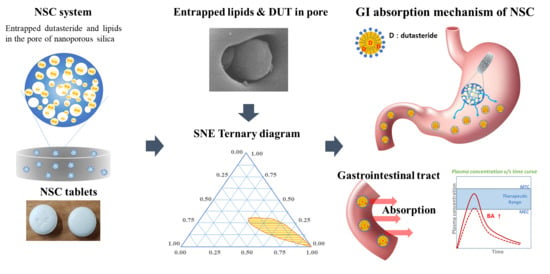
Nanoporous Silica Entrapped Lipid-Drug Complexes for the Solubilization and Absorption Enhancement of Poorly Soluble Drugs


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
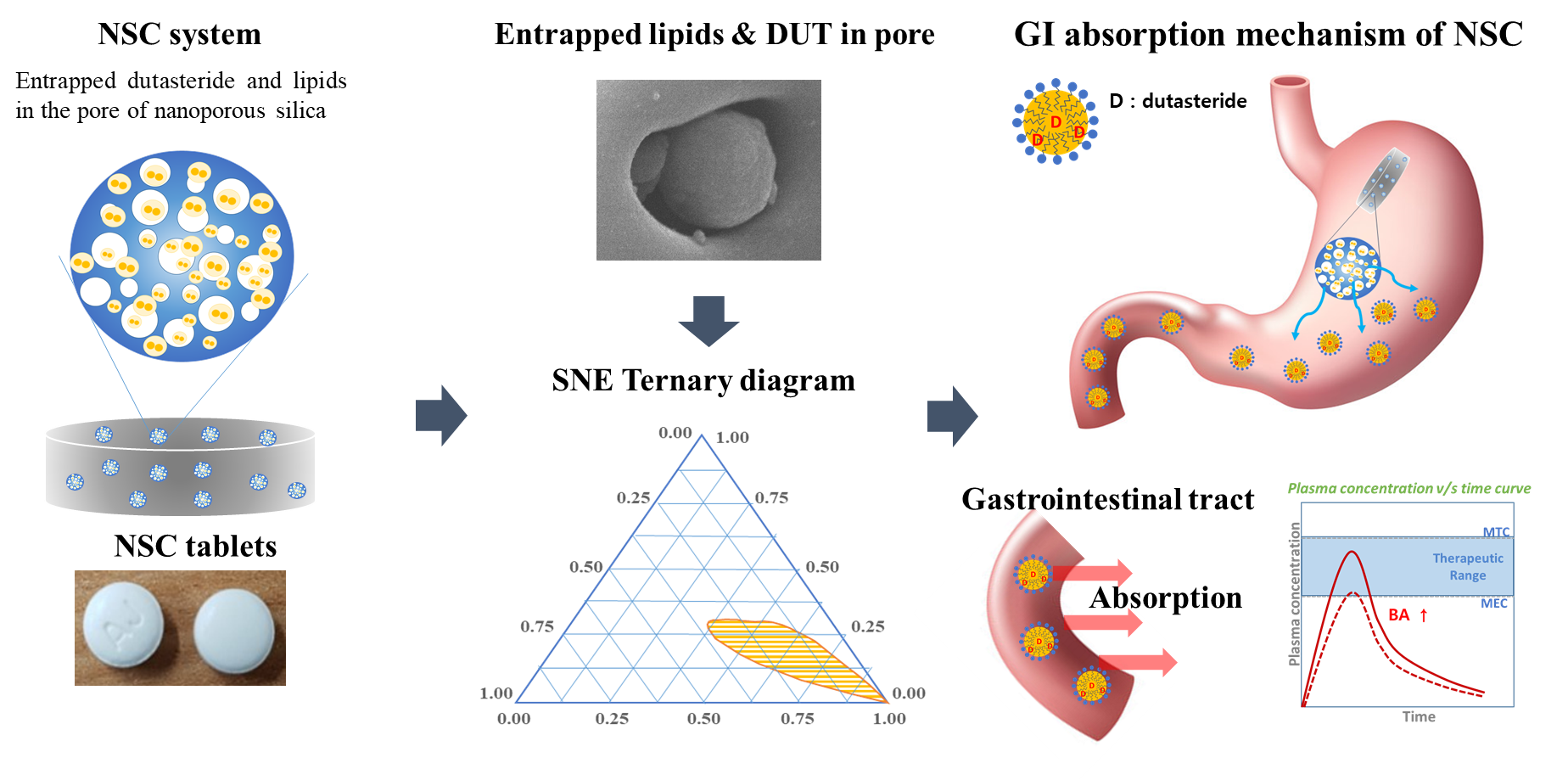
2.2. Ternary Phase Diagram
2.3. Preparation of NSCs
2.4. Preparation of NSC Tablets
2.5. Physicochemical Properties of NSC
2.5.1. Scanning Electron Microscopy
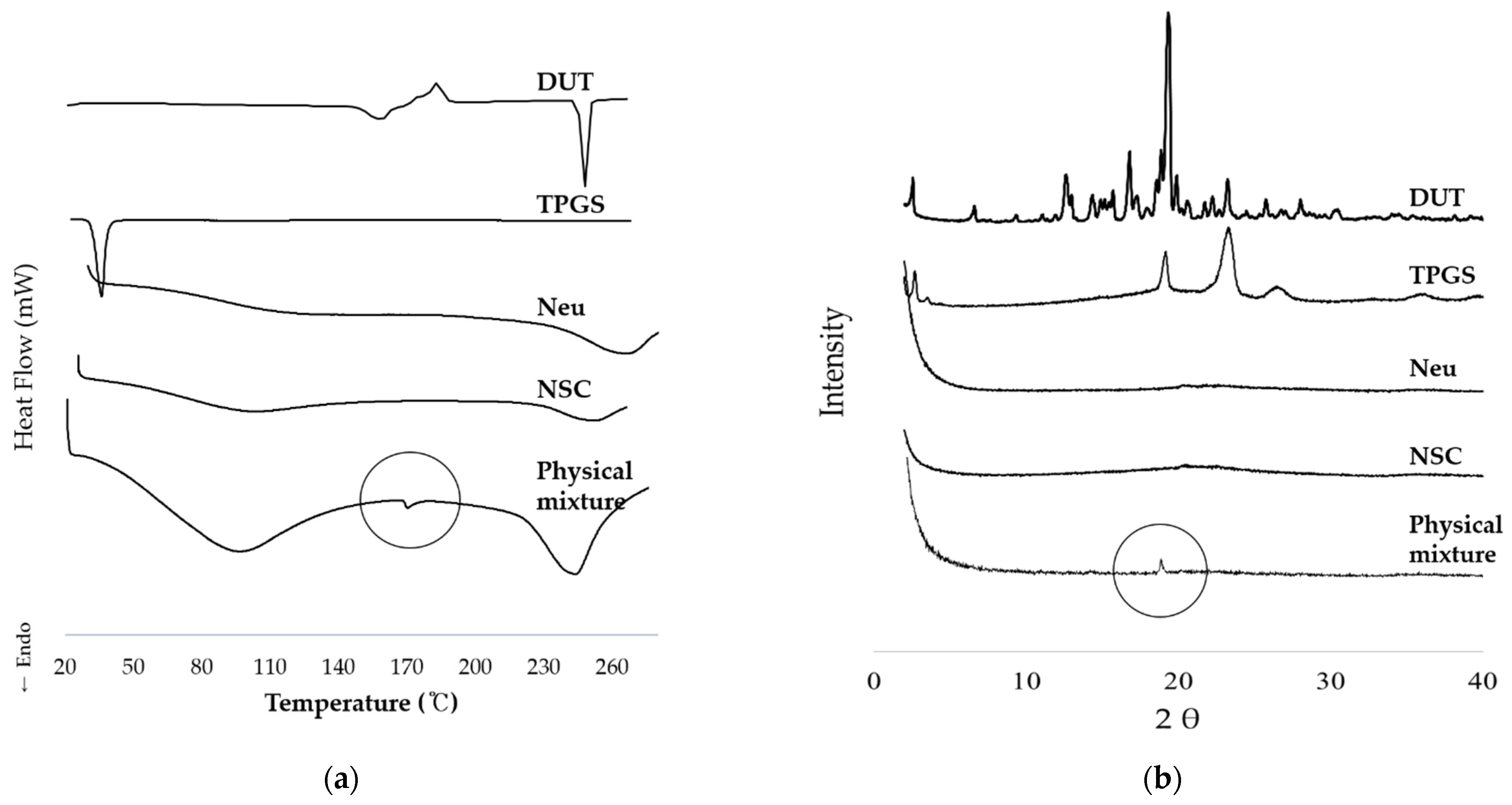
2.5.2. Differential Scanning Calorimetry
2.5.3. Powder-X-ray Diffraction
2.5.4. Flowability
2.5.5. Loading Capacity
2.5.6. Content Homogeneity
2.5.7. Equilibrium Solubility of DUT in NSC
2.6. In Vitro Release of NSC Tablets
2.6.1. In Vitro Release
2.6.2. HPLC Analysis
2.7. Pharmacokinetics Studies
2.8. Statistical Analysis
3. Results and Discussion
3.1. Ternary Phase Diagram
3.2. Physicochemical Properties of NSC
3.2.1. Morphology
3.2.2. Thermodynamic Analysis and Crystallinity
3.2.3. Flowability
3.2.4. Loading Capacity
3.3. Characterization of NSC and NSC Tablet
3.3.1. Drug Content Homogeneity in NSC
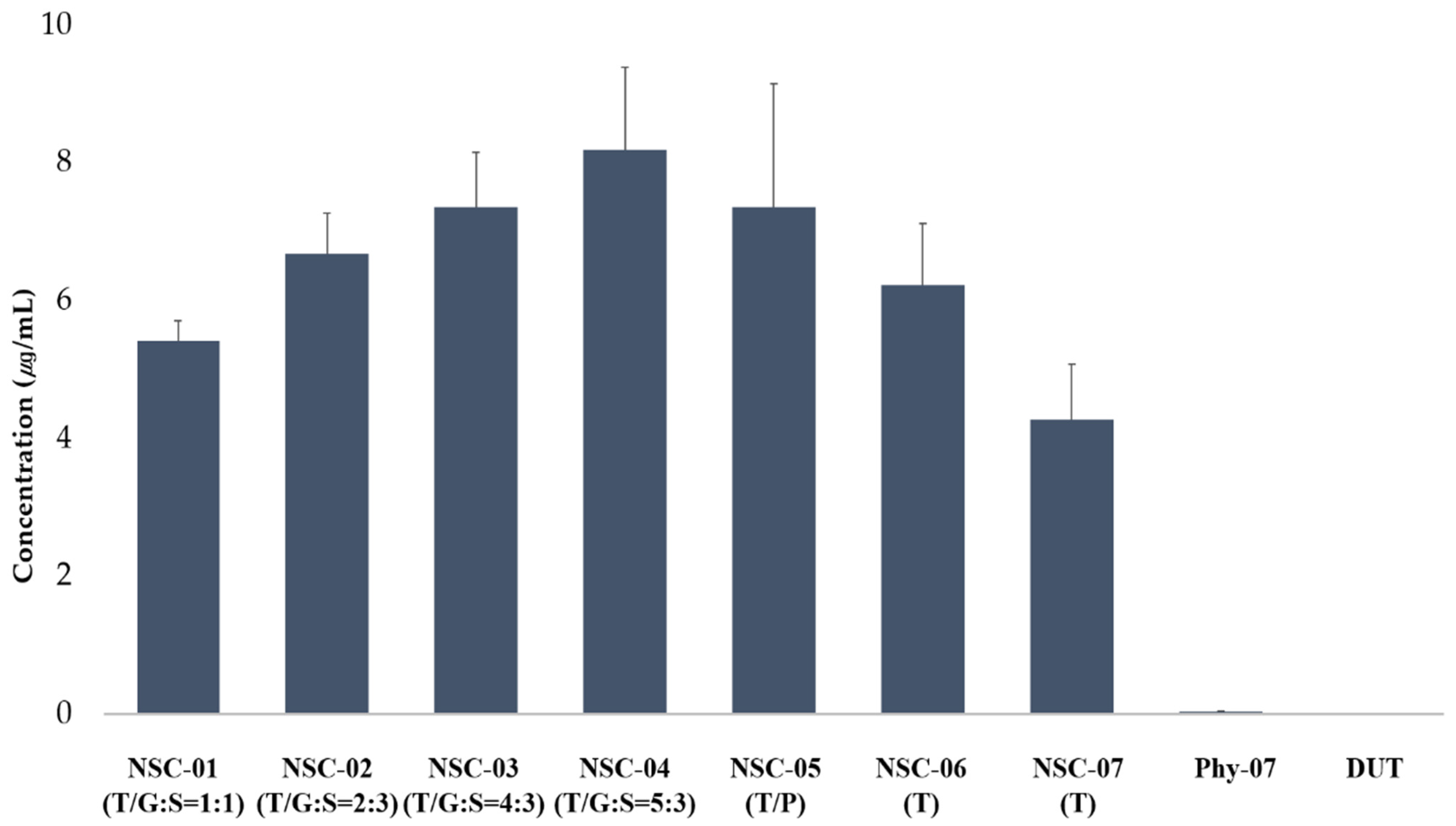
3.3.2. Solubility
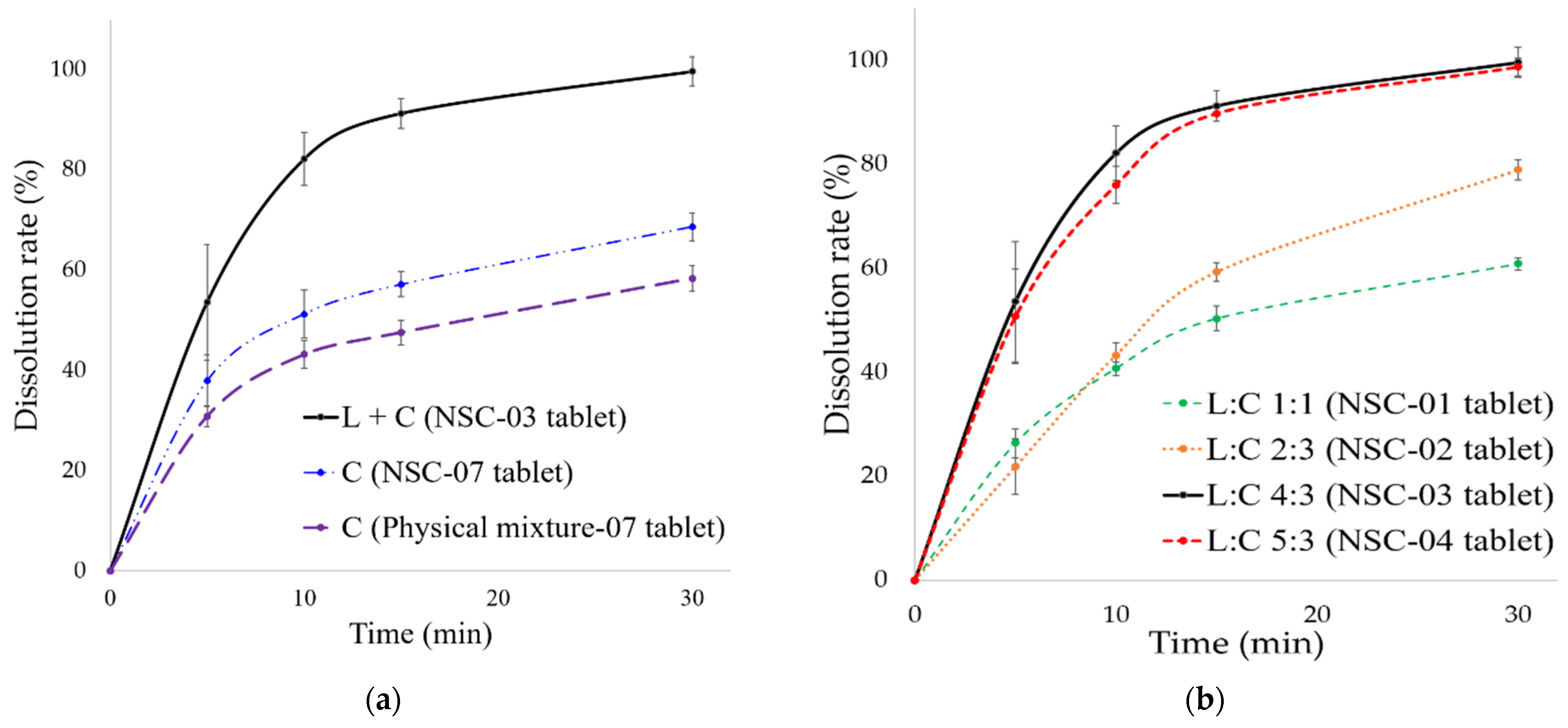
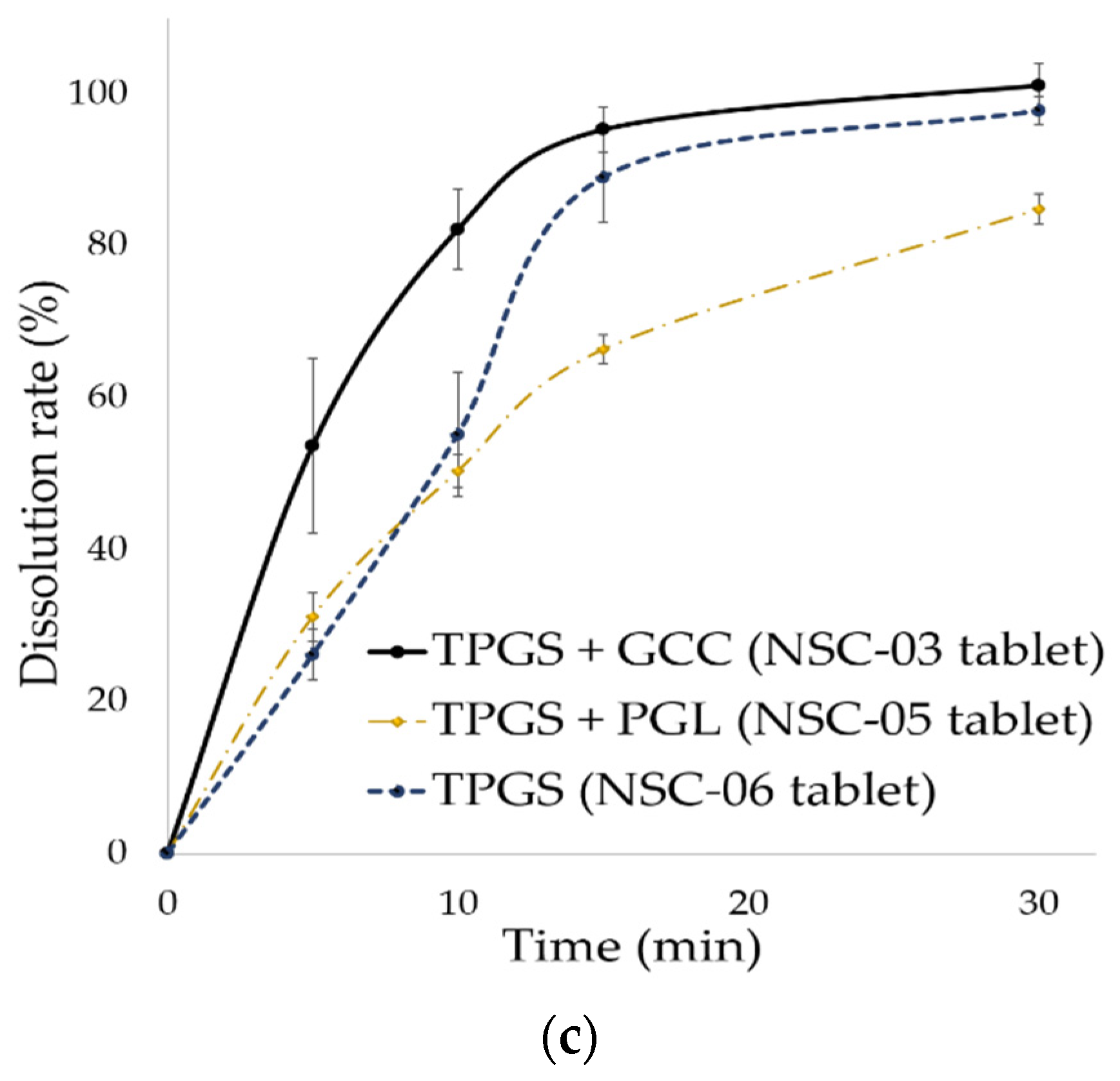
3.3.3. In Vitro Dissolution of NSC Tablet
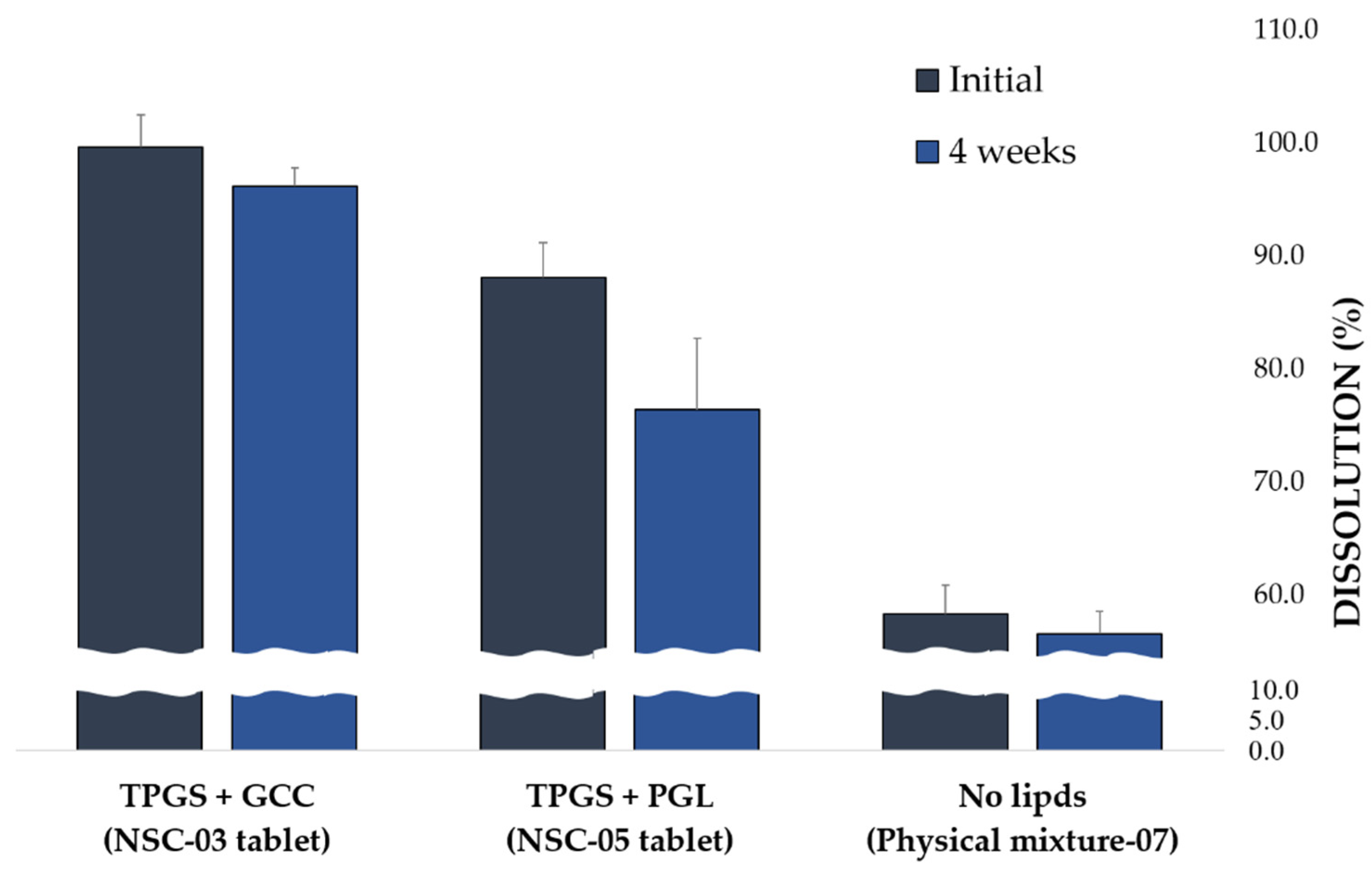
3.3.4. Stability Evaluation of NSC Tablets via Dissolution Rate
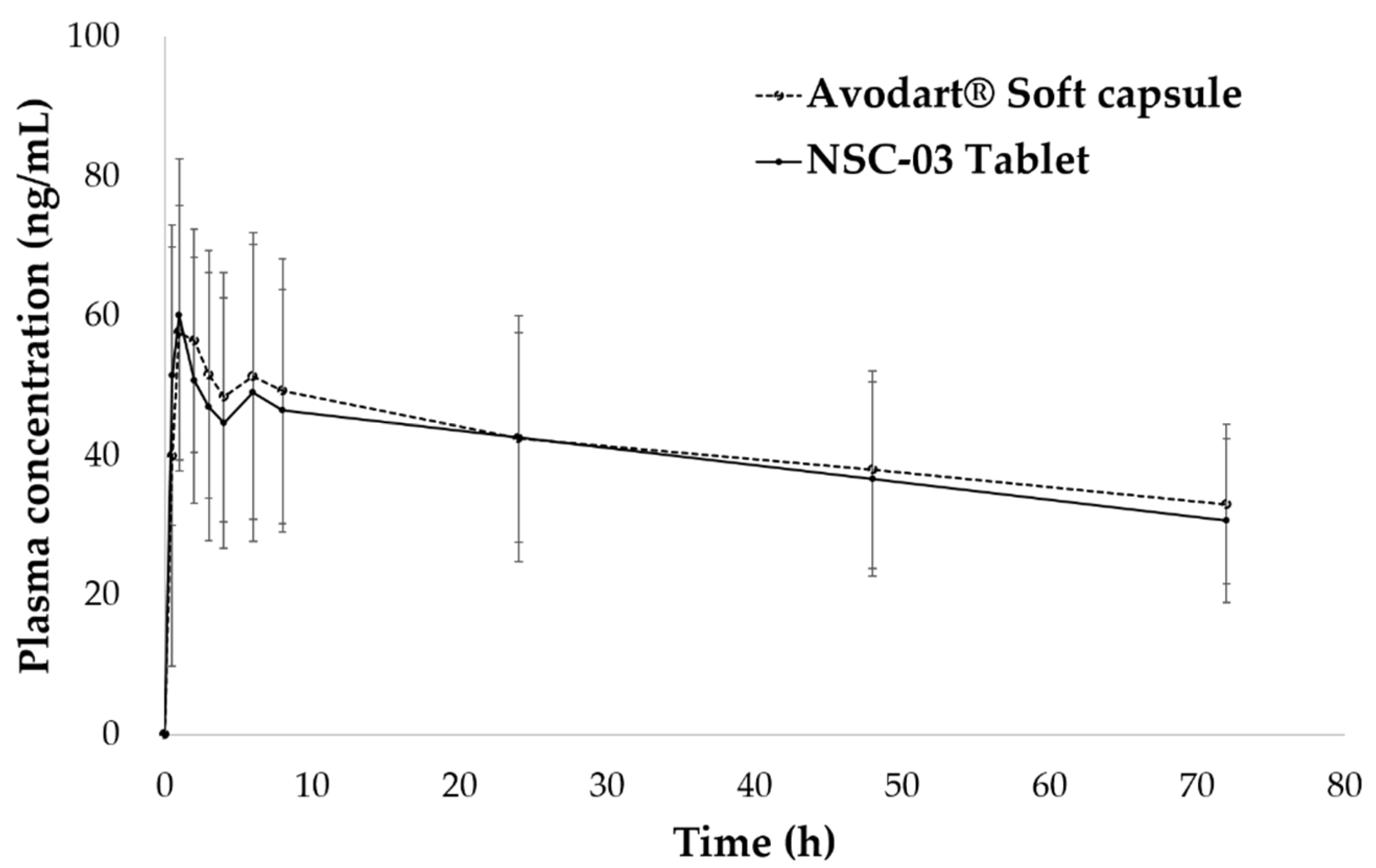
3.4. Pharmacokinetic Study of NSC Tablets in Beagle Dogs
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhong, H.; Chan, G.; Hu, Y.; Hu, H.; Ouyang, D. A comprehensive map of FDA-approved pharmaceutical products. Pharmaceutics 2018, 10, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhakay, A.; Rahman, M.; Dave, R.; Bilgili, E. Bioavailability enhancement of poorly water-soluble drugs via nanocomposites: Formulation–Processing aspects and challenges. Pharmaceutics 2018, 10, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, P.; Pyo, Y.-C.; Kim, D.-H.; Lee, S.-E.; Kim, J.-K.; Park, J.-S. Overview of the manufacturing methods of solid dispersion technology for improving the solubility of poorly water-soluble drugs and application to anticancer drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kale, A.R.; Kakade, S.; Bhosale, A. A Review on: Solubility Enhancement Techniques. J.Ceram. Process. Res. 2020, 10, 3630–3647. [Google Scholar]
- Godase, C.; Babar, A.; Gopal, A. A Concise Review on Methods of Solubility Enhancement. Int. Pharm. Sci. 2020, 11, 109. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.; Dai, M.; Nai, J.; Zhu, L.; Sheng, H. Solubility and bioavailability enhancement of oridonin: A review. Molecules 2020, 25, 332. [Google Scholar] [CrossRef] [Green Version]
- Malek, M.A.H.; Patel, P.M. Dendrimers for drug solubility enhancement–A review. Int. J. Pharm. Sci. Res. 2020, 11, 507–523. [Google Scholar]
- Mahapatra, A.P.; Patil, V.; Patil, R. Solubility Enhancement of Poorly soluble Drugs by using Novel Techniques: A Comprehensive Review. Int. J. PharmTech Res. 2020, 13, 80–93. [Google Scholar] [CrossRef]
- Singh, D.; Bedi, N.; Tiwary, A.K. Enhancing solubility of poorly aqueous soluble drugs: Critical appraisal of techniques. J. Pharm. Investig. 2018, 48, 509–526. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [Green Version]
- Kulyadi, G.P.; Mutalik, S.; Kulkarni, V.I.; Tippavajhala, V.K. Application of Hot Melt Extrusion for the Solubility Enhancement of a BCS Class II Drug. Res. J. Pharm. Technol. 2019, 12, 3751–3754. [Google Scholar] [CrossRef]
- Bidkar, S.; Kadam, D.; Dama, G.; Bidkar, J. A review: Factors affecting dissolution of BCS class II drug. World J. Pharm. Res. 2019, 8, 669–692. [Google Scholar] [CrossRef]
- Kumar, H.; Rajpoot, A.K.; Sharma, S.; Kumar, A. Pharmacological screening of Griseofulvin loaded solid lipid nanoparticles for improved oral delivery of poorly water soluble drug. Pharma Innov. J. 2019, 8, 55–58. [Google Scholar]
- Bhardwaj, P.; Tripathi, P.; Gupta, R.; Pandey, S. Niosomes: A review on niosomal research in the last decade. J. Drug Deliv. Sci. Technol. 2020, 56, 101581. [Google Scholar] [CrossRef]
- Marihart, S.; Harik, M.; Djavan, B. Dutasteride: A review of current data on a novel dual inhibitor of 5α reductase. Rev. Urol. 2005, 7, 203. [Google Scholar]
- Arif, T.; Dorjay, K.; Adil, M.; Sami, M. Dutasteride in androgenetic alopecia: An update. Curr. Clin. Pharm. 2017, 12, 31–35. [Google Scholar] [CrossRef]
- Gaines, K.K. Dutasteride (Avodart®): New 5-Alpha-Reductase Inhibitor for Treating BPH. Urol. Nurs. 2003, 23, 219. [Google Scholar]
- Noor, N.M.; Sheikh, K.; Somavarapu, S.; Taylor, K.M. Preparation and characterization of dutasteride-loaded nanostructured lipid carriers coated with stearic acid-chitosan oligomer for topical delivery. Eur. J. Pharm. Biopharm. 2017, 117, 372–384. [Google Scholar] [CrossRef]
- Foye, W.O. Foye’s Principles of Medicinal Chemistry; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2008. [Google Scholar]
- Ojeda-Serna, I.; Rocha-Guzmán, N.; Gallegos-Infante, J.; Cháirez-Ramírez, M.; Rosas-Flores, W.; Perez-Martinez, J.; Moreno-Jiménez, M.; González-Laredo, R. Water-in-oil organogel based emulsions as a tool for increasing bioaccessibility and cell permeability of poorly water-soluble nutraceuticals. Food Res. 2019, 120, 415–424. [Google Scholar] [CrossRef]
- Butt, S.; Hasan, S.M.F.; Hassan, M.M.; Alkharfy, K.M.; Neau, S.H. Directly compressed rosuvastatin calcium tablets that offer hydrotropic and micellar solubilization for improved dissolution rate and extent of drug release. Saudi Pharm. J. 2019, 27, 619–628. [Google Scholar] [CrossRef]
- Clark, S.T.; Arras, M.M.; Sarles, S.A.; Frymier, P.D. Lipid shape determination of detergent solubilization in mixed-lipid liposomes. Colloid Surf. B: Biointerfaces 2020, 187, 110609. [Google Scholar] [CrossRef]
- Shirsand, S.B.; Keshavshetti, G.G. Recent advances in niosomal drug delivery—A review. Res. J. Life Sci. Bioinform. Pharm. Chem. Sci. 2019, 3, 514–531. [Google Scholar]
- Patil, M.; Palde, S.; Deshmukh, A. A Review on Self Micro Emulsifying Drug Delivery System: An Approach to Enhance the Oral Bioavailability of Poorly Water Soluble Drug. Asian J. Pharm. Res. 2020, 8, 202–208. [Google Scholar] [CrossRef]
- Patel, V.; Lalani, R.; Bardoliwala, D.; Ghosh, S.; Misra, A. Lipid-based oral formulation strategies for lipophilic drugs. AAPS PharmSciTech 2018, 19, 3609–3630. [Google Scholar] [CrossRef]
- Holm, R. Bridging the gaps between academic research and industrial product developments of lipid-based formulations. Adv. Drug Deliv. Rev. 2019, 142, 118–127. [Google Scholar] [CrossRef]
- Tindal, S. Soft Capsules. In Pharmaceutical Formulation; CPI Group (UK) Ltd.: Croydon, UK, 2018; pp. 52–77. [Google Scholar]
- Roberson, A. liquid-filled capsules. In Tablet & Capsule; North Americas at ACG: South Plainfield, NJ, USA, 2017; pp. 1–5. [Google Scholar]
- Prasad, V.D. Formulation and modifying drug release from Hard and Soft Gelatin Capsules for Oral drug delivery. Int. J. Res. Dev. Pharm 2017, 6, 2663–2677. [Google Scholar] [CrossRef]
- Raj, A. Soft Gelatin Capsules (Softgels). PharmaTutor 2015, 3, 16–18. [Google Scholar]
- Betageri, G.V. Self-emulsifying Drug Delivery Systems and their Marketed Products: A Review. Asian J. Pharm. Free Full Text Artic. Asian J. Pharm. 2019, 13, 73–84. [Google Scholar] [CrossRef]
- Li, T.; Geng, T.; Md, A.; Banerjee, P.; Wang, B. Novel scheme for rapid synthesis of hollow mesoporous silica nanoparticles (HMSNs) and their application as an efficient delivery carrier for oral bioavailability improvement of poorly water-soluble BCS type II drugs. Colloid Surf. B 2019, 176, 185–193. [Google Scholar] [CrossRef]
- Waters, L.J.; Hanrahan, J.P.; Tobin, J.M.; Finch, C.V.; Parkes, G.M.; Ahmad, S.A.; Mohammad, F.; Saleem, M. Enhancing the dissolution of phenylbutazone using Syloid® based mesoporous silicas for oral equine applications. J. Pharm. Anal. 2018, 8, 181–186. [Google Scholar] [CrossRef]
- Hempel, N.-J.; Brede, K.; Olesen, N.E.; Genina, N.; Knopp, M.M.; Löbmann, K. A fast and reliable DSC-based method to determine the monomolecular loading capacity of drugs with good glass-forming ability in mesoporous silica. Int. J. Pharm. 2018, 544, 153–157. [Google Scholar] [CrossRef]
- Paseta, L.; Simón-Gaudó, E.; Gracia-Gorría, F.; Coronas, J. Encapsulation of essential oils in porous silica and MOFs for trichloroisocyanuric acid tablets used for water treatment in swimming pools. Chem. Eng. J. 2016, 292, 28–34. [Google Scholar] [CrossRef]
- Wei, Q.; Keck, C.M.; Müller, R.H. Preparation and tableting of long-term stable amorphous rutin using porous silica. Eur. J. Pharm. Biopharm. 2017, 113, 97–107. [Google Scholar] [CrossRef]
- Alhasani, K.F.; Kazi, M.; Ibrahim, M.A.; Shahba, A.A.; Alanazi, F.K. Self-nanoemulsifying ramipril tablets: A novel delivery system for the enhancement of drug dissolution and stability. Int. J. Nanotechnol. Nanomed. 2019, 14, 5435. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Sun, H.; Zhao, Q.; Han, N.; Bai, L.; Wang, Y.; Jiang, T.; Wang, S. Multilayer encapsulated mesoporous silica nanospheres as an oral sustained drug delivery system for the poorly water-soluble drug felodipine. Mater. Sci. Eng. C 2015, 47, 313–324. [Google Scholar] [CrossRef]
- Santosh, P.B. Increasing the oral bioavailability of poorly water-soluble valsartan using non-ordered mesoporous silica microparticles. Asian J. Pharm. (AJP) Free Full Text Artic. Asian J. Pharm. 2016. [Google Scholar] [CrossRef]
- Limnell, T.; Santos, H.A.; Mäkilä, E.; Heikkilä, T.; Salonen, J.; Murzin, D.Y.; Kumar, N.; Laaksonen, T.; Peltonen, L.; Hirvonen, J. Drug delivery formulations of ordered and nonordered mesoporous silica: Comparison of three drug loading methods. J. Pharm. Sci. 2011, 100, 3294–3306. [Google Scholar] [CrossRef]
- Jadhav, N.; Irny, P.; Patil, U. Solid state behavior of progesterone and its release from Neusilin US2 based liquisolid compacts. J. Drug Deliv. Sci. Technol. 2017, 38, 97–106. [Google Scholar] [CrossRef]
- Pandey, V.; Gilhotra, R.M.; Kohli, S. Granulated colloidal silicon dioxide-based self-microemulsifying tablets, as a versatile approach in enhancement of solubility and therapeutic potential of anti-diabetic agent: Formulation design and in vitro/in vivo evaluation. Drug Dev. Ind. Pharm. 2017, 43, 1023–1032. [Google Scholar] [CrossRef]
- Ibrahim, A.H.; Smått, J.-H.; Govardhanam, N.P.; Ibrahim, H.M.; Ismael, H.R.; Afouna, M.I.; Samy, A.M.; Rosenholm, J.M. Formulation and optimization of drug-loaded mesoporous silica nanoparticle-based tablets to improve the dissolution rate of the poorly water-soluble drug silymarin. Eur. J. Pharm. Sci. 2020, 142, 105103. [Google Scholar] [CrossRef]
- Zielińska, A.; Pereira, I.; Antunes, S.; Veiga, F.; Santos, A.; Nowak, I.; Silva, A.; Souto, E. Chapter 10 -Mesoporous silica nanoparticles as drug delivery systems against melanoma. In Design of Nanostructures for Theranostics Applications; Elsevier: Amsterdam, The Netherlands, 2018; pp. 437–466. [Google Scholar] [CrossRef]
- Karaman, D.Ş.; Kettiger, H. Silica-based nanoparticles as drug delivery systems: Chances and challenges. In Inorganic Frameworks as Smart Nanomedicines; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1–40. [Google Scholar]
- Ecetoc, J. Synthetic Amorphous Silica (CAS No. 7631-86-9); Report No. 51; European Centre for Ecotoxicology and Toxicology of Chemicals: Brussels, Belgium, 2006; pp. 1–231. [Google Scholar]
- Adler, C. New Lipid-Based Formulation Approaches and Characterization Tools for Hot-Melt Extrusion. Ph.D. Thesis, University of Basel, Basel, Switzerland, 2017. [Google Scholar]
- Han, C.; Zhang, S.; Huang, H.; Dong, Y.; Sui, X.; Jian, B.; Zhu, W. In vitro and in vivo evaluation of core–shell mesoporous silica as a promising water-insoluble drug delivery system: Improving the dissolution rate and bioavailability of celecoxib with needle-like crystallinity. J. Pharm. Sci. 2019, 108, 3225–3232. [Google Scholar] [CrossRef] [PubMed]
- Allgeier, M.C.; Piper, J.L.; Hinds, J.; Yates, M.H.; Kolodsick, K.J.; Meury, R.; Shaw, B.; Kulkarni, M.R.; Remick, D.M. Isolation and physical property optimization of an amorphous drug substance utilizing a high surface area magnesium aluminometasilicate (Neusilin® US2). J. Pharm. Sci. 2016, 105, 3105–3114. [Google Scholar] [CrossRef] [PubMed]
- Elkadi, S.; Elsamaligy, S.; Al-Suwayeh, S.; Mahmoud, H. The development of self-nanoemulsifying liquisolid tablets to improve the dissolution of simvastatin. AAPS PharmSciTech 2017, 18, 2586–2597. [Google Scholar] [CrossRef] [PubMed]
- Madan, J.R.; Patil, S.; Mathure, D.; Bahirat, S.P.; Awasthi, R.; Dua, K. Improving dissolution profile of poorly water-soluble drug using non-ordered mesoporous silica. Marmara Pharm. J. 2018, 22, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Schultz, H.B.; Thomas, N.; Rao, S.; Prestidge, C.A. Supersaturated silica-lipid hybrids (super-SLH): An improved solid-state lipid-based oral drug delivery system with enhanced drug loading. Eur. J. Pharm. Biopharm. 2018, 125, 13–20. [Google Scholar] [CrossRef] [PubMed]
- USP <1174> Powder Flow, USP43-NF38; The United States Pharmacopeial Convention: Rockville, MD, USA, 2020.
- USP <616> Bulk Density and Tapped Density, USP43-NF38; The United States Pharmacopeial Convention: Rockville, MD, USA, 2020.
- Attia, U.M.; Fones, A.; Trepleton, R.; Hamilton, H.; Davies, S.; Wimpenny, D. HIPing of Pd-doped titanium components: A study of mechanical and corrosion properties. In Proceedings of the 11th International Conference on Hot Isostatic Pressing, Stockholm, Sweden, 9–13 June 2014. [Google Scholar]
- Šimek, M.; Grünwaldová, V.; Kratochvíl, B. Comparison of Compression and Material Properties of Differently Shaped and Sized Paracetamols. KONA Powder Part J. 2017, 34, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Smeets, N.M.; McKenna, T.F. The synthesis of translucent polymer nanolatexes via microemulsion polymerization. J. Colloid Interface Sci. 2012, 383, 28–35. [Google Scholar] [CrossRef]
- Chatzidaki, M.D.; Mateos-Diaz, E.; Leal-Calderon, F.; Xenakis, A.; Carrière, F. Water-in-oil microemulsions versus emulsions as carriers of hydroxytyrosol: An in vitro gastrointestinal lipolysis study using the pHstat technique. Food Funct. 2016, 7, 2258–2269. [Google Scholar] [CrossRef]
- Lokhande, S.S. Microemulsions as Promising Delivery Systems: A Review. Asian J. Pharm. Res. 2019, 9, 90–96. [Google Scholar] [CrossRef]
- Min, M.-H.; Park, J.-H.; Choi, M.-R.; Hur, J.-H.; Ahn, B.-N.; Kim, D.-D. Formulation of a film-coated dutasteride tablet bioequivalent to a soft gelatin capsule (Avodart®): Effect of γ-cyclodextrin and solubilizers. Asian J. Pharm. Sci. 2019, 14, 313–320. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhi, Z.; Jiang, T.; Zhang, J.; Wang, Z.; Wang, S. Spherical mesoporous silica nanoparticles for loading and release of the poorly water-soluble drug telmisartan. J. Control. Release 2010, 145, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Lukášová, I.; Muselík, J.; Franc, A.; Goněc, R.; Mika, F.; Vetchý, D. Factor analysis in optimization of formulation of high content uniformity tablets containing low dose active substance. Eur. J. Pharm. Sci. 2017, 109, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Mahours, G.M.; Shaaban, D.E.Z.; Shazly, G.A.; Auda, S.H. The effect of binder concentration and dry mixing time on granules, tablet characteristics and content uniformity of low dose drug in high shear wet granulation. J. Drug Deliv. Sci. Technol. 2017, 39, 192–199. [Google Scholar] [CrossRef]
- Leung, S.S.Y.; Wong, J.; Guerra, H.V.; Samnick, K.; Prud’homme, R.K.; Chan, H.-K. Porous mannitol carrier for pulmonary delivery of cyclosporine A nanoparticles. AAPS J. 2017, 19, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-S.; Lee, S.-E.; Jang, W.S.; Byeon, J.C.; Park, J.-S. Solid dispersion of dutasteride using the solvent evaporation method: Approaches to improve dissolution rate and oral bioavailability in rats. Mater. Sci. Eng. C 2018, 90, 387–396. [Google Scholar] [CrossRef]
- Bahurupi, A.D.; Burange, P.J.; Tawar, M.G.; Ingole, S.R. Formulation and Characterization of Solid Lipid Microparticles. Am. J. Pharm. Health Res. 2019, 7, 10–27. [Google Scholar] [CrossRef]
- Kim, J.-S.; Ha, E.-S.; Park, H.; Choi, D.H.; Kim, M.-S.; Baek, I.-H. Pharmacokinetic Study of a Soft Gelatin Capsule and a Solid-Supersaturatable SMEDDS Tablet of Dutasteride in Beagle Dogs. Eur. J. Drug Metab. Pharm. 2019, 45, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Revathi, S.; Raju, M.D. Self-emulsifying drug delivery system: A review. World J. Pharm. Pharm. Sci. 2012, 2, 89–107. [Google Scholar]
- Jang, D.-J.; Kim, S.T.; Oh, E.; Ban, E. Enhanced oral bioavailability and controlled release of dutasteride by a novel dry elixir. Biomed. Mater. Eng. 2014, 24, 571–579. [Google Scholar] [CrossRef]
- Nikolakakis, I.; Partheniadis, I. Self-emulsifying granules and pellets: Composition and formation mechanisms for instant or controlled release. Pharmaceutics 2017, 9, 50. [Google Scholar] [CrossRef] [Green Version]
- Hasan, N.M.Y. Preparation of solid self-micro-emulsified lipid systems for the delivery of hydrophobic drugs. Int. J. Pharm. Res. 2015, 7, 75–84. [Google Scholar]
- Roehrborn, C.G.; Pérez, I.O.; Roos, E.P.; Calomfirescu, N.; Brotherton, B.; Wang, F.; Palacios, J.M.; Vasylyev, A.; Manyak, M.J. Efficacy and safety of a fixed-dose combination of dutasteride and tamsulosin treatment (D uodart®) compared with watchful waiting with initiation of tamsulosin therapy if symptoms do not improve, both provided with lifestyle advice, in the management of treatment-naïve men with moderately symptomatic benign prostatic hyperplasia: 2-year CONDUCT study results. BJU Int. 2015, 116, 450–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manyak, M.J.; Zhu, J.; Roehrborn, C.; McAleese, P.; Manyak, M. Impact of formulation on the pharmacokinetics of dutasteride: Results from two phase I studies. Clin. Drug Investig. 2016, 36, 763–767. [Google Scholar] [CrossRef]
- Kim, M.-S. Influence of hydrophilic additives on the supersaturation and bioavailability of dutasteride-loaded hydroxypropyl-β-cyclodextrin nanostructures. Int. J. Nanomed. 2013, 8, 2029–2039. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.; Tarter, T.H. Combination therapy with dutasteride and tamsulosin for the treatment of symptomatic enlarged prostate. Clin. Interv. Aging 2009, 4, 251. [Google Scholar]
- Keating, G.M. Dutasteride/Tamsulosin. Drugs Aging 2012, 29, 405–419. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DUT | TPGS | GCC | PGL | Carrier * | L:C | |
|---|---|---|---|---|---|---|
| NSC-01 | 1 | 5 | 5 | - | 10 | 1:1 |
| NSC-02 | 1 | 10 | 10 | - | 30 | 2:3 |
| NSC-03 | 1 | 20 | 20 | - | 30 | 4:3 |
| NSC-04 | 1 | 25 | 25 | - | 30 | 5:3 |
| NSC-05 | 1 | 20 | - | 20 | 30 | 4:3 |
| NSC-06 | 1 | 40 | - | - | 30 | 4:3 |
| NSC-07 | 1 | - | - | - | 30 | - |
| Physical mixture-07 | 1 | - | - | - | 30 | - |
| NSC-01 Tablet | NSC-02 Tablet | NSC-03 Tablet | NSC-04 Tablet | NSC-05 Tablet | NSC-06 Tablet | NSC-07 Tablet | Physical Mixture-07 Tablet | |
|---|---|---|---|---|---|---|---|---|
| NSC-01 | 7 | - | - | - | - | - | - | - |
| NSC-02 | - | 15 | - | - | - | - | - | - |
| NSC-03 | - | - | 21 | - | - | - | - | - |
| NSC-04 | - | - | - | 18 | - | - | - | - |
| NSC-05 | - | - | - | - | 21 | - | - | - |
| NSC-06 | - | - | - | - | - | 21 | - | - |
| NSC-07 | - | - | - | - | - | - | 10 | - |
| Physical mixture-07 | - | - | - | - | - | - | - | 10 |
| CCS | 9 | 8 | 8 | 7 | 8 | 8 | 9 | 9 |
| L-HPC | 21 | 21 | 18 | 18 | 18 | 18 | 21 | 21 |
| MCC | 21 | 20 | 18 | 18 | 18 | 18 | 21 | 21 |
| SMCC | 21 | 18 | 16 | 16 | 16 | 16 | 19 | 19 |
| IM | 21 | 18 | 18 | 16 | 18 | 18 | 21 | 21 |
| SSF | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Total weight | 150 mg | 160 mg | 170 mg | 220 mg | 170 mg | 170 mg | 150 mg | 150 mg |
| NSC | A.R. 1,2 (°) | Bulk Density 1 (g/mL) | Tap Density 1 (g/mL) | Carr’s Index 1 (%) | Hausner 1 Ratio 1 (H.R.) |
|---|---|---|---|---|---|
| NSC-01 | 34.1° ± 0.9 | 0.39 ± 0.00 | 0.46 ± 0.01 | 18.1 ± 2.32 | 1.18 ± 0.02 |
| NSC-02 | 36.1° ± 1.8 | 0.38 ± 0.02 | 0.46 ± 0.01 | 21.3 ± 2.4 | 1.21 ± 0.02 |
| NSC-03 | 33.2° ± 2.9 | 0.42 ± 0.01 | 0.48 ± 0.00 | 14.5 ± 3.7 | 1.14 ± 0.04 |
| NSC-04 | 36.7° ± 2.9 | 0.40 ± 0.01 | 0.46 ± 0.01 | 14.9 ± 0.5 | 1.15 ± 0.00 |
| NSC-05 | 44.5° ± 1.5 | 0.42 ± 0.01 | 0.55 ± 0.01 | 30.8 ± 2.4 | 1.31 ± 0.02 |
| NSC-06 | 56.1° ± 3.1 | 0.41 ± 0.03 | 0.57 ± 0.02 | 36.8 ± 3.8 | 1.37 ± 0.04 |
| NSC-07 | 33.1° ± 2.1 | 0.36 ± 0.01 | 0.42 ± 0.00 | 16.9 ± 2.7 | 1.17 ± 0.03 |
| Physical mixture-07 | 31.1° ± 0.8 | 0.33 ± 0.02 | 0.38 ± 0.01 | 15.4 ± 3.0 | 1.15 ± 0.03 |
| Assay (Unit: mg/g) | NSC-03 (TPGS + GCC) | NSC-05 (TPGS + PGL) | NSC-06 (TPGS) | Physical Mixture-07 (no Lipid) |
|---|---|---|---|---|
| Upper | 14.47 | 14.51 | 13.96 | 13.52 |
| Middle-Right | 14.47 | 14.26 | 14.16 | 13.94 |
| Middle-Left | 14.44 | 14.92 | 14.06 | 13.82 |
| Bottom-Right | 14.49 | 14.38 | 14.06 | 14.42 |
| Bottom-Left | 14.41 | 14.10 | 14.07 | 14.45 |
| Average | 14.46 | 14.43 | 14.06 | 14.03 |
| SD | 0.03 | 0.28 | 0.06 | 0.36 |
| RSD (%) | 0.21 | 1.94 | 0.43 | 2.57 |
| p value | <0.05 | <0.05 | <0.05 | >0.05 |
| NSC Tablets | Avodart® | 90% CI 4 | |
|---|---|---|---|
| AUCt 1 (ng·h/mL) | 2940.3 ± 1036.2 | 2843.7 ± 1106.9 | 0.8641, 1.0454 |
| Relative BA, % (to the Avodart®) | 103.4% | - | - |
| Cmax 2 (ng/mL) | 67.60 ± 16.37 | 63.73 ± 23.75 | 0.8132, 1.0024 |
| Tmax 3 (h) | 1.79 ± 1.50 | 1.75 ± 2.03 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, H.-W.; Kim, J.-E.; Park, Y.-J. Nanoporous Silica Entrapped Lipid-Drug Complexes for the Solubilization and Absorption Enhancement of Poorly Soluble Drugs. Pharmaceutics 2021, 13, 63. https://doi.org/10.3390/pharmaceutics13010063
Shin H-W, Kim J-E, Park Y-J. Nanoporous Silica Entrapped Lipid-Drug Complexes for the Solubilization and Absorption Enhancement of Poorly Soluble Drugs. Pharmaceutics. 2021; 13(1):63. https://doi.org/10.3390/pharmaceutics13010063
Chicago/Turabian StyleShin, Hey-Won, Joo-Eun Kim, and Young-Joon Park. 2021. "Nanoporous Silica Entrapped Lipid-Drug Complexes for the Solubilization and Absorption Enhancement of Poorly Soluble Drugs" Pharmaceutics 13, no. 1: 63. https://doi.org/10.3390/pharmaceutics13010063