The Influence of Polymers on the Supersaturation Potential of Poor and Good Glass Formers

 , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Determination of Thermal Stability
2.3. Determination of Glass Forming Ability by Melt Quenching
2.4. Determination of pKa
2.5. Preparation of Fasted State Simulated Intestinal Fluid
2.6. Determination of Equilibrium Solubility
2.7. Determination of the Supersaturation Potential
2.8. Statistical Analysis
3. Results
3.1. Determination of GFA of Drugs
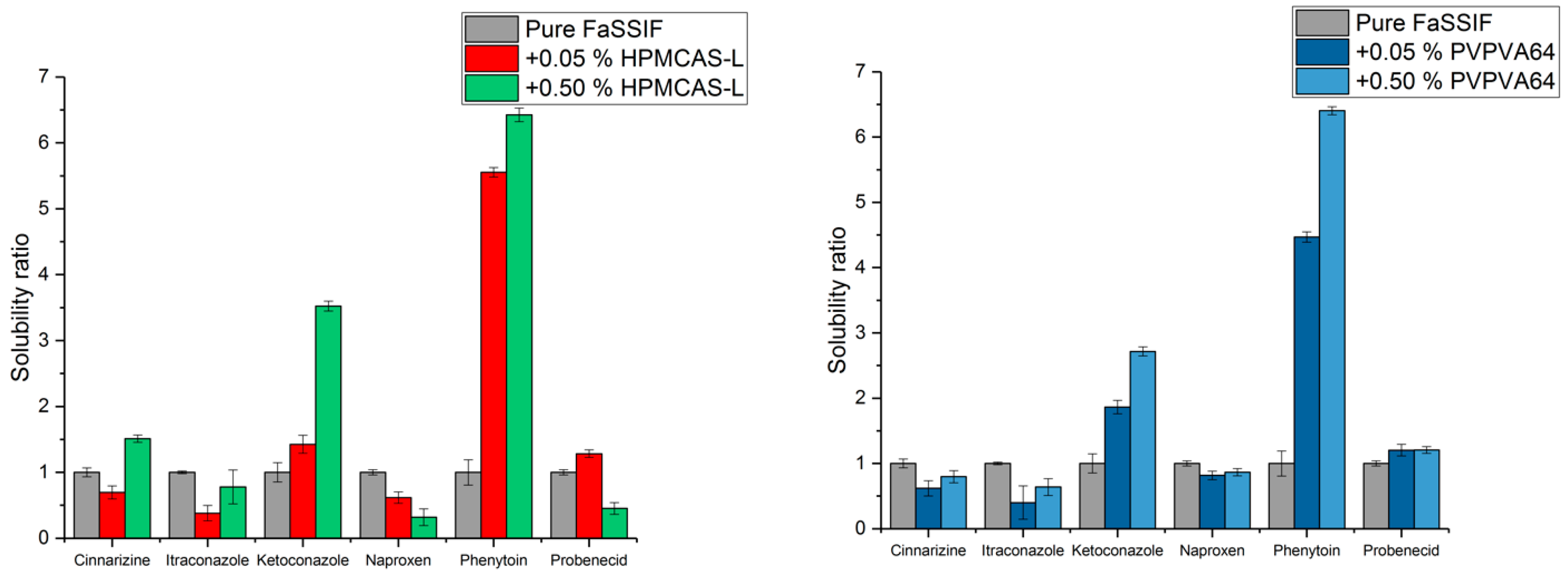
3.2. Determination of Equilibrium Solubility with and without Polymer
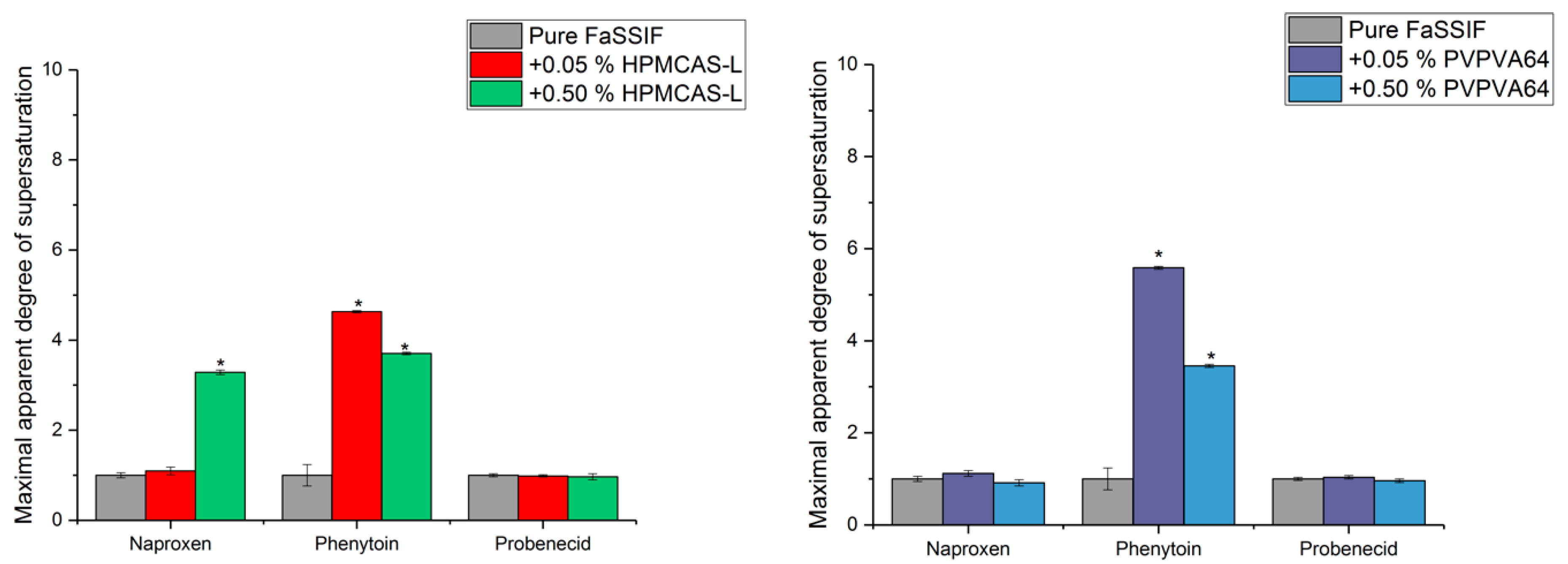
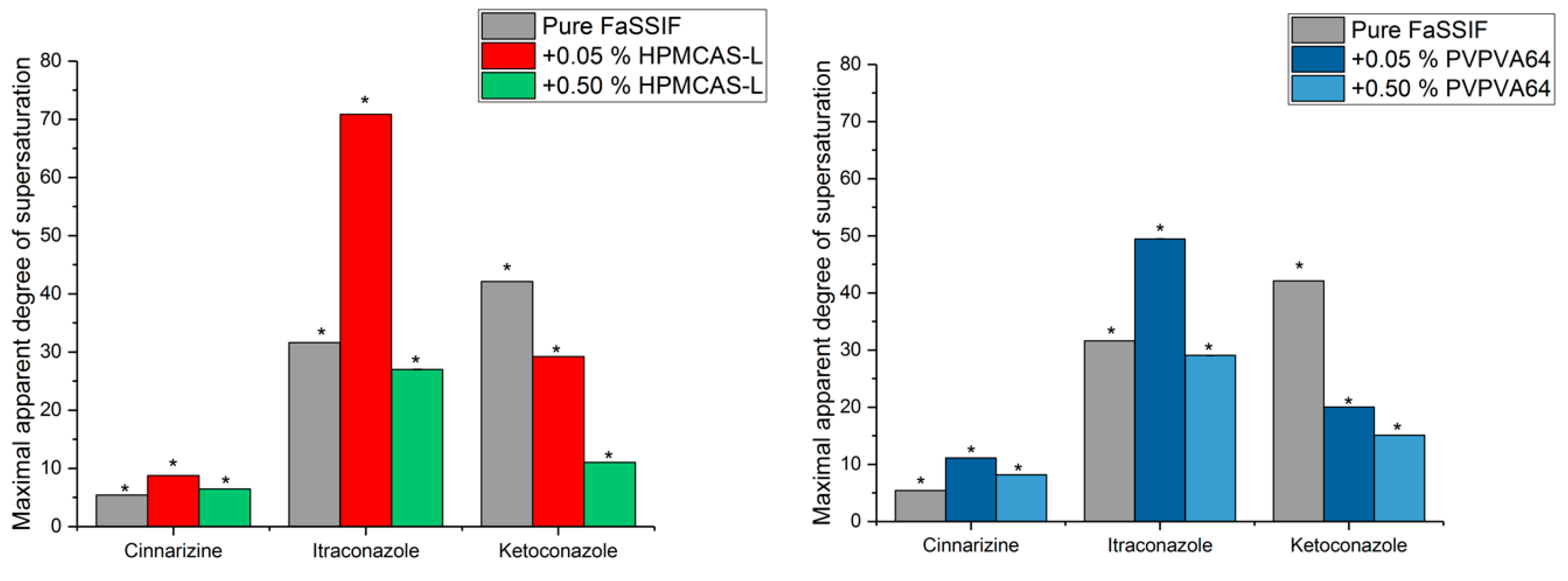
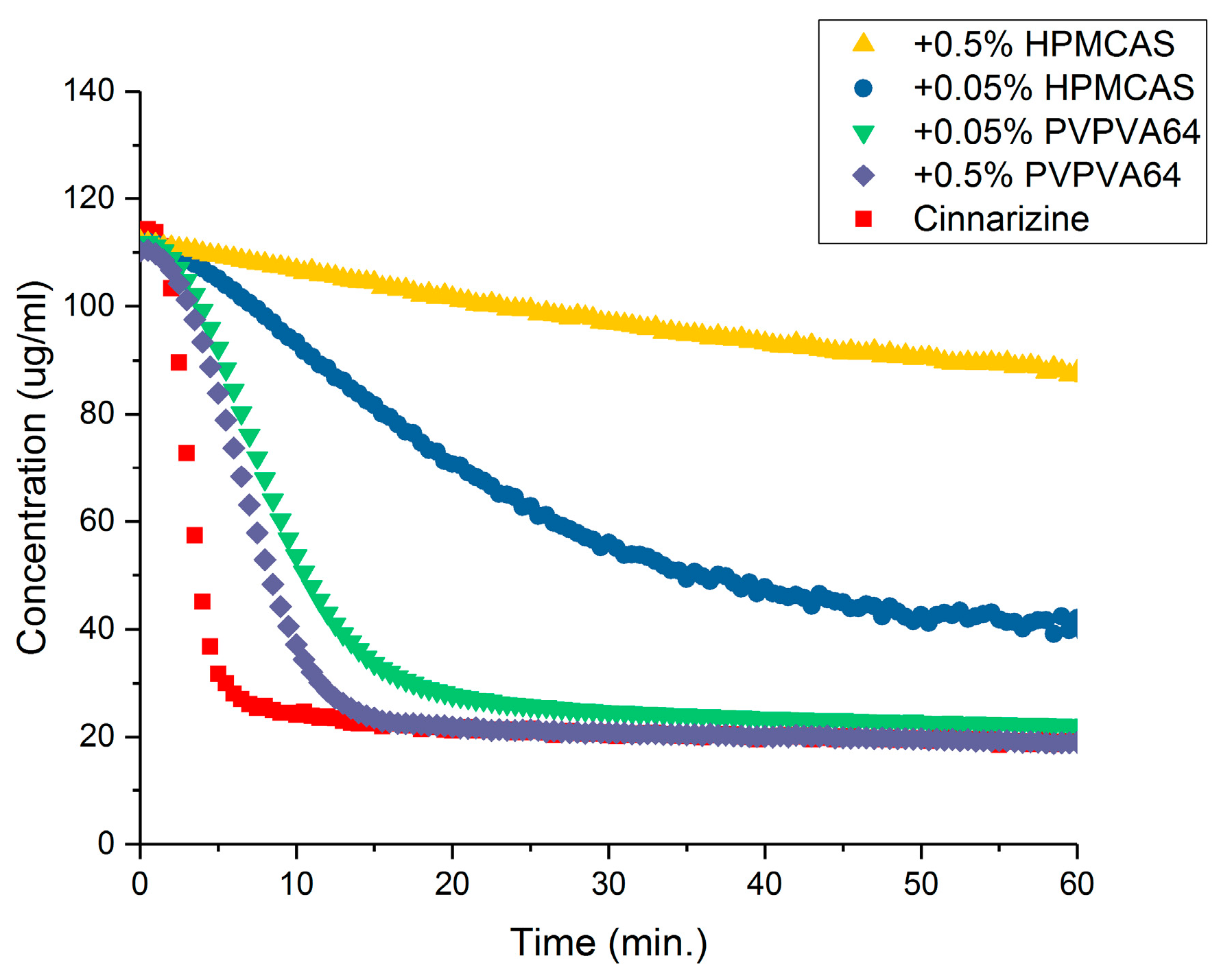
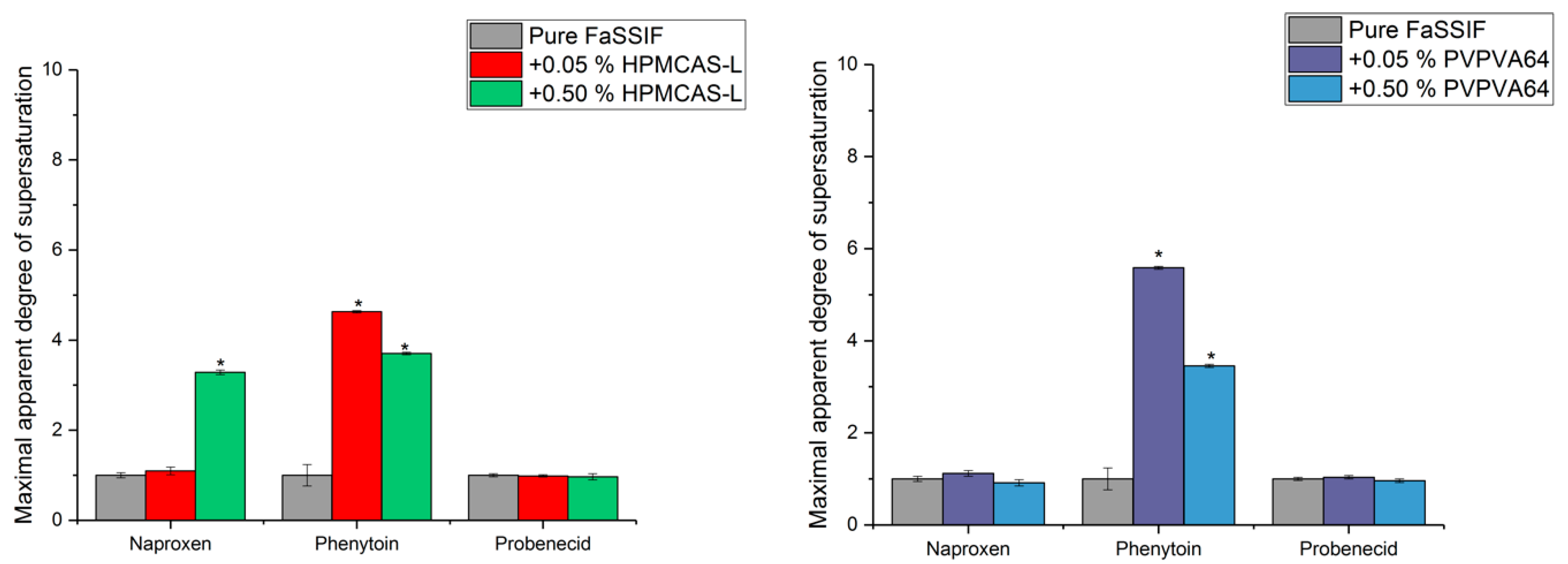
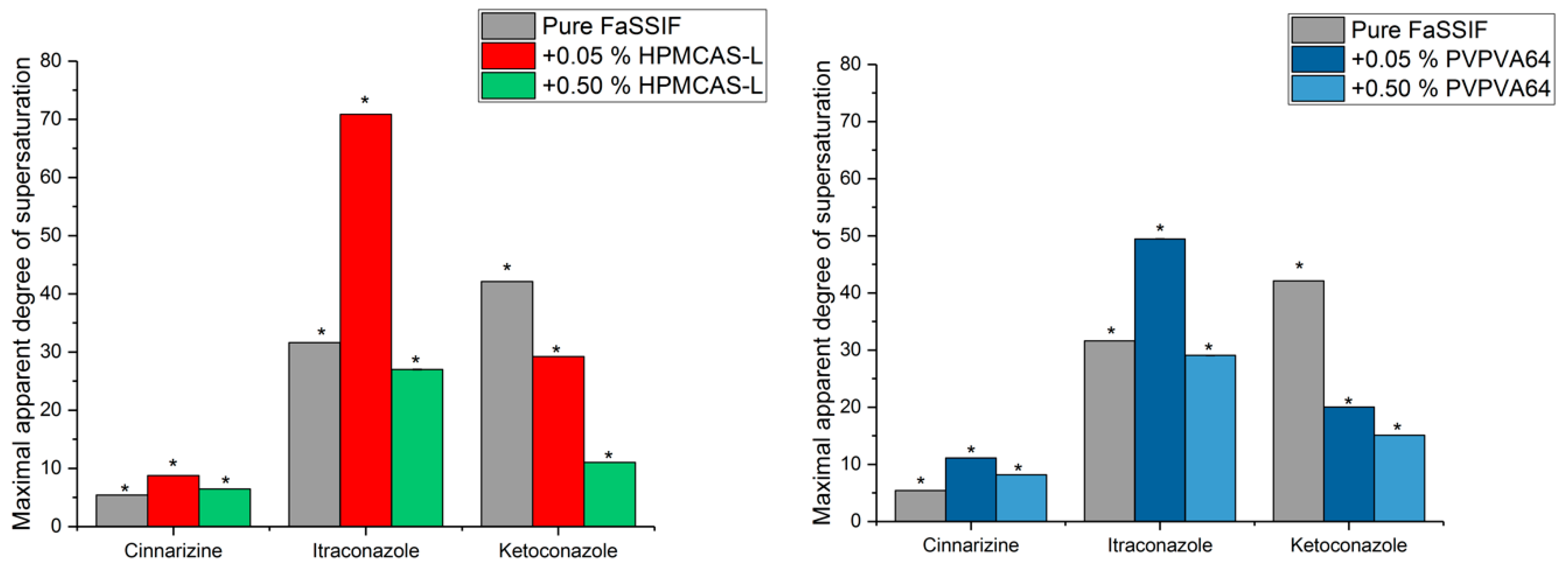
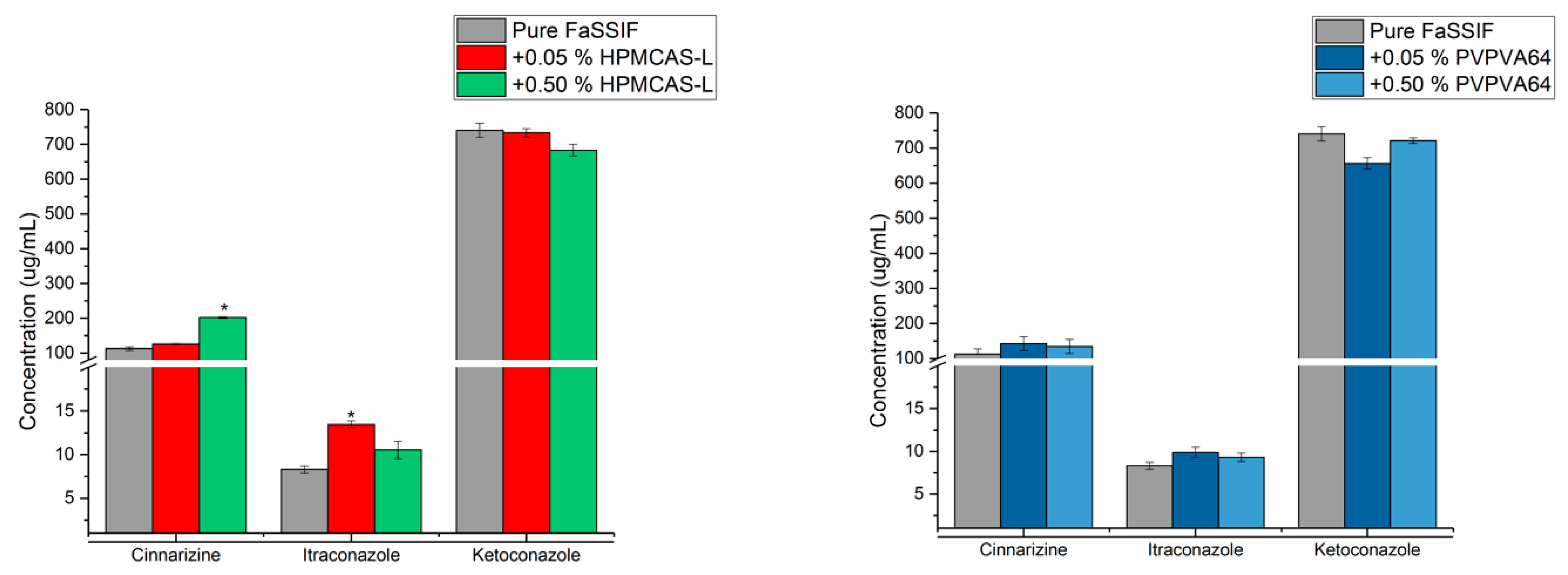
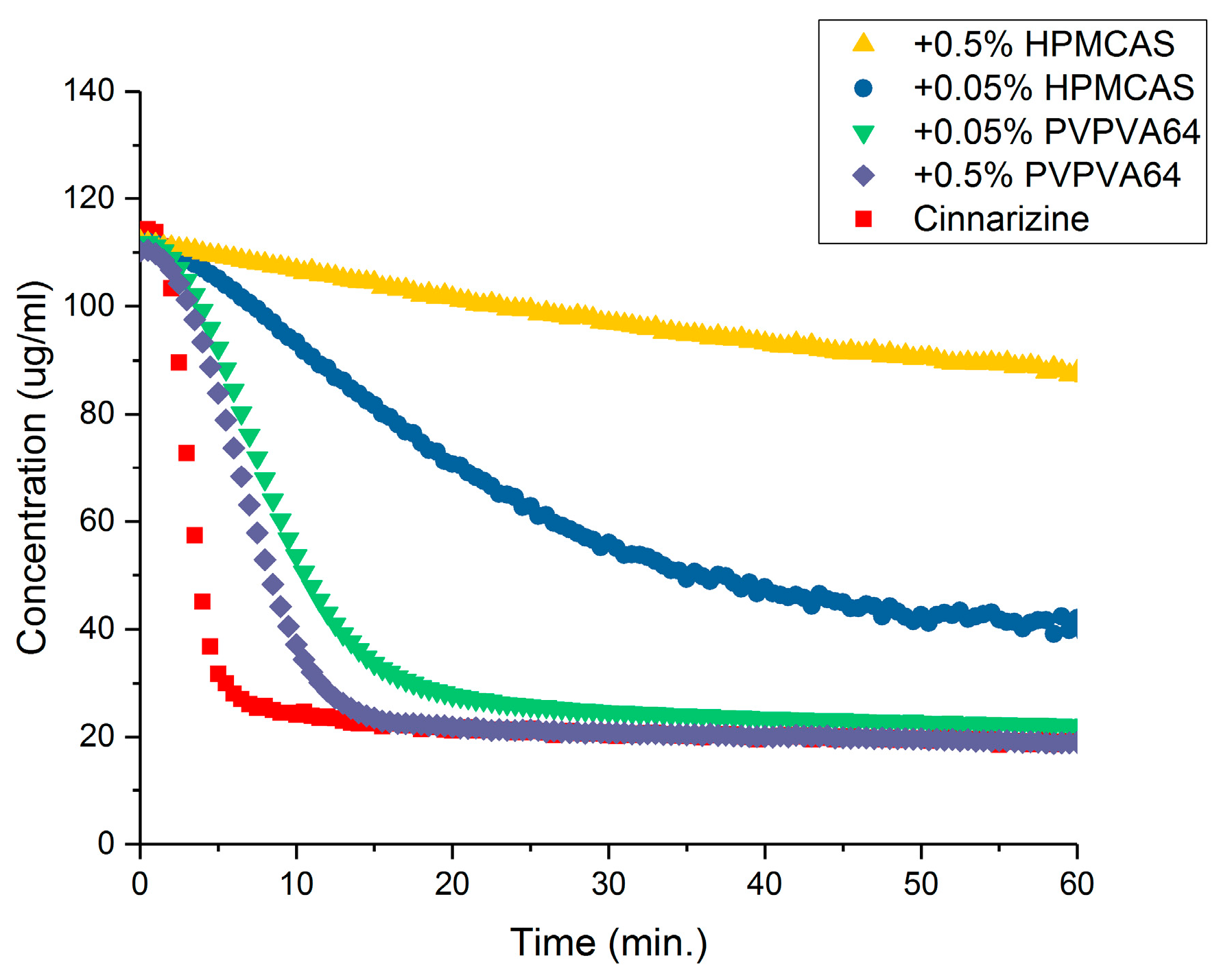
3.3. Determination of the Supersaturation Potential of Poor Glass Formers
3.4. Determination of the Supersaturation Potential of Good Glass Formers
3.5. Correlation between GFA and Supersaturation Potential of Drugs
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Applied Cooling Rates (K/min) |
|---|---|
| Naproxen | 750, 20 |
| Phenytoin | 750, 20 |
| Probenecid | 750, 20 |
| Cinnarizine | 750, 20, 5, 1, 0.5 |
| Itraconazole | 750, 20, 5, 1, 0.5 |
| Ketoconazole | 750, 20, 5, 1, 0.5 |
Appendix B
| Drug | Probe Tip Length (mm) |
|---|---|
| Naproxen | 1 |
| +0.05% HPMCAS-L | 1 |
| +0.50% HPMCAS-L | 1 |
| +0.05% PVPVA64 | 1 |
| +0.50% PVPVA64 | 1 |
| Phenytoin | 1 |
| +0.05% HPMCAS-L | 1 |
| +0.50% HPMCAS-L | 1 |
| +0.05% PVPVA64 | 1 |
| +0.50% PVPVA64 | 1 |
| Probenecid | 1 |
| +0.05% HPMCAS-L | 1 |
| +0.50% HPMCAS-L | 1 |
| +0.05% PVPVA64 | 1 |
| +0.50% PVPVA64 | 1 |
| Cinnarizine | 1 |
| +0.05% HPMCAS-L | 1 |
| +0.50% HPMCAS-L | 1 |
| +0.05% PVPVA64 | 1 |
| +0.50% PVPVA64 | 1 |
| Itraconazole | 5 |
| +0.05% HPMCAS-L | 5 |
| +0.50% HPMCAS-L | 5 |
| +0.05% PVPVA64 | 5 |
| +0.50% PVPVA64 | 5 |
| Ketoconazole | 2 |
| +0.05% HPMCAS-L | 2 |
| +0.50% HPMCAS-L | 2 |
| +0.05% PVPVA64 | 2 |
| +0.50% PVPVA64 | 2 |
Appendix C
| Medium | Naproxen (ug/mL) | Phenytoin (μg/mL) | Probenecid (μg/mL) | Cinnarizine (μg/mL) | Itraconazole (μg/mL) | Ketoconazole (μg/mL) |
|---|---|---|---|---|---|---|
| FaSSIF | 2412.0 ± 96.5 | 4.7 ± 0.9 | 2028.3 ± 79.3 | 20.7 ± 1.4 | 0.5 ± 0.1 | 17.6 ± 2.6 |
| pH (6.0) | pH (6.4) | pH (6.7) | pH (6.4) | pH (6.4) | pH (6.5) | |
| +0.05% HPMCAS-L | 768.5 ± 14.1 | 26.1 ± 1.6 | 2603.4 ± 126.6 | 14.4 ± 0.1 | 0.2 ± 0.1 | 25.1 ± 2.3 |
| pH (5.7) | pH (6.4) | pH (5.5) | pH (6.4) | pH (6.4) | pH (6.3) | |
| +0.50% HPMCAS-L | 1488.3 ± 87.1 | 30.2 ± 3.0 | 2499.0 ± 96.1 | 31.3 ± 1.0 | 0.4 ± 0.1 | 62.0 ± 3.9 |
| pH (5.1) | pH (6.4) | pH (5.2) | pH (5.8) | pH (5.7) | pH (5.6) | |
| +0.05% PVPVA64 | 1970.6 ± 81.6 | 21.0 ± 1.4 | 2440.1 ± 202.7 | 12.8 ± 0.5 | 0.2 ± 0.1 | 32.8 ± 2.2 |
| pH (6.0) | pH (6.4) | pH (6.0) | pH (6.4) | pH (6.4) | pH (6.4) | |
| +0.50% PVPVA64 | 2093.2 ± 64.8 | 30.1 ± 1.6 | 2299.0 ± 92.1 | 16.5 ± 0.6 | 0.3 ± 0. | 47.8 ± 2.0 |
| pH (5.8) | pH (6.4) | pH (5.8) | pH (6.4) | pH (6.4) | pH (6.4) |
References
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.A.; DiNunzio, J.C.; Yang, W.; McGinity, J.W.; Williams, R.O., III. Enhanced in vivo absorption of itraconazole via stabilization of supersaturation following acidic-to-neutral pH transition. Drug Dev. Ind. Pharm. 2008, 34, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.A.; Van Eerdenbrugh, B.; Taylor, L.S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef] [PubMed]
- Blaabjerg, L.I.; Lindenberg, E.; Löbmann, K.; Grohganz, H.; Rades, T. Glass Forming Ability of Amorphous Drugs Investigated by Continuous Cooling and Isothermal Transformation. Mol. Pharm. 2016, 13, 3318–3325. [Google Scholar] [CrossRef] [PubMed]
- Blaabjerg, L.I.; Lindenberg, E.; Rades, T.; Grohganz, H.; Löbmann, K. Influence of preparation pathway on the glass forming ability. Int. J. Pharm. 2017, 521, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Guzman, H.R.; Tawa, M.; Zhang, Z.; Ratanabanangkoon, P.; Shaw, P.; Gardner, C.R.; Chen, H.; Moreau, J.P.; Almarsson, O.; Remenar, J.F. Combined use of crystalline salt forms and precipitation inhibitors to improve oral absorption of celecoxib from solid oral formulations. J. Pharm. Sci. 2007, 96, 2686–2702. [Google Scholar] [CrossRef] [PubMed]
- Lindfors, L.; Forssén, S.; Westergren, J.; Olsson, U. Nucleation and crystal growth in supersaturated solutions of a model drug. J. Colloid Interface Sci. 2008, 325, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Bevernage, J.; Brouwers, J.; Annaert, P.; Augustijns, P. Drug precipitation–permeation interplay: Supersaturation in an absorptive environment. Eur. J. Pharm. Biopharm. 2012, 82, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Mullin, J.W.; Söhnel, O. Expressions of supersaturation in crystallization studies. Chem. Eng. Sci. 1977, 32, 683–686. [Google Scholar] [CrossRef]
- Madsen, C.M.; Feng, K.-I.; Leithead, A.; Canfield, N.; Jørgensen, S.A.; Müllertz, A.; Rades, T. Effect of composition of simulated intestinal media on the solubility of poorly soluble compounds investigated by design of experiments. Eur. J. Pharm. Sci. 2018, 111, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Trasi, N.S.; Taylor, L.S. Effect of polymers on nucleation and crystal growth of amorphous acetaminophen. CrystEngComm 2012, 14, 5188–5197. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Maintaining Supersaturation in Aqueous Drug Solutions: Impact of Different Polymers on Induction Times. Cryst. Growth Des. 2013, 13, 740–751. [Google Scholar] [CrossRef]
- Trasi, N.S.; Abbou Oucherif, K.; Litster, J.D.; Taylor, L.S. Evaluating the influence of polymers on nucleation and growth in supersaturated solutions of acetaminophen. CrystEngComm 2015, 17, 1242–1248. [Google Scholar] [CrossRef]
- Loftsson, T.; Fri, H.; Gu, T.K. The effect of water-soluble polymers on aqueous solubility of drugs. Int. J. Pharm. 1996, 127, 293–296. [Google Scholar] [CrossRef]
- Laitinen, R.; Löbmann, K.; Grohganz, H.; Priemel, P.; Strachan, C.J.; Rades, T. Supersaturating drug delivery systems: The potential of co-amorphous drug formulations. Int. J. Pharm. 2017, 532, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, N.; Kuentz, M. Glass-forming ability of compounds in marketed amorphous drug products. Eur. J. Pharm. Biopharm. 2017, 112, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Usui, F.; Maeda, K.; Kusai, A.; Nishimura, K.; Keiji, Y. Inhibitory effects of water-soluble polymers on precipitation of RS-8359. Int. J. Pharm. 1997, 154, 59–66. [Google Scholar] [CrossRef]
- Christfort, J.F.; Plum, J.; Madsen, C.M.; Nielsen, L.H.; Sandau, M.; Andersen, K.; Müllertz, A.; Rades, T. Development of a Video-Microscopic Tool To Evaluate the Precipitation Kinetics of Poorly Water Soluble Drugs: A Case Study with Tadalafil and HPMC. Mol. Pharm. 2017, 14, 4154–4160. [Google Scholar] [CrossRef] [PubMed]
- Schiller, C.; Fröhlich, C.P.; Giessmann, T.; Siegmund, W.; Mönnikes, H.; Hosten, N.; Weitschies, W. Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Aliment. Pharmacol. Ther. 2005, 22, 971–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Eerdenbrugh, B.; Raina, S.; Hsieh, Y.-L.; Augustijns, P.; Taylor, L.S. Classification of the crystallization behavior of amorphous active pharmaceutical ingredients in aqueous environments. Pharm. Res. 2014, 31, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Raina, S.A.; Eerdenbrugh, B.V.; Alonzo, D.E.; Mo, H.; Zhang, G.G.; Gao, Y.; Taylor, L.S. Trends in the Precipitation and Crystallization Behavior of Supersaturated Aqueous Solutions of Poorly Water-Soluble Drugs Assessed Using Synchrotron Radiation. J. Pharm. Sci. 2015, 104, 1981–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmelund, H.; Madsen, C.M.; Plum, J.; Müllertz, A.; Rades, T. Studying the Propensity of Compounds to Supersaturate: A Practical and Broadly Applicable Approach. J. Pharm. Sci. 2016, 105, 3021–3029. [Google Scholar] [CrossRef] [PubMed]
- Blaabjerg, L.I.; Lindenberg, E.; Löbmann, K.; Grohganz, H.; Rades, T. Is there a correlation between the glass forming ability of a drug and its supersaturation propensity? Int. J. Pharm. 2018, 538, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Van Speybroeck, M.; Mols, R.; Mellaerts, R.; Thi, T.D.; Martens, J.A.; Van Humbeeck, J.; Annaert, P.; Van den Mooter, G.; Augustijns, P. Combined use of ordered mesoporous silica and precipitation inhibitors for improved oral absorption of the poorly soluble weak base itraconazole. Eur. J. Pharm. Biopharm. 2010, 75, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Beyer, A.; Radi, L.; Grohganz, H.; Löbmann, K.; Rades, T.; Leopold, C.S. Preparation and recrystallization behavior of spray-dried co-amorphous naproxen–indomethacin. Eur. J. Pharm. Biopharm. 2016, 104 (Suppl. C), 72–81. [Google Scholar] [CrossRef] [PubMed]
- Doreth, M.; Löbmann, K.; Grohganz, H.; Holm, R.; De Diego, H.L.; Rades, T.; Priemel, P.A. Glass solution formation in water-In situ amorphization of naproxen and ibuprofen with Eudragit® E PO. J. Drug Deliv. Sci. Technol. 2016, 34, 32–40. [Google Scholar] [CrossRef]
- Jensen, K.; Löbmann, K.; Rades, T.; Grohganz, H. Improving Co-Amorphous Drug Formulations by the Addition of the Highly Water Soluble Amino Acid, Proline. Pharmaceutics 2014, 6, 416. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, T.; Ichikawa, T.; Sugawara, N.; Tatsuishi, K.; Kayano, M.; Machida, Y.; Hoshida, H.; Nagai, T. Kinetics of degradation of cinnarizine in aqueous solution. Chem. Pharm. Bull. 1985, 33, 2069–2072. [Google Scholar] [CrossRef] [PubMed]
- Box, K.; Comer, J.E.; Gravestock, T.; Stuart, M. New ideas about the solubility of drugs. Chem. Biodivers. 2009, 6, 1767–1788. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, N.; Ueda, K.; Ohyagi, N.; Shimizu, K.; Katakawa, K.; Kumamoto, T.; Higashi, K.; Yamamoto, K.; Moribe, K. An insight into different stabilization mechanisms of phenytoin derivatives supersaturation by HPMC and PVP. J. Pharm. Sci. 2015, 104, 2574–2582. [Google Scholar] [CrossRef] [PubMed]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Inhibition of solution crystal growth of ritonavir by cellulose polymers–factors influencing polymer effectiveness. CrystEngComm 2012, 14, 6503–6514. [Google Scholar] [CrossRef]

| Drug | Tm (K) a | Tg (K) a | GFA Class | Critical Cooling Rate (K/min) | pKa (Acidic/Basic) |
|---|---|---|---|---|---|
| Naproxen | 429 | 278 b | 1 | >750 | 4.2/- |
| Phenytoin | 569 | - | 1 | >750 | 8.0/- |
| Probenecid | 468 | - | 1 | >750 | 3.3/- |
| Cinnarizine | 393 | 288 | 3 | 1 | -/2.0 c, 7.6 |
| Itraconazole | 439 | 332 | 3 | 1 | -/4.0 |
| Ketoconazole | 419 | 319 | 3 | 1 | -/3.3, 6.2 |
| Drug | Solubility in FaSSIF (mM) | Solubility in pH Adjusted FaSSIF (mM) | Solubility in FaSSIF + 0.5% (w/v) HPMCAS-L (mM) |
|---|---|---|---|
| Naproxen | 10.5 ± 0.4 | 6.1 ± 0.3 (pH 6) | 6.5 ± 0.4 (pH 6) |
| Phenytoin | 0.02 ± 0.004 | 0.13 ± 0.002 (pH 6) | 0.12 ± 0.012 (pH 6) |
| Probenecid | 7.1 ± 0.3 | 8.0 ± 0.2 (pH 6) | 8.6 ± 0.3 (pH 6) |
| Cinnarizine | 0.06 ± 0.004 | 0.02 ± 0.001 (pH 7) | 0.08 ± 0.003 (pH 7) |
| Ketoconazole | 0.03 ± 0.005 | 0.084 ± 0.006 (pH 6) | 0.117 ± 0.007 (pH 6) |
| Itraconazole | 0.001 ± 0.0001 | 0.001 ± 0.0002 (pH 6) | 0.0006 ± 0.0001 (pH 6) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blaabjerg, L.I.; Grohganz, H.; Lindenberg, E.; Löbmann, K.; Müllertz, A.; Rades, T. The Influence of Polymers on the Supersaturation Potential of Poor and Good Glass Formers. Pharmaceutics 2018, 10, 164. https://doi.org/10.3390/pharmaceutics10040164
Blaabjerg LI, Grohganz H, Lindenberg E, Löbmann K, Müllertz A, Rades T. The Influence of Polymers on the Supersaturation Potential of Poor and Good Glass Formers. Pharmaceutics. 2018; 10(4):164. https://doi.org/10.3390/pharmaceutics10040164
Chicago/Turabian StyleBlaabjerg, Lasse I., Holger Grohganz, Eleanor Lindenberg, Korbinian Löbmann, Anette Müllertz, and Thomas Rades. 2018. "The Influence of Polymers on the Supersaturation Potential of Poor and Good Glass Formers" Pharmaceutics 10, no. 4: 164. https://doi.org/10.3390/pharmaceutics10040164