 5, 3634–3645. [Google Scholar] [CrossRef] [PubMed]
- Riedel, B.; Bjørke Monsen, A.-L.; Ueland, P.M.; Schneede, J. Effects of oral contraceptives and hormone replacement therapy on markers of cobalamin status. Clin. Chem. 2005, 51, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Shojania, A.M.; Wylie, B. The effect of oral contraceptives on vitamin B12 metabolism. Am. J. Obstet. Gynecol. 1979, 135, 129–134. [Google Scholar] [CrossRef]
- Brattström, L.; Israelsson, B.; Olsson, A.; Andersson, A.; Hultberg, B. Plasma homocysteine in women on oral oestrogen-containing contraceptives and in men with oestrogen-treated prostatic carcinoma. Scand. J. Clin. Lab. Investig. 1992, 52, 283–287. [Google Scholar] [CrossRef]
- Streiff, R.R. Folate Deficiency and Oral Contraceptives. JAMA 1970, 214, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Shojania, A.M.; Hornady, G.; Barnes, P.H. Oral contraceptives and serum-folate level. Lancet 1968, 1, 1376–1377. [Google Scholar] [CrossRef]
- Shere, M.; Bapat, P.; Nickel, C.; Kapur, B.; Koren, G. Association between Use of Oral Contraceptives and Folate Status: A Systematic Review and Meta-Analysis. J. Obstet. Gynaecol. Can. 2015, 37, 430–438. [Google Scholar] [CrossRef]
- Gambacciani, M.; Cappagli, B.; Lazzarini, V.; Ciaponi, M.; Fruzzetti, F.; Genazzani, A.R. Longitudinal evaluation of perimenopausal bone loss: Effects of different low dose oral contraceptive preparations on bone mineral density. Maturitas 2006, 54, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Kleerekoper, M.; Brienza, R.S.; Schultz, L.R.; Johnson, C.C. Oral contraceptive use may protect against low bone mass. Henry Ford Hospital Osteoporosis Cooperative Research Group. Arch. Intern. Med. 1991, 151, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Kuohung, W.; Borgatta, L.; Stubblefield, P. Low-dose oral contraceptives and bone mineral density: An evidence-based analysis. Contraception 2000, 61, 77–82. [Google Scholar] [CrossRef]
- Liu, S.L.; Lebrun, C.M. Effect of oral contraceptives and hormone replacement therapy on bone mineral density in premenopausal and perimenopausal women: A systematic review. Br. J. Sports Med. 2006, 40, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Hartard, M.; Kleinmond, C.; Kirchbichler, A.; Jeschke, D.; Wiseman, M.; Weissenbacher, E.R.; Felsenberg, D.; Erben, R.G. Age at first oral contraceptive use as a major determinant of vertebral bone mass in female endurance athletes. Bone 2004, 35, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Cobb, K.L.; Kelsey, J.L.; Sidney, S.; Ettinger, B.; Lewis, C.E. Oral contraceptives and bone mineral density in white and black women in CARDIA. Coronary Risk Development in Young Adults. Osteoporos. Int. 2002, 13, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Hartard, M.; Bottermann, P.; Bartenstein, P.; Jeschke, D.; Schwaiger, M. Effects on bone mineral density of low-dosed oral contraceptives compared to and combined with physical activity. Contraception 1997, 55, 87–90. [Google Scholar] [CrossRef]
- Burr, D.B.; Yoshikawa, T.; Teegarden, D.; Lyle, R.; McCabe, G.; McCabe, L.D.; Weaver, C.M. Exercise and oral contraceptive use suppress the normal age-related increase in bone mass and strength of the femoral neck in women 18-31 years of age. Bone 2000, 27, 855–863. [Google Scholar] [CrossRef]
- Weaver, C.M.; Teegarden, D.; Lyle, R.M.; McCabe, G.P.; McCabe, L.D.; Proulx, W.; Kern, M.; Sedlock, D.; Anderson, D.D.; Hillberry, B.M.; et al. Impact of exercise on bone health and contraindication of oral contraceptive use in young women. Med. Sci. Sports Exerc. 2001, 33, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Teegarden, D.; Legowski, P.; Gunther, C.W.; McCabe, G.P.; Peacock, M.; Lyle, R.M. Dietary calcium intake protects women consuming oral contraceptives from spine and hip bone loss. J. Clin. Endocrinol. Metab. 2005, 90, 5127–5133. [Google Scholar] [CrossRef] [PubMed]
- Hameed, A.; Majeed, T.; Rauf, S.; Ashraf, M.; Jalil, M.A.; Nasrullah, M.; Hussan, A.; Noreen, R. Effect of oral and injectable contraceptives on serum calcium, magnesium and phosphorus in women. J. Ayub Med. Coll. Abbottabad 2001, 13, 24–25. [Google Scholar] [PubMed]
- Olatunbosun, D.A.; Adeniyi, F.A.; Adadevoh, B.K. Effect of oral contraceptives on Serum magnesium levels. Int. J. Fertil. 1974, 19, 224–226. [Google Scholar] [PubMed]
- Akinloye, O.; Adebayo, T.O.; Oguntibeju, O.O.; Oparinde, D.P.; Ogunyemi, E.O. Effects of contraceptives on serum trace elements, calcium and phosphorus levels. West Indian Med. J. 2011, 60, 308–315. [Google Scholar] [PubMed]
- Stanton, M.F.; Lowenstein, F.W. Serum magnesium in women during pregnancy, while taking contraceptives, and after menopause. J. Am. Coll. Nutr. 1987, 6, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Kitai, E.; Ariel, Y.; Schnierer, M.; Bograd, H. Oral contraceptive lowers serum magnesium. Harefuah 1991, 121, 363–364. [Google Scholar] [PubMed]
- Prasad, A.S.; Oberleas, D.; Moghissi, K.S.; Lei, K.Y.; Stryker, J.C. Effect of oral contraceptive agents on nutrients: I. Minerals. Am. J. Clin. Nutr. 1975, 28, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Stegeman, B.H.; de Bastos, M.; Rosendaal, F.R.; van Vlieg, A.H.; Helmerhorst, F.M.; Stijnen, T.; Dekkers, O.M. Different combined oral contraceptives and the risk of venous thrombosis: Systematic review and network meta-analysis. BMJ 2013, 347, f5298. [Google Scholar] [CrossRef] [PubMed]
- Termanini, B.; Gibril, F.; Sutliff, V.E.; Yu, F.; Venzon, D.J.; Jensen, R.T. Effect of long-term gastric acid suppressive therapy on serum vitamin B12 levels in patients with Zollinger-Ellison syndrome. Am. J. Med. 1998, 104, 422–430. [Google Scholar] [CrossRef]
- Den Elzen, W.P.J.; Groeneveld, Y.; de Ruijter, W.; Souverijn, J.H.M.; le Cessie, S.; Assendelft, W.J.J.; Gussekloo, J. Long-term use of proton pump inhibitors and vitamin B12 status in elderly individuals. Aliment. Pharmacol. Ther. 2008, 27, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Jensen, R.T. Association of Long-term Proton Pump Inhibitor Therapy with Bone Fractures and effects on Absorption of Calcium, Vitamin B12, Iron, and Magnesium. Curr. Gastroenterol. Rep. 2010, 12, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Pennypacker, L.C.; Allen, R.H.; Kelly, J.P.; Matthews, L.M.; Grigsby, J.; Kaye, K.; Lindenbaum, J.; Stabler, S.P. High prevalence of cobalamin deficiency in elderly outpatients. J. Am. Geriatr. Soc. 1992, 40, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Shirai, N.; Sugimoto, M.; Nakamura, A.; Hishida, A.; Ishizaki, T. Influence of CYP2C19 Pharmacogenetic Polymorphism on Proton Pump Inhibitor-based Therapies. Drug Metab. Pharmacokinet. 2005, 20, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Kittang, E.; Aadland, E.; Schjønsby, H. Effect of omeprazole on the secretion of intrinsic factor, gastric acid and pepsin in man. Gut 1985, 26, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Freedberg, D.E.; Kim, L.S.; Yang, Y.-X. The Risks and Benefits of Long-term Use of Proton Pump Inhibitors: Expert Review and Best Practice Advice from the American Gastroenterological Association. Gastroenterology 2017, 152, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Tempel, M.; Chawla, A.; Messina, C.; Çeliker, M.Y. Effects of Omeprazole on Iron Absorption: Preliminary Study. Turk. J. Hematol. 2013, 30, 307–310. [Google Scholar] [CrossRef]
- Stewart, C.A.; Termanini, B.; Sutliff, V.E.; Serrano, J.; Yu, F.; Gibril, F.; Jensen, R.T. Iron absorption in patients with Zollinger-Ellison syndrome treated with long-term gastric acid antisecretory therapy. Aliment. Pharmacol. Ther. 1998, 12, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Sarzynski, E.; Puttarajappa, C.; Xie, Y.; Grover, M.; Laird-Fick, H. Association between proton pump inhibitor use and anemia: A retrospective cohort study. Dig. Dis. Sci. 2011, 56, 2349–2353. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.R.; Schneider, J.L.; Quesenberry, C.P.; Corley, D.A. Proton Pump Inhibitor and Histamine-2 Receptor Antagonist Use and Iron Deficiency. Gastroenterology 2017, 152, 821–829.e1. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, C.; Geissler, C.A.; Powell, J.J.; Bomford, A. Proton pump inhibitors suppress absorption of dietary non-haem iron in hereditary haemochromatosis. Gut 2007, 56, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Sipponen, P.; Härkönen, M. Hypochlorhydric stomach: A risk condition for calcium malabsorption and osteoporosis? Scand. J. Gastroenterol. 2010, 45, 133–138. [Google Scholar] [CrossRef] [PubMed]
- McGowan, B.; Bennett, K.; Barry, M. Prescribing of anti-osteoporotic therapies following the use of proton pump inhibitors in general practice. Pharmacoepidemiol. Drug Saf. 2010, 19, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Ngamruengphong, S.; Leontiadis, G.I.; Radhi, S.; Dentino, A.; Nugent, K. Proton pump inhibitors and risk of fracture: A systematic review and meta-analysis of observational studies. Am. J. Gastroenterol. 2011, 106, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Huang, Y.; Li, H.; Sun, W.; Liu, J. Proton-pump inhibitors and risk of fractures: An update meta-analysis. Osteoporos. Int. 2016, 27, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Graziani, G.; Como, G.; Badalamenti, S.; Finazzi, S.; Malesci, A.; Gallieni, M.; Brancaccio, D.; Ponticelli, C. Effect of gastric acid secretion on intestinal phosphate and calcium absorption in normal subjects. Nephrol. Dial. Transplant. 1995, 10, 1376–1380. [Google Scholar] [PubMed]
- Serfaty-Lacrosniere, C.; Wood, R.J.; Voytko, D.; Saltzman, J.R.; Pedrosa, M.; Sepe, T.E.; Russell, R.R. Hypochlorhydria from short-term omeprazole treatment does not inhibit intestinal absorption of calcium, phosphorus, magnesium or zinc from food in humans. J. Am. Coll. Nutr. 1995, 14, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.L.; LaCroix, A.Z.; Larson, J.; Robbins, J.; Cauley, J.A.; Manson, J.E.; Chen, Z. Proton pump inhibitor use, hip fracture, and change in bone mineral density in postmenopausal women: Results from the Women’s Health Initiative. Arch. Intern. Med. 2010, 170, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.W.; Blackwell, T.; Ensrud, K.E.; Hillier, T.A.; Lane, N.E.; Orwoll, E.; Bauer, D.C. Acid-suppressive medications and risk of bone loss and fracture in older adults. Calcif. Tissue Int. 2008, 83, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Targownik, L.E.; Lix, L.M.; Leung, S.; Leslie, W.D. Proton-pump inhibitor use is not associated with osteoporosis or accelerated bone mineral density loss. Gastroenterology 2010, 138, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Cosman, F.; de Beur, S.J.; LeBoff, M.S.; Lewiecki, E.M.; Tanner, B.; Randall, S.; Lindsay, R. Clinician’s Guide to Prevention and Treatment of Osteoporosis. Osteoporos. Int. 2014, 25, 2359–2381. [Google Scholar] [CrossRef] [PubMed]
- Sheen, E.; Triadafilopoulos, G. Adverse effects of long-term proton pump inhibitor therapy. Dig. Dis. Sci. 2011, 56, 931–950. [Google Scholar] [CrossRef] [PubMed]
- Sturniolo, G.C.; Montino, M.C.; Rossetto, L.; Martin, A.; D’Inca, R.; D’Odorico, A.; Naccarato, R. Inhibition of gastric acid secretion reduces zinc absorption in man. J. Am. Coll. Nutr. 1991, 10, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Ozutemiz, A.O.; Aydin, H.H.; Isler, M.; Celik, H.A.; Batur, Y. Effect of omeprazole on plasma zinc levels after oral zinc administration. Indian J. Gastroenterol. 2002, 21, 216–218. [Google Scholar] [PubMed]
- Farrell, C.P.; Morgan, M.; Rudolph, D.S.; Hwang, A.; Albert, N.E.; Valenzano, M.C.; Wang, X.; Mercogliano, G.; Mullin, J.M. Proton Pump Inhibitors Interfere with Zinc Absorption and Zinc Body Stores. Gastroenterol. Res. 2011, 4, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Serfaty-Lacrosniere, C.; Camilo, M.E.; Russell, R.M. Gastric acidity influences the blood response to a beta-carotene dose in humans. Am. J. Clin. Nutr. 1996, 64, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Sahud, M.A.; Cohen, R.J. Effect of aspirin ingestion on ascorbic-acid levels in rheumatoid arthritis. Lancet 1971, 1, 937–938. [Google Scholar] [CrossRef]
- Brzozowski, T.; Kwiecień, S.; Konturek, P.C.; Konturek, S.J.; Mitis-Musiol, M.; Duda, A.; Bielański, W.; Hahn, E.G. Comparison of nitric oxide-releasing NSAID and vitamin C with classic NSAID in healing of chronic gastric ulcers; involvement of reactive oxygen species. Med. Sci. Monit. 2001, 7, 592–599. [Google Scholar] [PubMed]
- Dammann, H.-G.; Saleki, M.; Torz, M.; Schulz, H.-U.; Krupp, S.; Schürer, M.; Timm, J.; Gessner, U. Effects of buffered and plain acetylsalicylic acid formulations with and without ascorbic acid on gastric mucosa in healthy subjects. Aliment. Pharmacol. Ther. 2004, 19, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Konturek, P.C.; Kania, J.; Hahn, E.G.; Konturek, J.W. Ascorbic acid attenuates aspirin-induced gastric damage: Role of inducible nitric oxide synthase. J. Physiol. Pharmacol. 2006, 57 (Suppl 5), 125–136. [Google Scholar] [PubMed]
- Pohle, T.; Brzozowski, T.; Becker, J.C.; Van der Voort, I.R.; Markmann, A.; Konturek, S.J.; Moniczewski, A.; Domschke, W.; Konturek, J.W. Role of reactive oxygen metabolites in aspirin-induced gastric damage in humans: Gastroprotection by vitamin C. Aliment. Pharmacol. Ther. 2001, 15, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Fisher, M.; Voelker, M.; Gessner, U. Gastrointestinal effects of the addition of ascorbic acid to aspirin. Pain Pract. 2012, 12, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.M.; Lichtenstein, D.R.; Singh, G. Gastrointestinal Toxicity of Nonsteroidal Antiinflammatory Drugs. N. Engl. J. Med. 1999, 340, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Hawkey, C.J. Review article: Aspirin and gastrointestinal bleeding. Aliment. Pharmacol. Ther. 1994, 8, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, R.; Torley, H.I.; McKinlay, A.W.; Sturrock, R.D.; Russell, R.I. Iron deficiency anaemia in patients with rheumatic disease receiving non-steroidal anti-inflammatory drugs: The role of upper gastrointestinal lesions. Ann. Rheum. Dis. 1990, 49, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Valkhoff, V.E.; Sturkenboom, M.C.J.M.; Hill, C.; van Zanten, S.V.; Kuipers, E.J. Low-dose acetylsalicylic acid use and the risk of upper gastrointestinal bleeding: A meta-analysis of randomized clinical trials and observational studies. Can. J. Gastroenterol. 2013, 27, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Al-Azzam, S.I.; AlMahasneh, F.; Mhaidat, N.; Alzoubi, K.H.; Khader, Y.S. Prophylactic use of aspirin does not induce anaemia among adults. J. Clin. Pharm. Ther. 2010, 35, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Leibovici, A.; Lavi, N.; Wainstok, S.; Herman, J.; Greene, V.W. Low-dose acetylsalicylic acid use and hemoglobin levels. Effects in a primary care population. Can. Fam. Physician 1995, 41, 64–68. [Google Scholar] [PubMed]
- Silagy, C.A.; McNeil, J.J.; Donnan, G.A.; Tonkin, A.M.; Worsam, B.; Campion, K. Adverse effects of low-dose aspirin in a healthy elderly population. Clin. Pharmacol. Ther. 1993, 54, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Milman, N.; Ovesen, L.; Byg, K.; Graudal, N. Iron status in Danes updated 1994. I: Prevalence of iron deficiency and iron overload in 1332 men aged 40–70 years. Influence Of blood donation, alcohol intake, and iron supplementation. Ann. Hematol. 1999, 78, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Friedman, P.A.; Bushinsky, D.A. Diuretic effects on calcium metabolism. Semin. Nephrol. 1999, 19, 551–556. [Google Scholar] [PubMed]
- Stier, C.T.; Itskovitz, H.D. Renal calcium metabolism and diuretics. Annu. Rev. Pharmacol. Toxicol. 1986, 26, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Bourdeau, J.E.; Buss, S.L.; Vurek, G.G. Inhibition of calcium absorption in the cortical thick ascending limb of Henle’s loop by furosemide. J. Pharmacol. Exp. Ther. 1982, 221, 815–819. [Google Scholar] [PubMed]
- Quamme, G.A. Effect of furosemide on calcium and magnesium transport in the rat nephron. Am. J. Physiol. 1981, 241, F340–F347. [Google Scholar] [CrossRef] [PubMed]
- Reichel, H.; Deibert, B.; Geberth, S.; Schmidt-Gayk, H.; Ritz, E. Frusemide therapy and intact parathyroid hormone plasma concentrations in chronic renal insufficiency. Nephrol. Dial. Transplant. 1992, 7, 8–15. [Google Scholar] [PubMed]
- Costanzo, L.S. Localization of diuretic action in microperfused rat distal tubules: Ca and Na transport. Am. J. Physiol. 1985, 248, F527–F535. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, L.S.; Windhager, E.E. Calcium and sodium transport by the distal convoluted tubule of the rat. Am. J. Physiol. 1978, 235, F492–F506. [Google Scholar] [CrossRef] [PubMed]
- Lamberg, B.A.; Kuhlback, B. Effect of chlorothiazide and hydrochlorothiazide on the excretion of calcium in urine. Scand. J. Clin. Lab. Invest. 1959, 11, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Nijenhuis, T.; Vallon, V.; van der Kemp, A.W.C.M.; Loffing, J.; Hoenderop, J.G.J.; Bindels, R.J.M. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J. Clin. Investig. 2005, 115, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Lemann, J.; Gray, R.W.; Maierhofer, W.J.; Cheung, H.S. Hydrochlorothiazide inhibits bone resorption in men despite experimentally elevated serum 1,25-dihydroxyvitamin D concentrations. Kidney Int. 1985, 28, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.L.; Fraser, R. Do diuretics cause magnesium deficiency? Br. J. Clin. Pharmacol. 1993, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A. Diuretic complications. Am. J. Med. Sci. 2000, 319, 10–24. [Google Scholar] [CrossRef]
- Sarafidis, P.A.; Georgianos, P.I.; Lasaridis, A.N. Diuretics in clinical practice. Part II: Electrolyte and acid-base disorders complicating diuretic therapy. Expert Opin. Drug Saf. 2010, 9, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Kroenke, K.; Wood, D.R.; Hanley, J.F. The value of serum magnesium determination in hypertensive patients receiving diuretics. Arch. Intern. Med. 1987, 147, 1553–1556. [Google Scholar] [CrossRef] [PubMed]
- Dørup, I.; Skajaa, K.; Thybo, N.K. Oral magnesium supplementation restores the concentrations of magnesium, potassium and sodium-potassium pumps in skeletal muscle of patients receiving diuretic treatment. J. Intern. Med. 1993, 233, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Lubetsky, A.; Winaver, J.; Seligmann, H.; Olchovsky, D.; Almog, S.; Halkin, H.; Ezra, D. Urinary thiamine excretion in the rat: Effects of furosemide, other diuretics, and volume load. J. Lab. Clin. Med. 1999, 134, 232–237. [Google Scholar] [CrossRef]
- Rieck, J.; Halkin, H.; Almog, S.; Seligman, H.; Lubetsky, A.; Olchovsky, D.; Ezra, D. Urinary loss of thiamine is increased by low doses of furosemide in healthy volunteers. J. Lab. Clin. Med. 1999, 134, 238–243. [Google Scholar] [CrossRef]
- Russell, R.M.; Suter, P.M. Vitamin requirements of elderly people: An update. Am. J. Clin. Nutr. 1993, 58, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Leary, W.P.; Reyes, A.J.; Wynne, R.D.; van der Byl, K. Renal excretory actions of furosemide, of hydrochlorothiazide and of the vasodilator flosequinan in healthy subjects. J. Int. Med. Res. 1990, 18, 120–141. [Google Scholar] [CrossRef] [PubMed]
- Mountokalakis, T.; Dourakis, S.; Karatzas, N.; Maravelias, C.; Koutselinis, A. Zinc deficiency in mild hypertensive patients treated with diuretics. J. Hypertens. Suppl. 1984, 2, S571–S572. [Google Scholar] [PubMed]
- Sica, D.A.; Carter, B.; Cushman, W.; Hamm, L. Thiazide and loop diuretics. J. Clin. Hypertens. 2011, 13, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.H.; Loffing, J. Thiazide Effects and Adverse Effects. Hypertension 2009, 54, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Velázquez, H.; Wright, F.S. Control by drugs of renal potassium handling. Annu. Rev. Pharmacol. Toxicol. 1986, 26, 293–309. [Google Scholar] [CrossRef] [PubMed]
- Zillich, A.J.; Garg, J.; Basu, S.; Bakris, G.L.; Carter, B.L. Thiazide Diuretics, Potassium, and the Development of Diabetes. Hypertension 2006, 48, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G. Diuretic therapy and ventricular arrhythmias in persons 65 years of age and older. Am. J. Cardiol. 1990, 65, 599–603. [Google Scholar] [CrossRef]
- Siscovick, D.S.; Raghunathan, T.E.; Psaty, B.M.; Koepsell, T.D.; Wicklund, K.G.; Lin, X.; Cobb, L.; Rautaharju, P.M.; Copass, M.K.; Wagner, E.H. Diuretic Therapy for Hypertension and the Risk of Primary Cardiac Arrest. N. Engl. J. Med. 1994, 330, 1852–1857. [Google Scholar] [CrossRef] [PubMed]
- Persson, S. Potassium supplements or potassium-sparing agents. Acta Pharmacol. Toxicol. 1984, 54 (Suppl. S1), 107–113. [Google Scholar] [CrossRef]
- Kaplan, N.M.; Carnegie, A.; Raskin, P.; Heller, J.A.; Simmons, M. Potassium supplementation in hypertensive patients with diuretic-induced hypokalemia. N. Engl. J. Med. 1985, 312, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Schalhorn, A.; Siegert, W.; Sauer, H.-J. Antifolate effect of triamterene on human leucocytes and on a human lymphoma cell line. Eur. J. Clin. Pharmacol. 1981, 20, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Morrow, L.E.; Grimsley, E.W. Long-term diuretic therapy in hypertensive patients: Effects on serum homocysteine, vitamin B6, vitamin B12, and red blood cell folate concentrations. South. Med. J. 1999, 92, 866–870. [Google Scholar] [CrossRef] [PubMed]
- Westphal, S.; Rading, A.; Luley, C.; Dierkes, J. Antihypertensive treatment and homocysteine concentrations. Metabolism 2003, 52, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Lowe, N.M.; Fekete, K.; Decsi, T. Methods of assessment of zinc status in humans: A systematic review. Am. J. Clin. Nutr. 2009, 89, 2040S–2051S. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J.; Vaughan, D.E. Angiotensin-Converting Enzyme Inhibitors. Circulation 1998, 97, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Raebel, M.A. Hyperkalemia associated with use of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. Cardiovasc. Ther. 2012, 30, e156–e166. [Google Scholar] [CrossRef] [PubMed]
- Tejnani, A.; Mani, A.; Sodhi, N.K.; Mehta, A.; Gourkhede, S.; Thorat, V.; Marawar, P. Incidence of amlodipine-induced gingival overgrowth in the rural population of Loni. J. Indian Soc. Periodontol. 2014, 18, 226–228. [Google Scholar] [CrossRef] [PubMed]
- Barclay, S.; Thomason, J.M.; Idle, J.R.; Seymour, R.A. The incidence and severity of nifedipine-induced gingival overgrowth. J. Clin. Periodontol. 1992, 19, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.; Arany, P. Mechanism of drug-induced gingival overgrowth revisited: A unifying hypothesis. Oral Dis. 2015, 21, e51–e61. [Google Scholar] [CrossRef] [PubMed]
- Arya, R.; Gulati, S.; Kabra, M.; Sahu, J.K.; Kalra, V. Folic acid supplementation prevents phenytoin-induced gingival overgrowth in children. Neurology 2011, 76, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.N.; Chawla, H.S.; Goyal, A.; Gauba, K.; Singhi, P. Folic acid and phenytoin induced gingival overgrowth—Is there a preventive effect. J. Indian Soc. Pedod. Prev. Dent. 2004, 22, 82–91. [Google Scholar] [PubMed]
- Brown, R.S.; Di Stanislao, P.T.; Beaver, W.T.; Bottomley, W.K. The administration of folic acid to institutionalized epileptic adults with phenytoin-induced gingival hyperplasia. A double-blind, randomized, placebo-controlled, parallel study. Oral Surg. Oral Med. Oral Pathol. 1991, 71, 565–568. [Google Scholar] [CrossRef]
- Bäckman, N.; Holm, A.-K.; Hänström, L.; Blomquist, H.K.; Heijbel, J.; Säfström, G. Folate treatment of diphenylhydantoin-induced gingival hyperplasia. Eur. J. Oral Sci. 1989, 97, 222–232. [Google Scholar] [CrossRef]
- Pepping, J. Coenzyme Q10. Am. J. Health. Syst. Pharm. 1999, 56, 519–521. [Google Scholar] [PubMed]
- Ghirlanda, G.; Oradei, A.; Manto, A.; Lippa, S.; Uccioli, L.; Caputo, S.; Greco, A.V.; Littarru, G.P. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: A double-blind, placebo-controlled study. J. Clin. Pharmacol. 1993, 33, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Folkers, K.; Langsjoen, P.; Willis, R.; Richardson, P.; Xia, L.J.; Ye, C.Q.; Tamagawa, H. Lovastatin decreases coenzyme Q levels in humans. Proc. Natl. Acad. Sci. USA 1990, 87, 8931–8934. [Google Scholar] [CrossRef] [PubMed]
- Bargossi, A.M.; Grossi, G.; Fiorella, P.L.; Gaddi, A.; Di Giulio, R.; Battino, M. Exogenous CoQ10 supplementation prevents plasma ubiquinone reduction induced by HMG-CoA reductase inhibitors. Mol. Asp. Med. 1994, 15, s187–s193. [Google Scholar] [CrossRef]
- Human, J.A.; Ubbink, J.B.; Jerling, J.J.; Delport, R.; Vermaak, W.J.; Vorster, H.H.; Lagendijk, J.; Potgieter, H.C. The effect of Simvastatin on the plasma antioxidant concentrations in patients with hypercholesterolaemia. Clin. Chim. Acta 1997, 263, 67–77. [Google Scholar] [CrossRef]
- De Pinieux, G.; Chariot, P.; Ammi-Saïd, M.; Louarn, F.; Lejonc, J.L.; Astier, A.; Jacotot, B.; Gherardi, R. Lipid-lowering drugs and mitochondrial function: Effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br. J. Clin. Pharmacol. 1996, 42, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Shouzu, A.; Nishikawa, M.; Yonemoto, T.; Shimizu, H.; Omoto, S.; Hayakawa, T.; Inada, M. Effect of treatment with 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors on serum coenzyme Q10 in diabetic patients. Arzneimittelforschung 1999, 49, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Jula, A.; Marniemi, J.; Huupponen, R.; Virtanen, A.; Rastas, M.; Rönnemaa, T. Effects of diet and simvastatin on serum lipids, insulin, and antioxidants in hypercholesterolemic men: A randomized controlled trial. JAMA 2002, 287, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Rundek, T.; Naini, A.; Sacco, R.; Coates, K.; DiMauro, S. Atorvastatin decreases the coenzyme Q10 level in the blood of patients at risk for cardiovascular disease and stroke. Arch. Neurol. 2004, 61, 889–892. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, H.; Higashikata, T.; Kawashiri, M.; Katsuda, S.; Mizuno, M.; Nohara, A.; Inazu, A.; Koizumi, J.; Kobayashi, J. Reduction of serum ubiquinol-10 and ubiquinone-10 levels by atorvastatin in hypercholesterolemic patients. J. Atheroscler. Thromb. 2005, 12, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Appelkvist, E.L.; Edlund, C.; Löw, P.; Schedin, S.; Kalén, A.; Dallner, G. Effects of inhibitors of hydroxymethylglutaryl coenzyme A reductase on coenzyme Q and dolichol biosynthesis. Clin. Investig. 1993, 71, S97–S102. [Google Scholar] [CrossRef] [PubMed]
- Laaksonen, R.; Jokelainen, K.; Laakso, J.; Sahi, T.; Harkonen, M.; Tikkanen, M.J.; Himberg, J.J. The effect of simvastatin treatment on natural antioxidants in low-density lipoproteins and high-energy phosphates and ubiquinone in skeletal muscle. Am. J. Cardiol. 1996, 77, 851–854. [Google Scholar] [CrossRef]
- Lamperti, C.; Naini, A.B.; Lucchini, V.; Prelle, A.; Bresolin, N.; Moggio, M.; Sciacco, M.; Kaufmann, P.; DiMauro, S. Muscle coenzyme Q10 level in statin-related myopathy. Arch. Neurol. 2005, 62, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, S.; Trevisson, E.; Salviati, L.; Aymé, S.; Rigal, O.; Redondo, A.G.; Mancuso, M.; Siciliano, G.; Tonin, P.; Angelini, C.; et al. Coenzyme Q10 is frequently reduced in muscle of patients with mitochondrial myopathy. Neuromuscul. Disord. 2010, 20, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Deichmann, R.; Lavie, C.; Andrews, S. Coenzyme Q10 and Statin-Induced Mitochondrial Dysfunction. Ochsner J. 2010, 10, 16–21. [Google Scholar] [PubMed]
- Mas, E.; Mori, T.A. Coenzyme Q(10) and statin myalgia: What is the evidence? Curr. Atheroscler. Rep. 2010, 12, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Marcoff, L.; Thompson, P.D. The Role of Coenzyme Q10 in Statin-Associated Myopathy: A Systematic Review. J. Am. Coll. Cardiol. 2007, 49, 2231–2237. [Google Scholar] [CrossRef] [PubMed]
- Thibault, A.; Samid, D.; Tompkins, A.C.; Figg, W.D.; Cooper, M.R.; Hohl, R.J.; Trepel, J.; Liang, B.; Patronas, N.; Venzon, D.J.; et al. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin. Cancer Res. 1996, 2, 483–491. [Google Scholar] [PubMed]
- Kim, W.S.; Kim, M.M.; Choi, H.J.; Yoon, S.-S.; Lee, M.H.; Park, K.; Park, C.H.; Kang, W.K. Phase II Study of High-Dose Lovastatin in Patients with Advanced Gastric Adenocarcinoma. Investig. New Drugs 2001, 19, 81–83. [Google Scholar] [CrossRef]
- Glossmann, H.H.; Blumthaler, M. Does rosuvastatin increase serum levels of 25-hydroxy-vitamin D? Dermato-Endocrinology 2012, 4, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. The statin D-lemma. Dermato-Endocrinology 2012, 4, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Grimnes, G.; Almaas, B.; Eggen, A.E.; Emaus, N.; Figenschau, Y.; Hopstock, L.A.; Hutchinson, M.S.; Methlie, P.; Mihailova, A.; Sneve, M.; et al. Effect of smoking on the serum levels of 25-hydroxyvitamin D depends on the assay employed. Eur. J. Endocrinol. 2010, 163, 339–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yavuz, B.; Ertugrul, D.T. Statins and vitamin D. Dermato-Endocrinology 2012, 4, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Cangemi, R.; Loffredo, L.; Carnevale, R.; Pignatelli, P.; Violi, F. Statins enhance circulating vitamin E. Int. J. Cardiol. 2008, 123, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Oranje, W.A.; Sels, J.P.; Rondas-Colbers, G.J.; Lemmens, P.J.; Wolffenbuttel, B.H. Effect of atorvastatin on LDL oxidation and antioxidants in normocholesterolemic type 2 diabetic patients. Clin. Chim. Acta 2001, 311, 91–94. [Google Scholar] [CrossRef]
- Rydén, M.; Leanderson, P.; Kastbom, K.-O.; Jonasson, L. Effects of simvastatin on carotenoid status in plasma. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Vasankari, T.; Ahotupa, M.; Viikari, J.; Nuotio, I.; Strandberg, T.; Vanhanen, H.; Gylling, H.; Miettinen, T.; Tikkanen, M.J. Effect of 12-month statin therapy on antioxidant potential of LDL and serum antioxidant vitamin concentrations. Ann. Med. 2004, 36, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.-J.; Chung, N.; Lee, J.H.; Jang, Y.; Park, E.; Jeon, K.-I.; Chung, J.H.; Seo, B.-Y. Effects of simvastatin on plasma antioxidant status and vitamins in hypercholesterolemic patients. Int. J. Cardiol. 2007, 118, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Calvo Romero, J.M.; Ramiro Lozano, J.M. Vitamin B(12) in type 2 diabetic patients treated with metformin. Endocrinol. Nutr. 2012, 59, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Kos, E.; Liszek, M.J.; Emanuele, M.A.; Durazo-Arvizu, R.; Camacho, P. Effect of metformin therapy on vitamin D and vitamin B12; levels in patients with type 2 diabetes mellitus. Endocr. Pract. 2012, 18, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Reinstatler, L.; Qi, Y.P.; Williamson, R.S.; Garn, J.V.; Oakley, G.P. Association of biochemical B12 deficiency with metformin therapy and vitamin B12 supplements: The National Health and Nutrition Examination Survey, 1999–2006. Diabetes Care 2012, 35, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Yun, J.-S.; Ko, S.-H.; Lim, T.-S.; Ahn, Y.-B.; Park, Y.-M.; Ko, S.-H. Higher prevalence of metformin-induced vitamin B12 deficiency in sulfonylurea combination compared with insulin combination in patients with type 2 diabetes: A cross-sectional study. PLoS ONE 2014, 9, e109878. [Google Scholar] [CrossRef] [PubMed]
- Yetley, E.A.; Pfeiffer, C.M.; Phinney, K.W.; Bailey, R.L.; Blackmore, S.; Bock, J.L.; Brody, L.C.; Carmel, R.; Curtin, L.R.; Durazo-Arvizu, R.A.; et al. Biomarkers of vitamin B-12 status in NHANES: A roundtable summary. Am. J. Clin. Nutr. 2011, 94, 313S–321S. [Google Scholar] [CrossRef] [PubMed]
- Wile, D.J.; Toth, C. Association of Metformin, Elevated Homocysteine, and Methylmalonic Acid Levels and Clinically Worsened Diabetic Peripheral Neuropathy. Diabetes Care 2010, 33, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Sparre Hermann, L.; Nilsson, B.; Wettre, S. Vitamin B12 status of patients treated with metformin: A cross-sectional cohort study. Br. J. Diabetes Vasc. Dis. 2004, 4, 401–406. [Google Scholar] [CrossRef]
- Pongchaidecha, M.; Srikusalanukul, V.; Chattananon, A.; Tanjariyaporn, S. Effect of metformin on plasma homocysteine, vitamin B12 and folic acid: A cross-sectional study in patients with type 2 diabetes mellitus. J. Med. Assoc. Thail. 2004, 87, 780–787. [Google Scholar]
- Wulffelé, M.G.; Kooy, A.; Lehert, P.; Bets, D.; Ogterop, J.C.; Borger van der Burg, B.; Donker, A.J.M.; Stehouwer, C.D.A. Effects of short-term treatment with metformin on serum concentrations of homocysteine, folate and vitamin B12 in type 2 diabetes mellitus: A randomized, placebo-controlled trial. J. Intern. Med. 2003, 254, 455–463. [Google Scholar] [CrossRef] [PubMed]
- De Jager, J.; Kooy, A.; Lehert, P.; Wulffelé, M.G.; van der Kolk, J.; Bets, D.; Verburg, J.; Donker, A.J.M.; Stehouwer, C.D.A. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: Randomised placebo controlled trial. BMJ 2010, 340, c2181. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.F.; Clark, J.S.; Ireland, J.T.; Kesson, C.M.; Watson, W.S. Malabsorption of vitamin B12 and intrinsic factor secretion during biguanide therapy. Diabetologia 1983, 24, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Andrès, E.; Federici, L. Vitamin B12 deficiency in patients receiving metformin: Clinical data. Arch. Intern. Med. 2007, 167, 729. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.S.H. Metformin-induced vitamin B12 deficiency presenting as a peripheral neuropathy. South. Med. J. 2010, 103, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Kumar, A.; Karmakar, D.; Jha, R.K. Association of B12 deficiency and clinical neuropathy with metformin use in type 2 diabetes patients. J. Postgrad. Med. 2013, 59, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.J.; Herman, W.H.; Leal, S.; Rhinehart, A.S.; Shubrook, J.H.; Skolnik, N.; Kalyani, R.R. Pharmacologic Therapy for Type 2 Diabetes: Synopsis of the 2017 American Diabetes Association Standards of Medical Care in Diabetes. Ann. Intern. Med. 2017, 166, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Kancherla, V.; Garn, J.V.; Zakai, N.A.; Williamson, R.S.; Cashion, W.T.; Odewole, O.; Judd, S.E.; Oakley, G.P. Multivitamin Use and Serum Vitamin B12 Concentrations in Older-Adult Metformin Users in REGARDS, 2003–2007. PLoS ONE 2016, 11, e0160802. [Google Scholar] [CrossRef] [PubMed]
- Lecka-Czernik, B. Bone as a target of type 2 diabetes treatment. Curr. Opin. Investig. 2009, 10, 1085–1090. [Google Scholar]
- Lecka-Czernik, B. Bone Loss in Diabetes: Use of Antidiabetic Thiazolidinediones and Secondary Osteoporosis. Curr. Osteoporos. Rep. 2010, 8, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Benvenuti, S.; Cellai, I.; Luciani, P.; Deledda, C.; Baglioni, S.; Giuliani, C.; Saccardi, R.; Mazzanti, B.; Dal Pozzo, S.; Mannucci, E.; et al. Rosiglitazone stimulates adipogenesis and decreases osteoblastogenesis in human mesenchymal stem cells. J. Endocrinol. Investig. 2007, 30, RC26-30. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.A.; Weinstein, R.S.; Stewart, S.A.; Parfitt, A.M.; Manolagas, S.C.; Jilka, R.L. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology 2005, 146, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Zabłocka-Słowińska, K.; Dzielska, E.; Gryszkin, I.; Grajeta, H. Dietary supplementation during diabetes therapy and the potential risk of interactions. Adv. Clin. Exp. Med. 2014, 23, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.K.; Thomsen, K.; Eriksen, E.F.; Charles, P.; Storm, T.; Mosekilde, L. The effects of high-dose glucocorticoid administration on serum bone gamma carboxyglutamic acid-containing protein, serum alkaline phosphatase and vitamin D metabolites in normal subjects. Bone Miner. 1988, 4, 105–113. [Google Scholar] [PubMed]
- Ton, F.N.; Gunawardene, S.C.; Lee, H.; Neer, R.M. Effects of low-dose prednisone on bone metabolism. J. Bone Miner. Res. 2005, 20, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Mazziotti, G.; Giustina, A.; Bilezikian, J.P. Glucocorticoid-induced osteoporosis: Pathophysiology and therapy. Osteoporos. Int. 2007, 18, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E. Clinical review 83: Mechanisms of glucocorticoid action in bone: Implications to glucocorticoid-induced osteoporosis. J. Clin. Endocrinol. Metab. 1996, 81, 3441–3447. [Google Scholar] [CrossRef] [PubMed]
- Blahos, J.; Care, A.D.; Sommerville, B.A. The effect of betamethasone on duodenal calcium absorption and 1,25-dihydroxy vitamin D3 production in the chick. Horm. Metab. Res. 1983, 15, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-H.; Lee, G.-S.; Jung, E.-M.; Choi, K.-C.; Jeung, E.-B. The negative effect of dexamethasone on calcium-processing gene expressions is associated with a glucocorticoid-induced calcium-absorbing disorder. Life Sci. 2009, 85, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-H.; Lee, G.-S.; Jung, E.-M.; Choi, K.-C.; Oh, G.-T.; Jeung, E.-B. Dexamethasone differentially regulates renal and duodenal calcium-processing genes in calbindin-D9k and -D28k knockout mice. Exp. Physiol. 2009, 94, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Huybers, S.; Naber, T.H.J.; Bindels, R.J.M.; Hoenderop, J.G.J. Prednisolone-induced Ca2+ malabsorption is caused by diminished expression of the epithelial Ca2+ channel TRPV6. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G92–G97. [Google Scholar] [CrossRef] [PubMed]
- Hahn, T.J.; Halstead, L.R.; Haddad, J.G. Serum 25-hydroxyvitamin D concentrations in patients receiving chronic corticosteroid therapy. J. Lab. Clin. Med. 1977, 90, 399–404. [Google Scholar] [PubMed]
- Allen, C.S.; Yeung, J.H.; Vandermeer, B.; Homik, J. Bisphosphonates for steroid-induced osteoporosis. Cochrane Database Syst. Rev. 2016, 10, CD001347. [Google Scholar] [CrossRef] [PubMed]
- Warriner, A.; Saag, K.G. Prevention and Treatment of Bone Changes Associated with Exposure to Glucocorticoids. Curr. Osteoporos. Rep. 2013, 11, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Grossman, J.M.; Gordon, R.; Ranganath, V.K.; Deal, C.; Caplan, L.; Chen, W.; Curtis, J.R.; Furst, D.E.; McMahon, M.; Patkar, N.M.; et al. American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res. 2010, 62, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- McKay, L.I.; Cidlowski, J.A. Physiologic and Pharmacologic Effects of Corticosteroids. In Holland-Frei Cancer Medicine, 6th ed.; BC Decker: Hamilton, ON, Cancada, 2003. [Google Scholar]
- Goodwin, J.E.; Geller, D.S. Glucocorticoid-induced hypertension. Pediatr. Nephrol. 2012, 27, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Bia, M.J.; Tyler, K.; DeFronzo, R. The effect of dexamethasone on renal potassium excretion and acute potassium tolerance. Endocrinology 1983, 113, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- Campen, T.J.; Vaughn, D.A.; Fanestil, D.D. Mineralo- and glucocorticoid effects on renal excretion of electrolytes. Pflugers Arch. 1983, 399, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Saruta, T. Mechanism of glucocorticoid-induced hypertension. Hypertens. Res. 1996, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shenfield, G.M.; Knowles, G.K.; Thomas, N.; Paterson, J.W. Potassium supplements in patients treated with corticosteroids. Br. J. Dis. Chest 1975, 69, 171–176. [Google Scholar] [CrossRef]
- Ravina, A.; Slezak, L.; Mirsky, N.; Bryden, N.A.; Anderson, R.A. Reversal of corticosteroid-induced diabetes mellitus with supplemental chromium. Diabet. Med. 1999, 16, 164–167. [Google Scholar] [CrossRef] [PubMed]
- De Vries, F.; Pouwels, S.; Bracke, M.; Leufkens, H.G.M.; Cooper, C.; Lammers, J.-W.J.; van Staa, T.-P. Use of beta-2 agonists and risk of hip/femur fracture: A population-based case-control study. Pharmacoepidemiol. Drug Saf. 2007, 16, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Tattersfield, A.E.; Town, G.I.; Johnell, O.; Picado, C.; Aubier, M.; Braillon, P.; Karlström, R. Bone mineral density in subjects with mild asthma randomised to treatment with inhaled corticosteroids or non-corticosteroid treatment for two years. Thorax 2001, 56, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Halpern, M.T.; Schmier, J.K.; Van Kerkhove, M.D.; Watkins, M.; Kalberg, C.J. Impact of long-term inhaled corticosteroid therapy on bone mineral density: Results of a meta-analysis. Ann. Allergy Asthma Immunol. 2004, 92, 201–207. [Google Scholar] [CrossRef]
- Vanfleteren, L.E.G.W.; Spruit, M.A.; Groenen, M.; Gaffron, S.; van Empel, V.P.M.; Bruijnzeel, P.L.B.; Rutten, E.P.A.; Op’t Roodt, J.; Wouters, E.F.M.; Franssen, F.M.E. Clusters of comorbidities based on validated objective measurements and systemic inflammation in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2013, 187, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Chan, V.; Cave, A.J.; Banh, H.L. Self-reported osteoporosis prevention in inhaled corticosteroid users in community pharmacy setting. SAGE Open Med. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Bencaz, A.F.; Hentz, J.G.; Crowell, M.D. Selective serotonin reuptake inhibitor treatment and risk of fractures: A meta-analysis of cohort and case-control studies. Osteoporos. Int. 2012, 23, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Tsapakis, E.M.; Gamie, Z.; Tran, G.T.; Adshead, S.; Lampard, A.; Mantalaris, A.; Tsiridis, E. The adverse skeletal effects of selective serotonin reuptake inhibitors. Eur. Psychiatry 2012, 27, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P. The influence of oestrogens on tryptophan metabolism in man. Clin. Sci. 1966, 31, 265–272. [Google Scholar] [PubMed]
- Luhby, A.L.; Brin, M.; Gordon, M.; Davis, P.; Murphy, M.; Spiegel, H. Vitamin B6 metabolism in users of oral contraceptive agents. I. Abnormal urinary xanthurenic acid excretion and its correction by pyridoxine. Am. J. Clin. Nutr. 1971, 24, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Donald, E.A.; Bossé, T.R. The vitamin B6 requirement in oral contraceptive users. II. Assessment by tryptophan metabolites, vitamin B6, and pyridoxic acid levels in urine. Am. J. Clin. Nutr. 1979, 32, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Price, J.M.; Thornton, M.J.; Mueller, L.M. Tryptophan metabolism in women using steroid hormones for ovulation control. Am. J. Clin. Nutr. 1967, 20, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Aly, H.E.; Donald, E.A.; Simpson, M.H. Oral contraceptives and vitamin B6 metabolism. Am. J. Clin. Nutr. 1971, 24, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.R.; Rose, D.P.; Leklem, J.E.; Linkswiler, H.M. Effects of oral contraceptives on tryptophan metabolism and vitamin B6 requirements in women. Acta Vitaminol. Enzymol. 1975, 29, 151–157. [Google Scholar] [PubMed]
- Leklem, J.E.; Brown, R.R.; Rose, D.P.; Linkswiler, H.M. Vitamin B6 requirements of women using oral contraceptives. Am. J. Clin. Nutr. 1975, 28, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, L.; Cleary, R.E.; Li, T.K. Effect of oral contraceptives on the plasma concentration of pyridoxal phosphate. Am. J. Clin. Nutr. 1974, 27, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Lussana, F.; Zighetti, M.L.; Bucciarelli, P.; Cugno, M.; Cattaneo, M. Blood levels of homocysteine, folate, vitamin B6 and B12 in women using oral contraceptives compared to non-users. Thromb. Res. 2003, 112, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Bossé, T.R.; Donald, E.A. The vitamin B6 requirement in oral contraceptive users. I. Assessment by pyridoxal level and transferase activity in erythrocytes. Am. J. Clin. Nutr. 1979, 32, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Salkeld, R.M.; Knörr, K.; Körner, W.F. The effect of oral contraceptives on vitamin B6 status. Clin. Chim. Acta Int. J. Clin. Chem. 1973, 49, 195–199. [Google Scholar] [CrossRef]
- Vir, S.C.; Love, A.H. Effect of oral contraceptives on vitamin B6 nutriture of young women. Int. J. Vitam. Nutr. Res. 1980, 50, 29–34. [Google Scholar] [PubMed]
- Leklem, J.E. Vitamin B-6 requirement and oral contraceptive use—A concern? J. Nutr. 1986, 116, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Bermond, P. Therapy of side effects of oral contraceptive agents with vitamin B6. Acta Vitaminol. Enzymol. 1982, 4, 45–54. [Google Scholar] [PubMed]
- Villegas-Salas, E.; Ponce de León, R.; Juárez-Perez, M.A.; Grubb, G.S. Effect of vitamin B6 on the side effects of a low-dose combined oral contraceptive. Contraception 1997, 55, 245–248. [Google Scholar] [CrossRef]
- Gardyn, J.; Mittelman, M.; Zlotnik, J.; Sela, B.A.; Cohen, A.M. Oral contraceptives can cause falsely low vitamin B(12) levels. Acta Haematol. 2000, 104, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S.; Lei, K.Y.; Moghissi, K.S.; Stryker, J.C.; Oberleas, D. Effect of oral contraceptives on nutrients. III. Vitamins B6, B12, and folic acid. Am. J. Obstet. Gynecol. 1976, 125, 1063–1069. [Google Scholar] [CrossRef]
- Barone, C.; Bartoloni, C.; Ghirlanda, G.; Gentiloni, N. Megaloblastic anemia due to folic acid deficiency after oral contraceptives. Haematologica 1979, 64, 190–195. [Google Scholar] [PubMed]
- Holmes, R.P. Megaloblastic anemia precipitated by the use of oral contraceptive. A case report. N. C. Med. J. 1970, 31, 17–18. [Google Scholar] [PubMed]
- Ryser, J.E.; Farquet, J.J.; Petite, J. Megaloblastic Anemia due to Folic Acid Deficiency in a Young Woman on Oral Contraceptives. Acta Haematol. 1971, 45, 319–324. [Google Scholar] [CrossRef] [PubMed]
- McLean, F.W.; Heine, M.W.; Held, B.; Streiff, R.R. Relationship between the oral contraceptive and folic acid metabolism. Am. J. Obstet. Gynecol. 1969, 104, 745–747. [Google Scholar] [CrossRef]
- Shojania, A.M. Oral contraceptives: Effect of folate and vitamin B12 metabolism. Can. Med. Assoc. J. 1982, 126, 244–247. [Google Scholar] [CrossRef]
- Castrén, O.M.; Rossi, R.R. Effect of oral contraceptives on serum folic acid content. J. Obstet. Gynaecol. Br. Commonw. 1970, 77, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Green, T.J.; Houghton, L.A.; Donovan, U.; Gibson, R.S.; O’Connor, D.L. Oral contraceptives did not affect biochemical folate indexes and homocysteine concentrations in adolescent females. J. Am. Diet. Assoc. 1998, 98, 49–55. [Google Scholar] [CrossRef]
- Paine, C.J.; Grafton, W.D.; Dickson, V.L.; Eichner, E.R. Oral contraceptives, serum folate, and hematologic status. JAMA 1975, 231, 731–733. [Google Scholar] [CrossRef] [PubMed]
- Palmery, M.; Saraceno, A.; Vaiarelli, A.; Carlomagno, G. Oral contraceptives and changes in nutritional requirements. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 1804–1813. [Google Scholar] [PubMed]
- Pietarinen, G.J.; Leichter, J.; Pratt, R.F. Dietary folate intake and concentration of folate in serum and erythrocytes in women using oral contraceptives. Am. J. Clin. Nutr. 1977, 30, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Castaño, P.M.; Aydemir, A.; Sampson-Landers, C.; Lynen, R. The folate status of reproductive-aged women in a randomised trial of a folate-fortified oral contraceptive: Dietary and blood assessments. Public Health Nutr. 2014, 17, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Bart, S.; Marr, J.; Diefenbach, K.; Trummer, D.; Sampson-Landers, C. Folate status and homocysteine levels during a 24-week oral administration of a folate-containing oral contraceptive: A randomized, double-blind, active-controlled, parallel-group, US-based multicenter study. Contraception 2012, 85, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Garnero, P.; Sornay-Rendu, E.; Delmas, P.D. Decreased bone turnover in oral contraceptive users. Bone 1995, 16, 499–503. [Google Scholar] [CrossRef]
- Zittermann, A. Decreased urinary calcium loss and lower bone turnover in young oral contraceptive users. Metabolism. 2000, 49, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Goulding, A.; McChesney, R. Oestrogen-progestogen oral contraceptives and urinary calcium excretion. Clin. Endocrinol. 1977, 6, 449–454. [Google Scholar] [CrossRef]
- Jankun, J.; Skrzypczak-Jankun, E.; Lipinski, B. Experimental immunology Complex function of magnesium in blood clot formation and lysis. Cent. Eur. J. Immunol. 2013, 38, 149–153. [Google Scholar] [CrossRef]
- Seelig, M.S. Increased need for magnesium with the use of combined oestrogen and calcium for osteoporosis treatment. Magnes. Res. 1990, 3, 197–215. [Google Scholar] [PubMed]
- McLeroy, V.J.; Schendel, H.E. Influence of oral contraceptives on ascorbic acid concentrations in healthy, sexually mature women. Am. J. Clin. Nutr. 1973, 26, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Briggs, M.; Briggs, M. Vitamin C requirements and oral contraceptives. Nature 1972, 238, 277. [Google Scholar] [CrossRef] [PubMed]
- Pincemail, J.; Vanbelle, S.; Gaspard, U.; Collette, G.; Haleng, J.; Cheramy-Bien, J.P.; Charlier, C.; Chapelle, J.P.; Giet, D.; Albert, A.; et al. Effect of different contraceptive methods on the oxidative stress status in women aged 40–48 years from the ELAN study in the province of Liege, Belgium. Hum. Reprod. 2007, 22, 2335–2343. [Google Scholar] [CrossRef] [PubMed]
- Zal, F.; Mostafavi-Pour, Z.; Amini, F.; Heidari, A. Effect of vitamin E and C supplements on lipid peroxidation and GSH-dependent antioxidant enzyme status in the blood of women consuming oral contraceptives. Contraception 2012, 86, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Palan, P.R.; Magneson, A.T.; Castillo, M.; Dunne, J.; Mikhail, M.S. Effects of menstrual cycle and oral contraceptive use on serum levels of lipid-soluble antioxidants. Am. J. Obstet. Gynecol. 2006, 194, e35–e38. [Google Scholar] [CrossRef] [PubMed]
- De Groote, D.; d’Hauterive, S.P.; Pintiaux, A.; Balteau, B.; Gerday, C.; Claesen, J.; Foidart, J.-M. Effects of oral contraception with ethinylestradiol and drospirenone on oxidative stress in women 18–35 years old. Contraception 2009, 80, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kotani, K. Oral Contraceptive Therapy Increases Oxidative Stress in Pre-Menopausal Women. Int. J. Prev. Med. 2012, 3, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, K.; Milnerowicz, H. Pro/antioxidant status in young healthy women using oral contraceptives. Environ. Toxicol. Pharmacol. 2016, 43, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cauci, S.; Buligan, C.; Marangone, M.; Francescato, M.P. Oxidative Stress in Female Athletes Using Combined Oral Contraceptives. Sports Med. Open 2016, 2, 40. [Google Scholar] [CrossRef] [PubMed]
- Massafra, C.; Buonocore, G.; Berni, S.; Gioia, D.; Giuliani, A.; Vezzosi, P. Antioxidant erythrocyte enzyme activities during oral contraception. Contraception 1993, 47, 590–596. [Google Scholar] [CrossRef]
- Lerner, V.; Kanevsky, M.; Dwolatzky, T.; Rouach, T.; Kamin, R.; Miodownik, C. Vitamin B12 and folate serum levels in newly admitted psychiatric patients. Clin. Nutr. Edinb. Scotl. 2006, 25, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Abou-Saleh, M.T.; Coppen, A. Serum and red blood cell folate in depression. Acta Psychiatr. Scand. 1989, 80, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Bottiglieri, T.; Laundy, M.; Crellin, R.; Toone, B.K.; Carney, M.W.; Reynolds, E.H. Homocysteine, folate, methylation, and monoamine metabolism in depression. J. Neurol. Neurosurg. Psychiatry 2000, 69, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.S.; Fava, M.; Jacques, P.F.; Selhub, J.; Rosenberg, I.H. Depression and folate status in the US Population. Psychother. Psychosom. 2003, 72, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Tolmunen, T.; Voutilainen, S.; Hintikka, J.; Rissanen, T.; Tanskanen, A.; Viinamäki, H.; Kaplan, G.A.; Salonen, J.T. Dietary folate and depressive symptoms are associated in middle-aged Finnish men. J. Nutr. 2003, 133, 3233–3236. [Google Scholar] [CrossRef] [PubMed]
- Penninx, B.W.; Guralnik, J.M.; Ferrucci, L.; Fried, L.P.; Allen, R.H.; Stabler, S.P. Vitamin B(12) deficiency and depression in physically disabled older women: Epidemiologic evidence from the Women’s Health and Aging Study. Am. J. Psychiatry 2000, 157, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Serum Vitamin B12, C and Folate Concentrations in the New Mexico Elder Health Survey: Correlations with Cognitive and Affective Functions. Available online: https://ncbi.nlm.nih.gov/labs/articles/10682878/ (accessed on 12 May 2017).
- Fava, M.; Davidson, K.G. Definition and epidemiology of treatment-resistant depression. Psychiatr. Clin. North Am. 1996, 19, 179–200. [Google Scholar] [CrossRef]
- Fava, M. Augmenting antidepressants with folate: A clinical perspective. J. Clin. Psychiatry 2007, 68 (Suppl. S10), 4–7. [Google Scholar] [PubMed]
- Sebastian, J.L.; McKinney, W.P.; Kaufman, J.; Young, M.J. Angiotensin-converting enzyme inhibitors and cough. Prevalence in an outpatient medical clinic population. Chest 1991, 99, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Werner-Felmayer, G.; Werner, E.R.; Grünewald, K.; Wachter, H.; Hentze, M.W. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J. Exp. Med. 1994, 180, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Park, S.W.; Kim, D.K.; Lee, S.H.; Hong, K.P. Iron supplementation inhibits cough associated with ACE inhibitors. Hypertens. Dallas Tex 1979 2001, 38, 166–170. [Google Scholar] [CrossRef]
- Qato, D.M.; Wilder, J.; Schumm, L.P.; Gillet, V.; Alexander, G.C. Changes in Prescription and Over-the-Counter Medication and Dietary Supplement Use Among Older Adults in the United States, 2005 vs. 2011. JAMA Intern. Med. 2016, 176, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Reider, C.; Brooks, J.R.; Fulgoni, V.L. 3rd. Comparison of Prevalence of Inadequate Nutrient Intake Based on Body Weight Status of Adults in the United States: An Analysis of NHANES 2001–2008. J. Am. Coll. Nutr. 2015, 34, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.J.B.; Suchindran, C.M.; Roggenkamp, K.J. Micronutrient intakes in two US populations of older adults: Lipid research clinics program prevalence study findings. J. Nutr. Health Aging 2009, 13, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.L.; Akabas, S.R.; Paxson, E.E.; Thuppal, S.V.; Saklani, S.; Tucker, K.L. Total Usual Intake of Shortfall Nutrients Varies With Poverty Among US Adults. J. Nutr. Educ. Behav. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kantor, E.D.; Rehm, C.D.; Du, M.; White, E.; Giovannucci, E.L. Trends in Dietary Supplement Use among US Adults from 1999–2012. JAMA 2016, 316, 1464–1474. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.C.; McBurney, M.; Fulgoni, V.L. Multivitamin/mineral supplement contribution to micronutrient intakes in the United States, 2007–2010. J. Am. Coll. Nutr. 2014, 33, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.L.; Gahche, J.J.; Miller, P.E.; Thomas, P.R.; Dwyer, J.T. Why US adults use dietary supplements. JAMA Intern. Med. 2013, 173, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Bird, J.K.; Murphy, R.A.; Ciappio, E.D.; McBurney, M.I. Risk of Deficiency in Multiple Concurrent Micronutrients in Children and Adults in the United States. Nutrients 2017, 9, 655. [Google Scholar] [CrossRef] [PubMed]
- McKay, D.L.; Perrone, G.; Rasmussen, H.; Dallal, G.; Blumberg, J.B. Multivitamin/Mineral Supplementation Improves Plasma B-Vitamin Status and Homocysteine Concentration in Healthy Older Adults Consuming a Folate-Fortified Diet. J. Nutr. 2000, 130, 3090–3096. [Google Scholar] [CrossRef] [PubMed]
- Ward, E. Addressing nutritional gaps with multivitamin and mineral supplements. Nutr. J. 2014, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Gaziano, J.M.; Sesso, H.D.; Christen, W.G.; Bubes, V.; Smith, J.P.; MacFadyen, J.; Schvartz, M.; Manson, J.E.; Glynn, R.J.; Buring, J.E. Multivitamins in the prevention of cancer in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2012, 308, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Massa, J.; Cho, E.; Orav, E.J.; Willett, W.C.; Wu, K.; Giovannucci, E.L. Long-term use of multivitamins and risk of colorectal adenoma in women. Br. J. Cancer 2014, 110, 249–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christen, W.G.; Glynn, R.J.; Manson, J.E.; MacFadyen, J.; Bubes, V.; Schvartz, M.; Buring, J.E.; Sesso, H.D.; Gaziano, J.M. Effects of multivitamin supplement on cataract and age-related macular degeneration in a randomized trial of male physicians. Ophthalmology 2014, 121, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Rautiainen, S.; Rist, P.M.; Glynn, R.J.; Buring, J.E.; Gaziano, J.M.; Sesso, H.D. Multivitamin Use and the Risk of Cardiovascular Disease in Men. J. Nutr. 2016, jn227884. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.L.; Fakhouri, T.H.; Park, Y.; Dwyer, J.T.; Thomas, P.R.; Gahche, J.J.; Miller, P.E.; Dodd, K.W.; Sempos, C.T.; Murray, D.M. Multivitamin-Mineral Use Is Associated with Reduced Risk of Cardiovascular Disease Mortality among Women in the United States. J. Nutr. 2015. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.D.; Weed, D.L.; Chang, E.T.; Miller, P.E.; Mohamed, M.A.; Elkayam, L. A systematic review of multivitamin-multimineral use and cardiovascular disease and cancer incidence and total mortality. J. Am. Coll. Nutr. 2013, 32, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Lamas, G.A.; Boineau, R.; Goertz, C.; Mark, D.B.; Rosenberg, Y.; Stylianou, M.; Rozema, T.; Nahin, R.L.; Lindblad, L.; Lewis, E.F.; et al. Oral high-dose multivitamins and minerals after myocardial infarction: A randomized trial. Ann. Intern. Med. 2013, 159, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Biesalski, H.K.; Tinz, J. Multivitamin/mineral supplements: Rationale and safety—A systematic review. Nutrition 2017, 33, 76–82. [Google Scholar] [CrossRef] [PubMed]

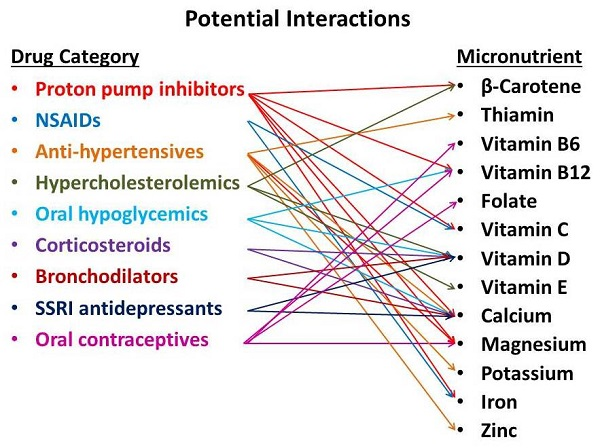{kind=link}
| Drug Category | Name | Nutrient | Effect on Nutrient Status or Function | Human Studies 1 | Risk Factors | References |
|---|---|---|---|---|---|---|
| Acid-Suppressing Drugs | Proton Pump Inhibitors | Vitamin B12 Vitamin C Iron CalciumMagnesium Zinc β-Carotene | Decrease Decrease Decrease Decrease Decrease Decrease Decrease | 5 observational 5 intervention 1 observation 4 intervention 2 case reports 1 observational 2 intervention >10 observational 4 intervention 30 case reports 2 intervention 1 intervention | Advanced age H. pylori infection Genetics (slow metabolizers Low dietary intake (vegetarians) H. pylori infection Pre-existing iron deficiency Vegetarians Advanced age Women Advanced age Duration of drug use Women Undetermined Undetermined | [10,11,12,13,14,15,16,17] [18,19,20,21,22] [23] [24,25,26,27,28] [29,30,31] -- -- |
| Non-Steroidal Anti-Inflammatory Drugs | Aspirin | Vitamin C Iron | Decrease Decrease | 1 observational 4 intervention 6 observational 8 intervention | Absence of cold virus Advanced age H. pylori infection | [32,33,34,35] [36,37,38,39] |
| Anti-Hypertensives | Diuretics (loop, thiazide) Diuretics (potassium-sparing) Angiotensin-Converting Enzyme Inhibitors Calcium Channel Blockers | Calcium Magnesium Thiamin Zinc Potassium Folate Zinc Potassium Iron 2 Folate Potassium | Decrease (loop) Increase (thiazide) Decrease (loop and thiazide) Decrease (loop) Decrease (thiazide) Decrease (thiazide) Decrease Decrease Increase N/A Decrease Increase | >20 observational 9 intervention >10 observational 1 intervention 4 observational 2 intervention 3 observational 6 intervention 3 observational >100 intervention 2 case reports 1 observational 3 observational 6 intervention 2 case reports 3 observational 1 intervention 1 intervention 6 case reports 3 observational 2 case reports 2 observational | Dose/duration of drug use Form of loop diuretic Advanced age Women Advanced age Heart failure Low Mg intake Alcohol use Long-term use Coronary heart failure Advanced age Low dietary thiamin intake Hepatic cirrhosis Diabetes mellitus Heart failure Gastro-intestinal disorders Renal disease Low dietary zinc intake Dose Form of thiazide used Low folate status Impaired liver function Liver cirrhosis (alcoholics) Use of captopril Heart failure Renal disease Age (elderly) Renal disease Diabetes mellitus Congestive heart failure Potassium supplement use Undetermined Presence of dental plaque Poor oral hygiene Gender (men) Dose Low folate intakes Concurrent use of beta-blockers | [40,41,42,43,44,45,46,47,48] loop [42,49,50,51,52,53,54] thiazide [55,56,57] [58,59,60,61,62] [63,64,65,66,67,68,69] [70,71,72,73,74] [75,76] [77,78,79,80,81,82,83,84,85] [86,87,88,89,90,91] -- [92,93,94,95,96,97,98,99] [100,101,102,103] |
| Hypercholesterolemics | Statins | Coenzyme Q10 Vitamin D Vitamin E/β-Carotene | Decrease Increase/Decrease Increase/Decrease | 7 observational >10 intervention >10 observational 4 intervention 1 observational 6 intervention | Dose Advanced age Statin-associated myopathy Heart disease Vitamin D deficiency Statin-associated myopathy Undetermined | [104,105,106,107,108,109,110,111,112,113,114] [115,116,117,118,119,120,121,122,123,124,125,126,127,128] -- |
| Hypoglycemics | Biguanides (Metformin) Thiazolidinediones | Vitamin B12 Calcium/Vitamin D | Decrease Decrease | >10 observational >10 intervention 3 observational >10 intervention | Dose/duration of drug use Advanced age Vegetarians Advanced age Women Low calcium/vitamin D intake | [129,130,131,132,133,134,135,136,137,138,139,140] [141,142,143,144,145] |
| Corticosteroids | Glucocorticoids (oral) | Calcium/Vitamin D Sodium/Potassium Chromium | Decrease Increase (sodium) Decrease (potassium) Decrease | >80 observational >10 intervention ~5 case reports/observational 1 intervention 1 intervention | Low calcium/vitamin D intake At risk for bone fracture/loss Undetermined Undetermined | [146,147,148,149,150,151,152,153,154] -- -- |
| Bronchodilators | Corticosteroids (inhaled) | Calcium/Vitamin D | Decrease | >10 observational >10 intervention | Presence of COPDSmoking At risk for bone fracture/loss Low calcium/vitamin D intake | [155,156,157,158,159] |
| Antidepressants | Selective Serotonin Reuptake Inhibitors | Folate 3 Calcium/Vitamin D | Increase 3 Decrease | 5 observational 2 intervention >10 observational | Low folate intake Genetics (MTHFR variants) Alcoholism At risk for bone fracture/loss Low calcium/vitamin D intake | [160,161,162,163,164,165,166,167] [168,169,170,171] |
| Oral Contraceptives | Estrogen and/or Progesterone | Vitamin B6 Vitamin B12/Folate Calcium Magnesium Vitamin C/Vitamin E | Decrease Decrease Increase/decrease Decrease Decrease | >10 observational 5 intervention 4 case reports >30 observational 5 intervention 7 observational 6 intervention >20 observational >10 observational 2 intervention | Undetermined Vegetarians Low folate intake Genetics (folate) Duration of drug use Duration of drug use Physical activity level Low calcium intake Age at first use Race Type of combined OC used Undetermined | -- [172,173,174,175,176,177,178,179,180,181,182,183] [184,185,186,187,188,189,190,191,192,193] [194,195,196,197,198,199,200] -- |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohn, E.S.; Kern, H.J.; Saltzman, E.; Mitmesser, S.H.; McKay, D.L. Evidence of Drug–Nutrient Interactions with Chronic Use of Commonly Prescribed Medications: An Update. Pharmaceutics 2018, 10, 36. https://doi.org/10.3390/pharmaceutics10010036
Mohn ES, Kern HJ, Saltzman E, Mitmesser SH, McKay DL. Evidence of Drug–Nutrient Interactions with Chronic Use of Commonly Prescribed Medications: An Update. Pharmaceutics. 2018; 10(1):36. https://doi.org/10.3390/pharmaceutics10010036
Chicago/Turabian StyleMohn, Emily S., Hua J. Kern, Edward Saltzman, Susan H. Mitmesser, and Diane L. McKay. 2018. "Evidence of Drug–Nutrient Interactions with Chronic Use of Commonly Prescribed Medications: An Update" Pharmaceutics 10, no. 1: 36. https://doi.org/10.3390/pharmaceutics10010036
APA StyleMohn, E. S., Kern, H. J., Saltzman, E., Mitmesser, S. H., & McKay, D. L. (2018). Evidence of Drug–Nutrient Interactions with Chronic Use of Commonly Prescribed Medications: An Update. Pharmaceutics, 10(1), 36. https://doi.org/10.3390/pharmaceutics10010036