Oral Bioavailability of Kinsenoside in Beagle Dogs Measured by LC-MS/MS: Improvement of Ex Vivo Stability of a Lactone-Containing Compound

 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. LC-MS/MS Conditions
2.3. Preparation of Calibration Standards, Quality Control (QC) Samples, and Esterase Inhibitor Solutions
2.4. Sample Preparation
2.5. Stability Evaluation in Dog Whole Blood and Plasma
2.6. Method Validation
2.7. Pharmacokinetic and Bioavailability Study of KD in Beagle Dogs
2.8. Pharmacokinetic Parameter Calculation
2.9. Statistical Analyses
3. Results and Discussion
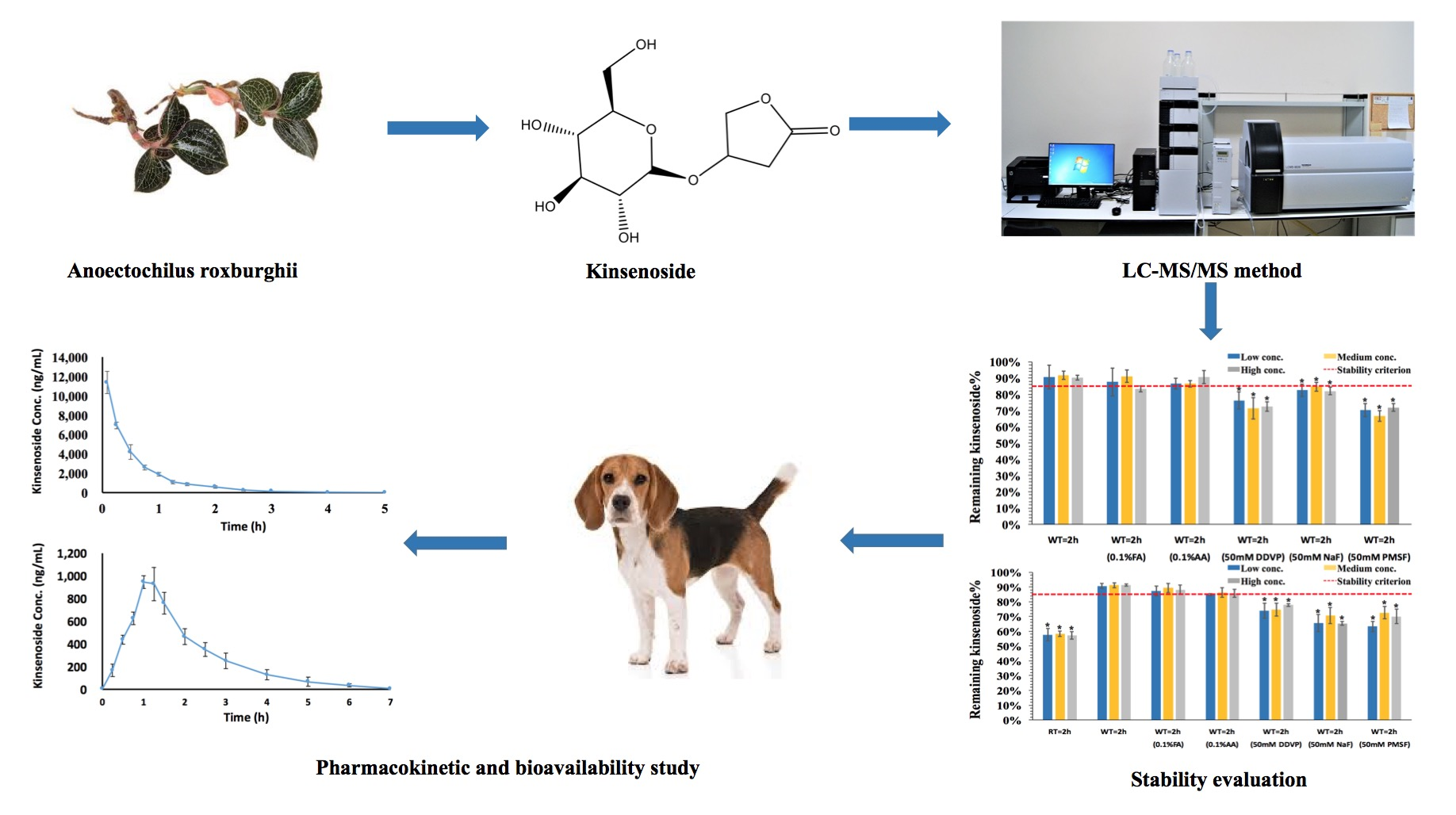
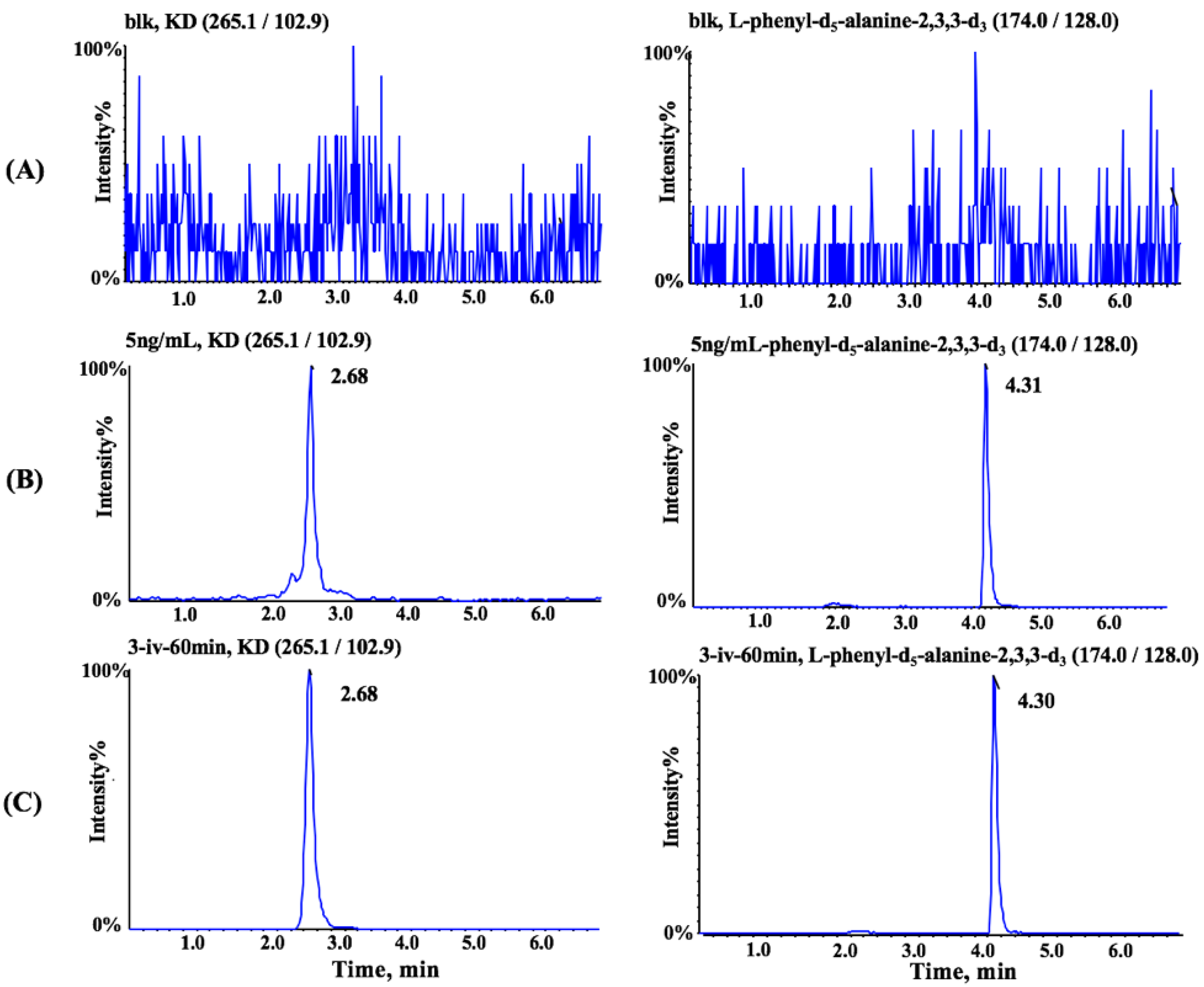
3.1. Method Optimization
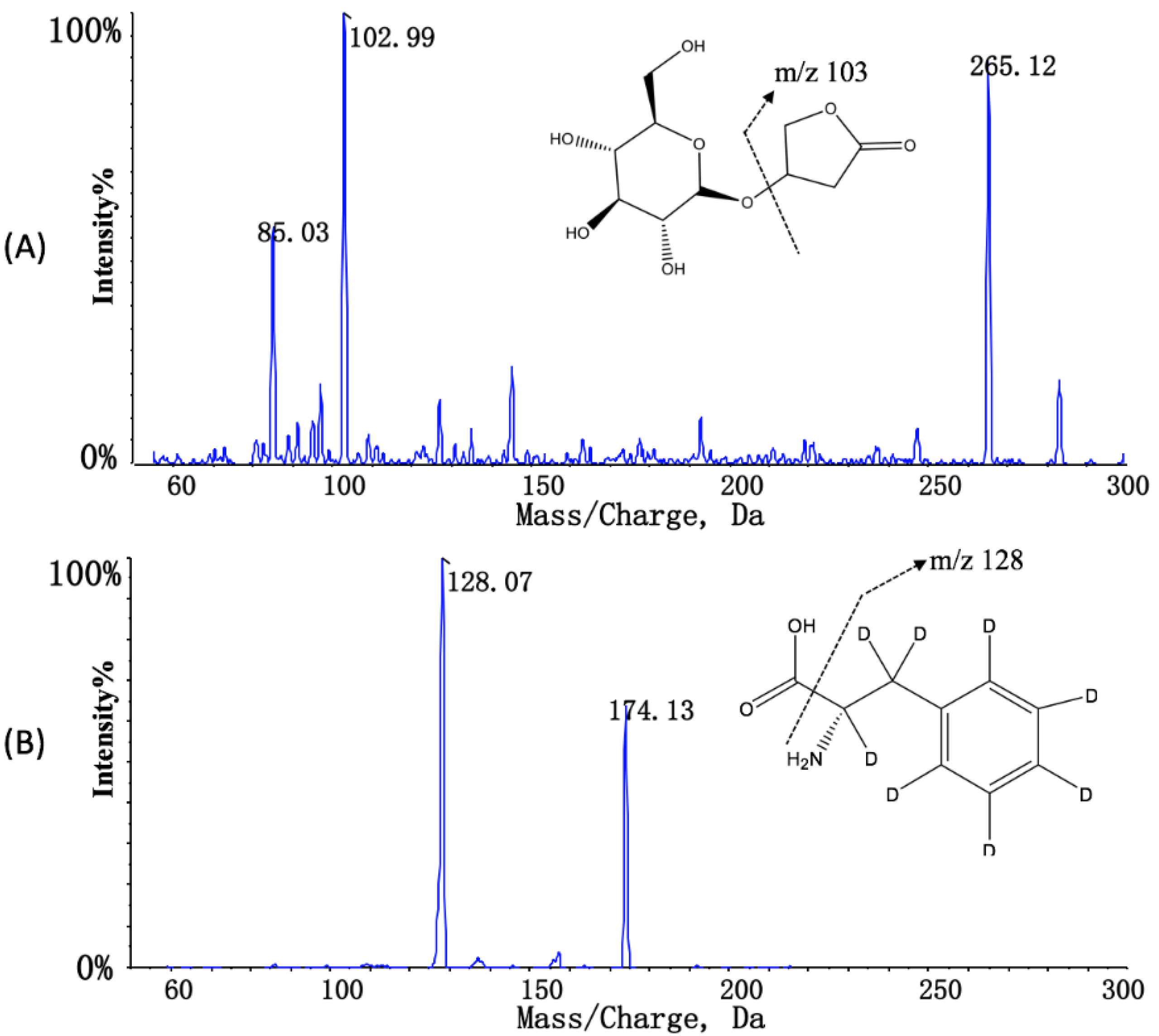
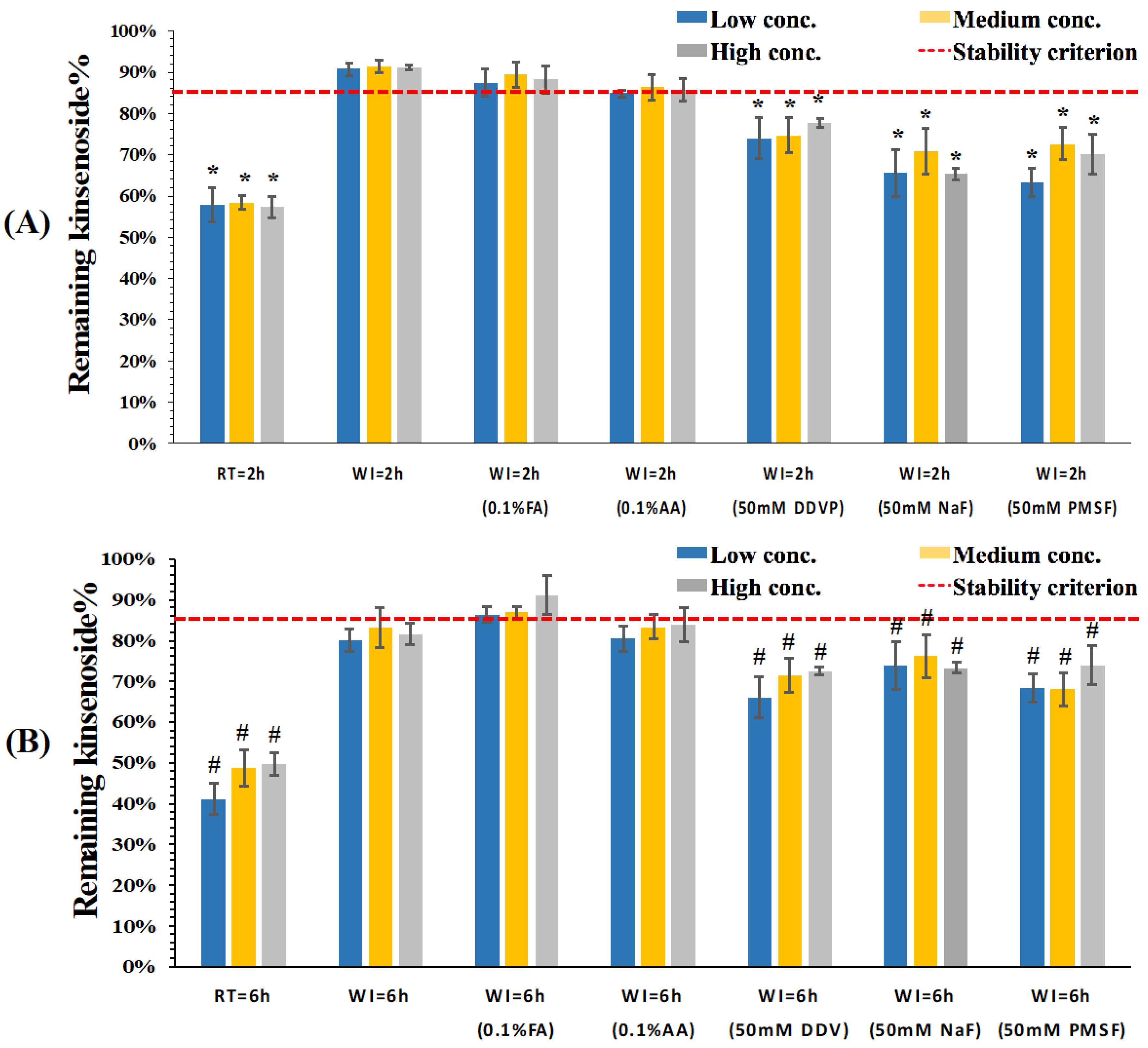
3.2. Stability Evaluation of KD in Dog Whole Blood and Plasma
3.3. Method Validation
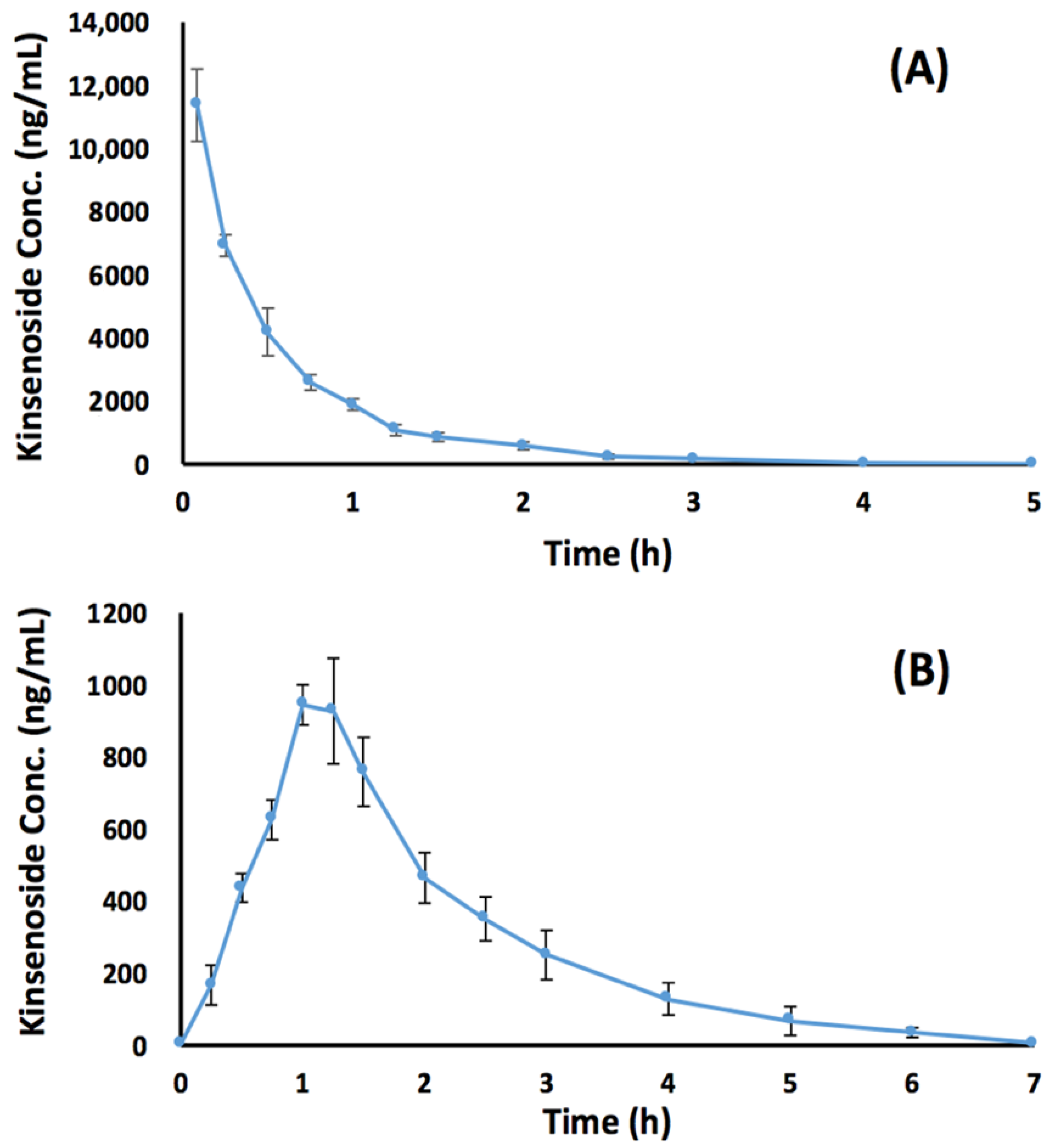
3.4. Preclinical Bioavailability Study
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ye, S.; Shao, Q.; Zhang, A. Anoectochilus roxburghii: A review of its phytochemistry, pharmacology, and clinical applications. J. Ethnopharmacol. 2017, 209, 184–202. [Google Scholar] [CrossRef] [PubMed]
- Du, X.M.; Irino, N.; Furusho, N.; Hayashi, J.; Shoyama, Y. Pharmacologically active compounds in the Anoectochilus and Goodyera species. J. Nat. Med. 2008, 62, 132–148. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.B.; Lin, W.L.; Hsieh, C.C.; Ho, H.Y.; Tsay, H.S.; Lin, W.C. The hepatoprotective activity of kinsenoside from Anoectochilus formosanus. Phytother. Res. 2007, 21, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cai, J.; Ruan, H.; Pi, H.; Wu, J. Antihyperglycemic activity of kinsenoside, a high yielding constituent from Anoectochilus roxburghii in streptozotocin diabetic rats. J. Ethnopharmacol. 2007, 114, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.B.; Wu, J.B.; Lin, H.; Lin, W.C. Kinsenoside isolated from Anoectochilus formosanus suppresses LPS-stimulated inflammatory reactions in macrophages and endotoxin shock in mice. Shock 2011, 35, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Bing, W.; Di, Y.; Hua, L.; Shi-He, L.; Yu-Hua, Z.; Xiu-Guo, H.; Yu-Gang, W.; Qi-Ming, F.; Shih-Mo, Y.; et al. Kinsenoside screening with a microfluidic chip attenuates gouty arthritis through inactivating NF-kappaB signaling in macrophages and protecting endothelial cells. Cell Death Dis. 2016, 7, e2350. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.L.; Liu, Q.; Xiao, B.; Zhou, J.; Zhang, J.G.; Li, Y. The vascular protective properties of kinsenoside isolated from Anoectochilus roxburghii under high glucose condition. Fitoterapia 2013, 86, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Liu, T.; Tan, W.; Ren, H.; Li, H.; Liu, J.; Cao, H.; Cheng, Q.; Liu, X.; Zhu, H.; et al. Effects of kinsenoside, a potential immunosuppressive drug for autoimmune hepatitis, on dendritic cells/CD8+T cells communication in mice. Hepatology 2016, 64, 2135–2150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xia, Y.; Lai, Y.; Tang, F.; Luo, Z.; Xue, Y.; Yao, G.; Zhang, Y.; Zhang, J. Efficient synthesis of kinsenoside and goodyeroside a by a chemo-enzymatic approach. Molecules 2014, 19, 16950–16958. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Kim, N.S.; Choi, M.S.; Luo, Z.; Yao, G.; Xue, Y.; Zhang, Y.; Yoo, H.H. Evaluation of metabolic stability of kinsenoside, an antidiabetic candidate, in rat and human liver microsomes. Mass Spectrom. Lett. 2015, 6, 48–51. [Google Scholar] [CrossRef]
- Rehman, S.U.; Kim, I.S.; Choi, M.S.; Luo, Z.; Yao, G.; Xue, Y.; Zhang, Y.; Yoo, H.H. Development of a hydrophilic interaction liquid chromatography-tandem mass spectrometric method for the determination of kinsenoside, an antihyperlipidemic candidate, in rat plasma and its application to pharmacokinetic studies. J. Pharm. Biomed. Anal. 2016, 120, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Freisleben, A.; Brudny-Kloppel, M.; Mulder, H.; de Vries, R.; de Zwart, M.; Timmerman, P. Blood stability testing: European bioanalysis forum view on current challenges for regulated bioanalysis. Bioanalysis 2011, 3, 1333–1336. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Li, G.; Huang, L.; Zhao, S.; Hu, Y.; Qin, L.; Qiu, L.; Zhu, W.; Si, L.; Huang, J. Screening of stabilizers for LC-MS/MS analysis of clevidipine and its primary metabolite in dog whole blood. Bioanalysis 2015, 7, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Health and Human Services Food and Drug Administration. Guidance for Industry, Bioanalytical Methods Validation. 2013. Available online: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070107.Pdf (accessed on 6 June 2018).
- Loukotkova, L.; VonTungeln, L.S.; Vanlandingham, M.; da Costa, G.G. A simple and highly sensitive UPLC-ESI-MS/MS method for the simultaneous quantification of nicotine, cotinine, and the tobacco-specific carcinogens N′-nitrosonornicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in serum samples. J. Chromatogr. B 2018, 1072, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. The role of nitrogen oxidation in the excretion of drugs and foreign compounds. Xenobiotica 1971, 1, 399–418. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, J.; Song, L.; Lynn, B.C.; Burke, T.G. Degradation of camptothecin-20(S)-glycinate ester prodrug under physiological conditions. J. Pharm. Biomed. Anal. 2004, 35, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Billecke, S.; Draganov, D.; Counsell, R.; Stetson, P.; Watson, C.; Hsu, C.; La Du, B.N. Human serum paraoxonase (PON1) isozymes Q and R hydrolyze lactones and cyclic carbonate esters. Drug Metab. Dispos. 2000, 28, 1335–1342. [Google Scholar] [PubMed]
- Xue, K.; Li, G.; Sun, X.; Hu, Y.; Hu, L.; Huang, J.; Si, L. Simultaneous quantification of fosinopril and its active metabolite fosinoprilat in rat plasma by UFLC-MS/MS: Application of formic acid in the stabilization of an ester-containing drug. J. Chromatogr. B 2015, 990, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Fung, E.N.; Buzescu, A.; Arnold, M.E.; Zeng, J. Esterase inhibitors as ester-containing drug stabilizers and their hydrolytic products: Potential contributors to the matrix effects on bioanalysis by liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2012, 26, 1291–1304. [Google Scholar] [CrossRef] [PubMed]
- Lindup, W.E.; Orme, M.C. Clinical pharmacology: Plasma protein binding of drugs. Br. Med. J. 1981, 282, 212–214. [Google Scholar] [CrossRef]
- Yang, J.; Sun, X.; Feng, Z.; Hao, D.; Wang, M.; Zhao, X.; Sun, C. Metabolomic analysis of the toxic effects of chronic exposure to low-level dichlorvos on rats using ultra-performance liquid chromatography—Mass spectrometry. Toxicol. Lett. 2011, 206, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Fung, E.N.; Zheng, N.; Arnold, M.E.; Zeng, J. Effective screening approach to select esterase inhibitors used for stabilizing ester-containing prodrugs analyzed by LC-MS/MS. Bioanalysis 2010, 2, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Hostetler, G.L.; Ralston, R.A.; Schwartz, S.J. Flavones: Food sources, bioavailability, metabolism, and bioactivity. Adv. Nutr. 2017, 8, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.Y.; Fan, M.X.; Zhao, H.Y.; Li, M.X.; Wu, X.; Gao, W.Y. Pharmacokinetics and bioavailability of the isoflavones formononetin and ononin and their in vitro absorption in ussing chamber and Caco-2 cell models. J. Agric. Food Chem. 2018, 66, 2917–2924. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Lin, L.C.; Lin, C.H.; Tsai, T.H. Comparative oral bioavailability of geniposide following oral administration of geniposide, Gardenia jasminoides Ellis fruits extracts and gardenia herbal formulation in rats. J. Pharm. Pharmacol. 2014, 66, 705–712. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | Nominal Conc. (ng/mL) | Calculated Conc. (ng/mL) | Accuracy (%) | Precision (% RSD) |
|---|---|---|---|---|
| Intra-day (n = 6) | 5 | 4.87 ± 0.19 | 97.43 | 3.82 |
| 15 | 15.55 ± 0.71 | 103.67 | 4.54 | |
| 150 | 154.50 ± 7.76 | 103.00 | 5.02 | |
| 1500 | 1595.00 ± 41.93 | 100.78 | 4.67 | |
| Inter-day (n = 18) | 5 | 4.84 ± 0.27 | 96.78 | 5.52 |
| 15 | 15.61 ± 0.68 | 104.07 | 4.34 | |
| 150 | 152.61 ± 7.62 | 101.74 | 4.99 | |
| 1500 | 1547.78 ± 66.13 | 103.19 | 4.27 |
| Compound | Nominal Conc. (ng/mL) | Extraction Recovery (n = 6) | Matrix Effect (n = 6) | ||
|---|---|---|---|---|---|
| Mean (%) | RSD (%) | Mean (%) | RSD (%) | ||
| kinsenoside | 15 | 79.92 | 3.67 | 93.01 | 5.66 |
| 150 | 86.14 | 5.81 | 101.49 | 3.71 | |
| 1500 | 80.90 | 3.17 | 95.27 | 3.08 | |
| IS | 500 | 84.75 | 4.88 | 97.55 | 4.44 |
| Stability | Nominal Conc. (ng/mL) | Calculated Conc. (ng/mL) | Accuracy (%) |
|---|---|---|---|
| Plasma samples on wet ice for 2 h | 15 | 14.27 ± 0.74 | 95.11 |
| 150 | 140.80 ± 2.98 | 93.87 | |
| 1500 | 1374.13 ± 24.63 | 91.61 | |
| Plasma samples stored at −80 °C for 21 days | 15 | 13.63 ± 0.36 | 90.89 |
| 150 | 137.67 ± 2.62 | 91.78 | |
| 1500 | 1406.67 ± 26.25 | 93.78 | |
| Plasma samples after three freeze–thaw cycles | 15 | 13.43 ± 0.46 | 89.53 |
| 150 | 136.33 ± 4.50 | 90.89 | |
| 1500 | 1386.67 ± 24.94 | 92.44 | |
| Post-preparative samples stored at 4 °C for 24 h | 15 | 13.42 ± 0.37 | 89.47 |
| 150 | 137.50 ± 4.99 | 91.67 | |
| 1500 | 1333.33 ± 34.60 | 88.89 |
| Pharmacokinetic Parameter | Intravenous Group | Oral Group |
|---|---|---|
| AUC0–t (ng·h/mL) | 6820 ± 295 | 1880 ± 17.6 |
| AUC0–∞ (ng·h/mL) | 6830 ± 295 | 1910 ± 42.5 |
| Cmax (ng/mL) | 11,400 ± 996 | 965 ± 59.7 |
| Tmax (h) | 0.0833 | 1.04 ± 0.102 |
| λz (1/h) | 1.47 ± 0.0404 | 0.764 ± 0.0737 |
| t1/2 (h) | 0.47 ± 0.0130 | 0.915 ± 0.0940 |
| CL (mL/h/kg) | 440 ± 18.7 | NA |
| Vd (mL/kg) | 300 ± 17.1 | NA |
| Bioavailability | NA | 27.6% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Jin, M.; Liu, Y.; Chen, Q.; Si, L.; Li, G.; Zhang, Y.; Huang, J. Oral Bioavailability of Kinsenoside in Beagle Dogs Measured by LC-MS/MS: Improvement of Ex Vivo Stability of a Lactone-Containing Compound. Pharmaceutics 2018, 10, 87. https://doi.org/10.3390/pharmaceutics10030087
Zhang X, Jin M, Liu Y, Chen Q, Si L, Li G, Zhang Y, Huang J. Oral Bioavailability of Kinsenoside in Beagle Dogs Measured by LC-MS/MS: Improvement of Ex Vivo Stability of a Lactone-Containing Compound. Pharmaceutics. 2018; 10(3):87. https://doi.org/10.3390/pharmaceutics10030087
Chicago/Turabian StyleZhang, Xin, Ming Jin, Yuping Liu, Qimingxing Chen, Luqin Si, Gao Li, Yonghui Zhang, and Jiangeng Huang. 2018. "Oral Bioavailability of Kinsenoside in Beagle Dogs Measured by LC-MS/MS: Improvement of Ex Vivo Stability of a Lactone-Containing Compound" Pharmaceutics 10, no. 3: 87. https://doi.org/10.3390/pharmaceutics10030087