Comparison of Target Features for Predicting Drug-Target Interactions by Deep Neural Network Based on Large-Scale Drug-Induced Transcriptome Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
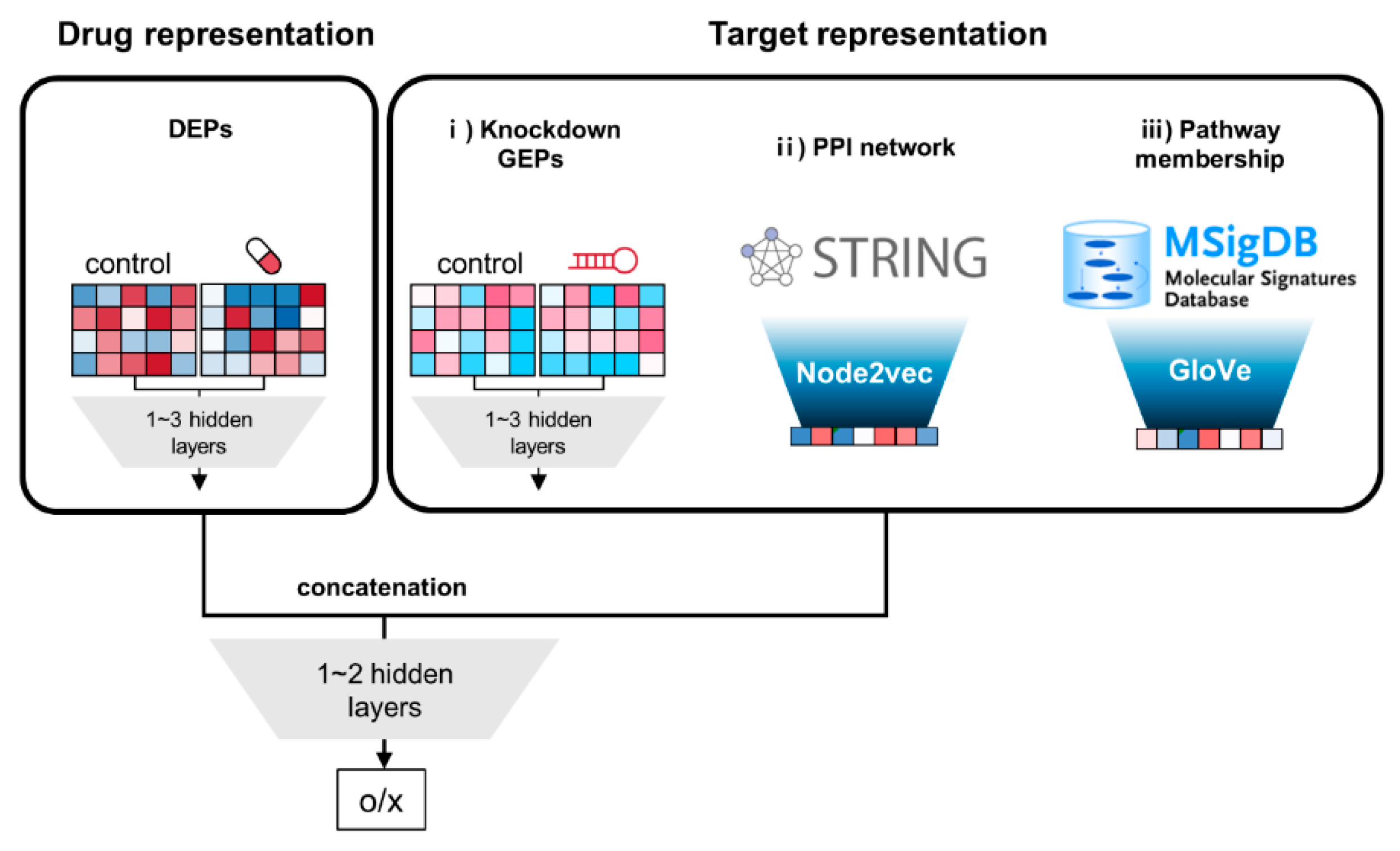
2.1. Extraction of Drug Features
2.2. Extraction of Target Features
2.2.1. Expression Profiles by Gene Knockdown (GEPs)-Based Target Features
2.2.2. Target Features by Protein–Protein Interaction (PPI) Network
2.2.3. Pathway Membership (PM)-Based Target Features
2.3. Construction of Deep Neural Networks (DNNs) and Machine Learning Models
3. Results
3.1. Extraction of Drug and Target Features
3.2. Overall Architecture of DNNs
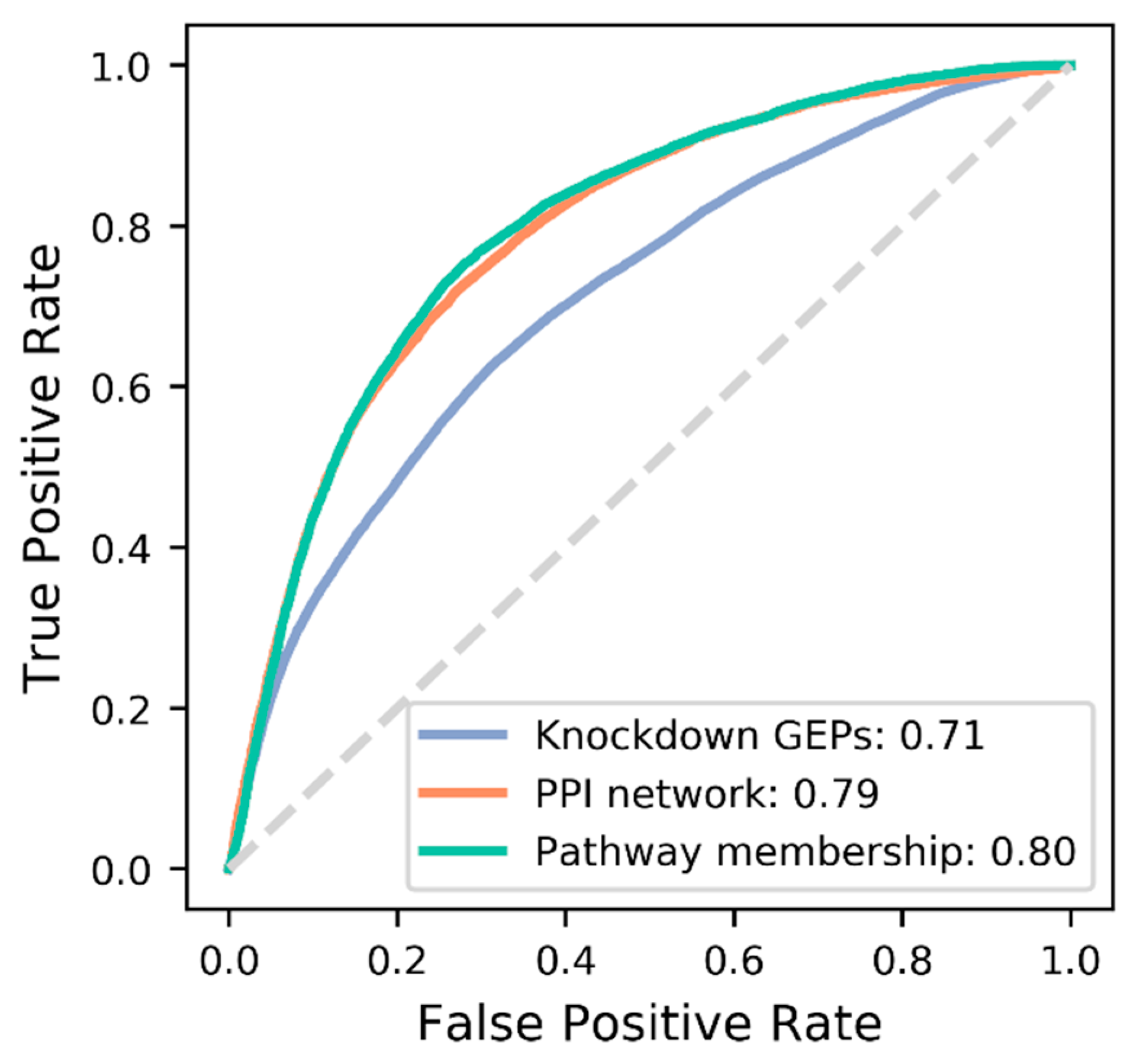
3.3. Comparison of Target Features
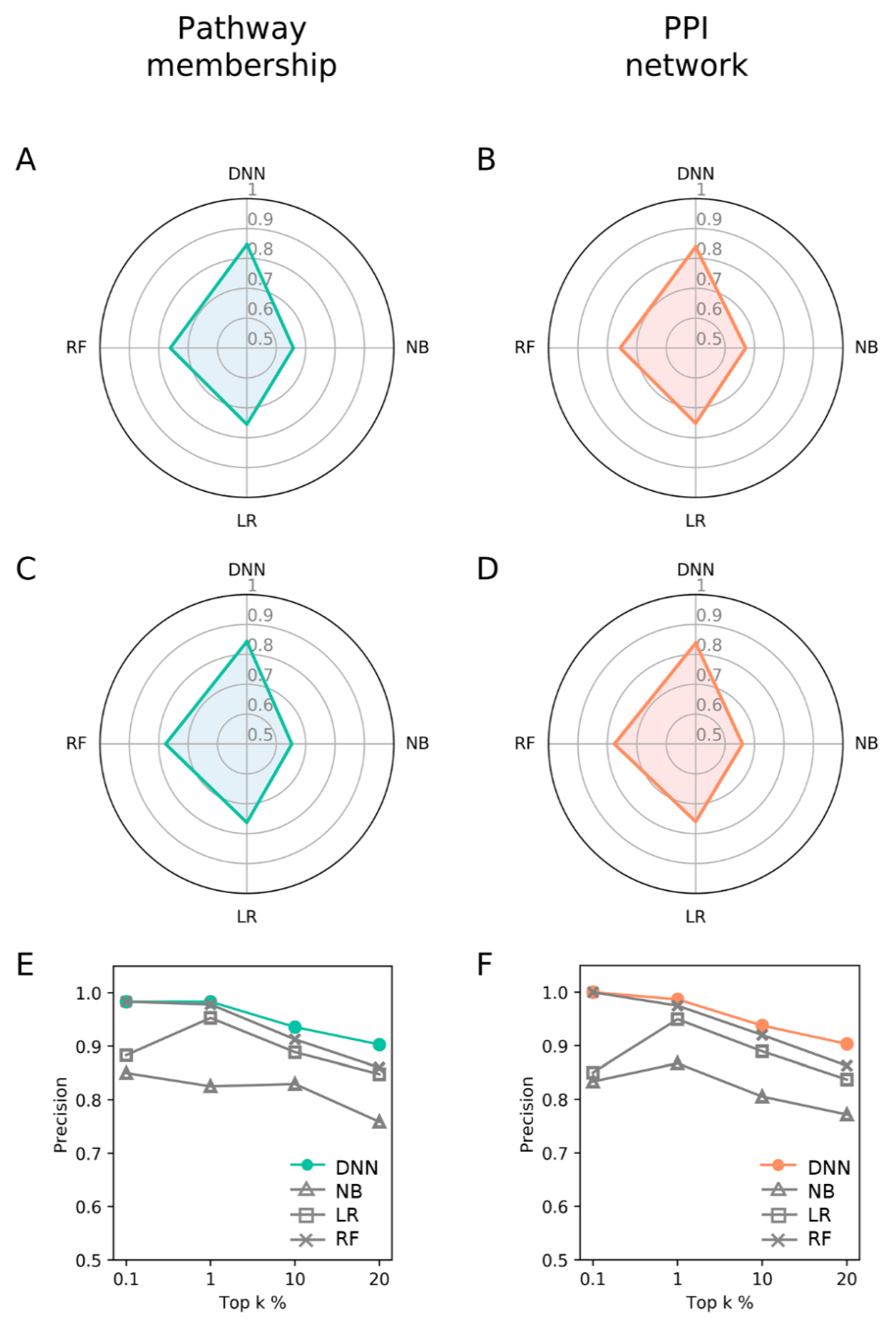
3.4. Comparison with DNN and Other Machine Learning Methods
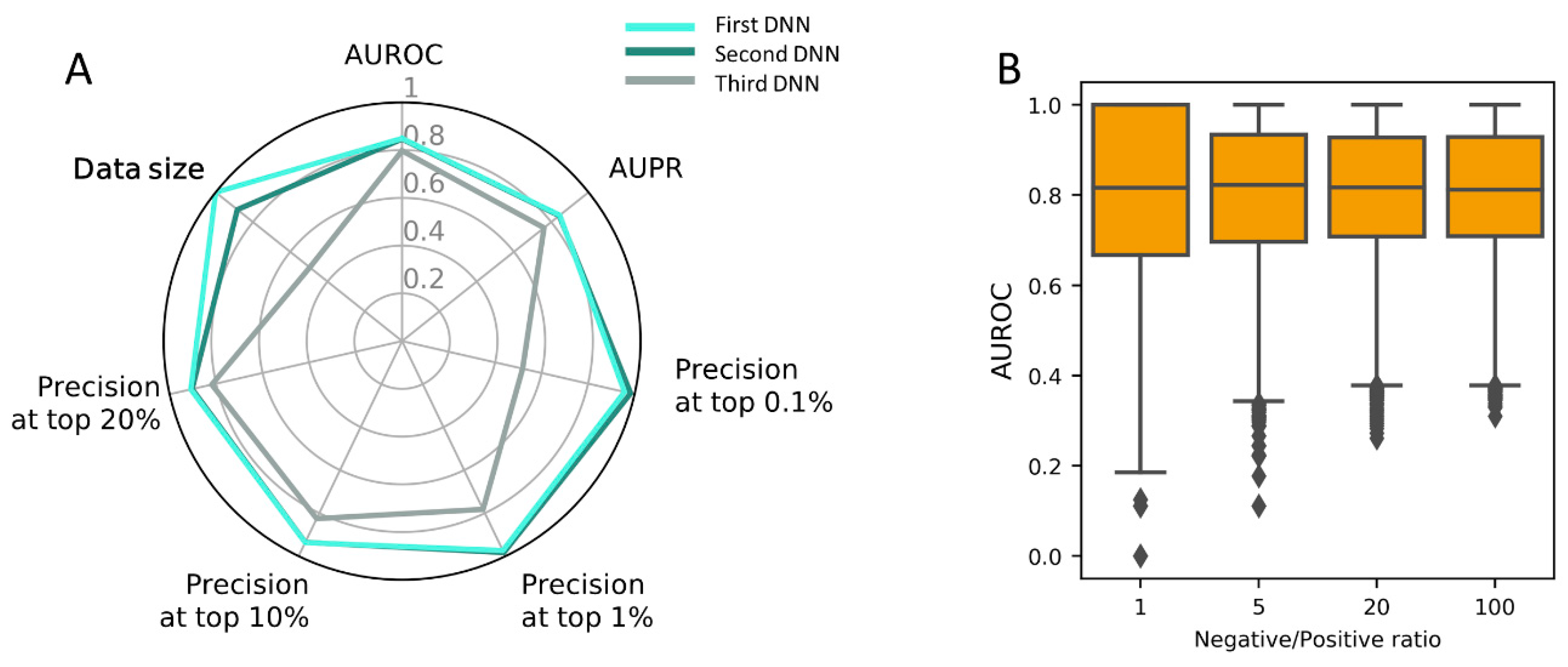
3.5. Construction of PM-Based DNN Model Using the Full Dataset
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Senior, A.; Jumper, J.; Hassabis, D. AlphaFold: Using AI for Scientific Discovery. Available online: https://deepmind.com/blog/alphafold/ (accessed on 15 July 2019).
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Do, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, C.C.; Zhang, X.; Zhang, X.; Dai, F.; Yin, J.; Zhang, Y. Drug–target interaction prediction: Databases, web servers and computational models. Brief. Bioinform. 2016, 17, 696–712. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Takigawa, I.; Mamitsuka, H.; Zhu, S. Similarity-based machine learning methods for predicting drug–target interactions: A brief review. Brief. Bioinform. 2014, 15, 734–747. [Google Scholar] [CrossRef] [PubMed]
- Ripphausen, P.; Nisius, B.; Bajorath, J. State-of-the-art in ligand-based virtual screening. Drug Discov. Today 2011, 16, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Griffith, R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013, 10, e395–e401. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, A.; Wu, M.; Li, X.-L.; Kwoh, C.-K. Computational prediction of drug–target interactions using chemogenomic approaches: An empirical survey. Brief. Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Liu, X.; Jin, S.; Lin, J.; Liu, J. Machine Learning for Drug-Target Interaction Prediction. Molecules 2018, 23, 2208. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Musa, A.; Ghoraie, L.S.; Zhang, S.-D.; Glazko, G.; Yli-Harja, O.; Dehmer, M.; Haibe-Kains, B.; Emmert-Streib, F. A review of connectivity map and computational approaches in pharmacogenomics. Brief. Bioinform. 2018, 19, 506–523. [Google Scholar] [PubMed]
- Iorio, F.; Bosotti, R.; Scacheri, E.; Belcastro, V.; Mithbaokar, P.; Ferriero, R.; Murino, L.; Tagliaferri, R.; Brunetti-Pierri, N.; Isacchi, A.; et al. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA 2010, 107, 14621–14626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Sun, J.; Zhou, S.; Wan, C.; Qin, S.; Li, C.; He, L.; Yang, L. Prediction of drug-target interactions for drug repositioning only based on genomic expression similarity. PLoS Comput. Biol. 2013, 9, e1003315. [Google Scholar] [CrossRef]
- Hizukuri, Y.; Sawada, R.; Yamanishi, Y. Predicting target proteins for drug candidate compounds based on drug-induced gene expression data in a chemical structure-independent manner. BMC Med. Genom. 2015, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Isik, Z.; Baldow, C.; Cannistraci, C.V.; Schroeder, M. Drug target prioritization by perturbed gene expression and network information. Sci. Rep. 2015, 5, 17417. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef] [PubMed]
- Van Noort, V.; Scholch, S.; Iskar, M.; Zeller, G.; Ostertag, K.; Schweitzer, C.; Werner, K.; Weitz, J.; Koch, M.; Bork, P. Novel Drug Candidates for the Treatment of Metastatic Colorectal Cancer through Global Inverse Gene-Expression Profiling. Cancer Res. 2014, 74, 5690–5699. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-D.; Guan, Y.; Burmeister, M.; Zhang, H.; Wall, D.P.; Duda, M. Brain-specific functional relationship networks inform autism spectrum disorder gene prediction. Transl. Psychiatry 2018, 8, 56. [Google Scholar]
- Brum, A.M.; van de Peppel, J.; van der Leije, C.S.; Schreuders-Koedam, M.; Eijken, M.; van der Eerden, B.C.J.; van Leeuwen, J.P.T.M. Connectivity Map-based discovery of parbendazole reveals targetable human osteogenic pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 12711–12716. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Kang, S.; Kim, W. Drug Repositioning for Cancer Therapy Based on Large-Scale Drug-Induced Transcriptional Signatures. PLoS ONE 2016, 11, e0150460. [Google Scholar] [CrossRef]
- Iwata, M.; Sawada, R.; Iwata, H.; Kotera, M.; Yamanishi, Y. Elucidating the modes of action for bioactive compounds in a cell-specific manner by large-scale chemically-induced transcriptomics. Sci. Rep. 2017, 7, 40164. [Google Scholar] [CrossRef]
- Xie, L.; He, S.; Song, X.; Bo, X.; Zhang, Z. Deep learning-based transcriptome data classification for drug-target interaction prediction. BMC Genom. 2018, 19, 667. [Google Scholar] [CrossRef]
- Grover, A.; Leskovec, J. Node2vec; Association for Computing Machinery (ACM): New York, NY, USA, 2016; pp. 855–864. [Google Scholar]
- Pennington, J.; Socher, R.; Manning, C. Glove: Global Vectors for Word Representation. In Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP), Doha, Qatar, 25–29 October 2014; Association for Computational Linguistics: Stroudsburg, PA, USA, 2014; pp. 1532–1543. [Google Scholar]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Distributed Systems. arXiv 2016, arXiv:1603.04467. [Google Scholar]
- Chollet, F. Keras. 2015. Available online: https://github.com/fchollet/keras (accessed on Day Month Year).
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Salakhutdinov, R. Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Peng, J.; Guan, J.; Shang, X. Predicting Parkinson’s Disease Genes Based on Node2vec and Autoencoder. Front. Genet. 2019, 10, 226. [Google Scholar] [CrossRef]
- Smaili, F.Z.; Gao, X.; Hoehndorf, R. Onto2Vec: Joint vector-based representation of biological entities and their ontology-based annotations. Bioinformatics 2018, 34, i52–i60. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Chen, L.; Chu, C.; Lu, J.; Kong, X.; Huang, T.; Cai, Y.-D. Gene Ontology and KEGG Pathway Enrichment Analysis of a Drug Target-Based Classification System. PLoS ONE 2015, 10, e0126492. [Google Scholar] [CrossRef]
- Du, J.; Jia, P.; Dai, Y.; Tao, C.; Zhao, Z.; Zhi, D. Gene2vec: Distributed representation of genes based on co-expression. BMC Genom. 2019, 20, 82. [Google Scholar] [CrossRef]
- Chen, X.; Ji, Z.L.; Chen, Y.Z. TTD: Therapeutic Target Database. Nucleic Acids Res. 2002, 30, 412–415. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL web services: Streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef]
- Gunther, S.; Kuhn, M.; Dunkel, M.; Campillos, M.; Senger, C.; Petsalaki, E.; Ahmed, J.; Urdiales, E.G.; Gewiess, A.; Jensen, L.J.; et al. SuperTarget and Matador: Resources for exploring drug-target relationships. Nucleic Acids Res. 2007, 36, D919–D922. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Sharman, J.L.; Benson, H.E.; Pawson, A.J.; Lukito, V.; Mpamhanga, C.P.; Bombail, V.; Davenport, A.P.; Peters, J.A.; Spedding, M.; Harmar, A.J.; et al. IUPHAR-DB: Updated database content and new features. Nucleic Acids Res. 2013, 41, D1083–D1088. [Google Scholar] [CrossRef]
- Thorn, C.F.; Klein, T.E.; Altman, R.B. PharmGKB: The Pharmacogenomics Knowledge Base. Methods Mol. Biol. 2013, 1015, 311–320. [Google Scholar] [Green Version]
- Roth, B.L.; Lopez, E.; Patel, S.; Kroeze, W.K. The Multiplicity of Serotonin Receptors: Uselessly Diverse Mol ecules or an Embarrassment of Riches? Neuroscientist 2000, 6, 252–262. [Google Scholar] [CrossRef]
- Ahmed, A.; Smith, R.D.; Clark, J.J.; Dunbar, J.B.; Carlson, H.A. Recent improvements to Binding MOAD: A resource for protein–ligand binding affinities and structures. Nucleic Acids Res. 2015, 43, D465–D469. [Google Scholar] [CrossRef]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef]
- Davis, A.P.; Murphy, C.G.; Johnson, R.; Lay, J.M.; Lennon-Hopkins, K.; Saraceni-Richards, C.; Sciaky, D.; King, B.L.; Rosenstein, M.C.; Wiegers, T.C.; et al. The Comparative Toxicogenomics Database: Update 2013. Nucleic Acids Res. 2013, 41, D1104–D1114. [Google Scholar] [CrossRef]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef]
- Webb, S. Deep learning for biology. Nature 2018, 554, 555–557. [Google Scholar] [CrossRef] [Green Version]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep drug-target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef]
- Feng, Q.; Dueva, E.; Cherkasov, A.; Ester, M. PADME: A Deep Learning-based Framework for Drug-Target Interaction Prediction. arXiv 2018, arXiv:1807.09741. [Google Scholar]
- Wen, M.; Zhang, Z.; Niu, S.; Sha, H.; Yang, R.; Yun, Y.; Lu, H. Deep-Learning-Based Drug−Target Interaction Prediction. J. Proteome Res. 2017, 16, 1401–1409. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.; Kim, W. Comparison of Target Features for Predicting Drug-Target Interactions by Deep Neural Network Based on Large-Scale Drug-Induced Transcriptome Data. Pharmaceutics 2019, 11, 377. https://doi.org/10.3390/pharmaceutics11080377
Lee H, Kim W. Comparison of Target Features for Predicting Drug-Target Interactions by Deep Neural Network Based on Large-Scale Drug-Induced Transcriptome Data. Pharmaceutics. 2019; 11(8):377. https://doi.org/10.3390/pharmaceutics11080377
Chicago/Turabian StyleLee, Hanbi, and Wankyu Kim. 2019. "Comparison of Target Features for Predicting Drug-Target Interactions by Deep Neural Network Based on Large-Scale Drug-Induced Transcriptome Data" Pharmaceutics 11, no. 8: 377. https://doi.org/10.3390/pharmaceutics11080377
APA StyleLee, H., & Kim, W. (2019). Comparison of Target Features for Predicting Drug-Target Interactions by Deep Neural Network Based on Large-Scale Drug-Induced Transcriptome Data. Pharmaceutics, 11(8), 377. https://doi.org/10.3390/pharmaceutics11080377