
Nano DNA Vaccine Encoding Toxoplasma gondii Histone Deacetylase SIR2 Enhanced Protective Immunity in Mice

 ,
,  ,
, 
Abstract
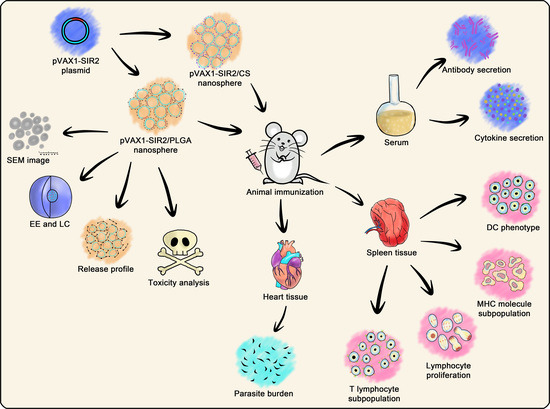
:
1. Introduction
2. Materials and Methods
2.1. Cells, Parasites, and Animals
2.2. Construction of the Recombinant Plasmid
2.3. Plasmid Preparation and Transfection In Vitro
2.4. Generation of Anti-rTgSIR2 Polyclonal Antibodies
2.5. Immunofluorescence Staining
2.6. Nanospheres Formulation
2.7. Characterizations and Release Characteristics of Nanospheres
2.8. Animals Immunization Schedule and Challenge
2.9. Determination of Antibodies and Cytokines
2.10. Lymphocyte Proliferation and Flow Cytometry
2.11. T. gondii Burdens in Animals
2.12. Statistical Analysis
3. Results
3.1. Construction of the Recombinant Plasmid and Its Expression
3.2. Physical Characterization and Release Characteristics
3.3. Toxicity Analysis in Animals
3.4. Modulations of Antibodies and Cytokines Secretions in Animals
3.5. Phenotype Analysis of Dendritic Cells
3.6. Lymphocyte Proliferation
3.7. Proportions of T Lymphocytes
3.8. T. gondii Burdens in Animals
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tenter, A.M.; Heckeroth, A.R.; Weiss, L.M. Toxoplasma gondii: From animals to humans. Int. J. Parasitol. 2000, 30, 1217–1258. [Google Scholar] [CrossRef] [Green Version]
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- McCabe, R.; Chirurgi, V. Issues in toxoplasmosis. Infect. Dis. Clin. N. Am. 1993, 7, 587–604. [Google Scholar] [CrossRef]
- Weiss, L.M.; Dubey, J.P. Toxoplasmosis: A history of clinical observations. Int. J. Parasitol. 2009, 39, 895–901. [Google Scholar] [CrossRef] [Green Version]
- Sroka, J.; Bilska-Zajac, E.; Wojcik-Fatla, A.; Zajac, V.; Dutkiewicz, J.; Karamon, J.; Piotrowska, W.; Cencek, T. Detection and Molecular Characteristics of Toxoplasma gondii DNA in Retail Raw Meat Products in Poland. Foodborne Pathog. Dis. 2019, 16, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Marques, C.S.; Sousa, S.; Castro, A.; da Costa, J.M.C. Detection of Toxoplasma gondii oocysts in fresh vegetables and berry fruits. Parasites Vectors 2020, 13, 180. [Google Scholar] [CrossRef]
- Hernandez-Cortazar, I.B.; Acosta-Viana, K.Y.; Guzman-Marin, E.; Ortega-Pacheco, A.; Segura-Correa, J.C.; Jimenez-Coello, M. Presence of Toxoplasma gondii in Drinking Water from an Endemic Region in Southern Mexico. Foodborne Pathog. Dis. 2017, 14, 288–292. [Google Scholar] [CrossRef]
- Aguirre, A.A.; Longcore, T.; Barbieri, M.; Dabritz, H.; Hill, D.; Klein, P.N.; Lepczyk, C.; Lilly, E.L.; McLeod, R.; Milcarsky, J.; et al. The One Health Approach to Toxoplasmosis: Epidemiology, Control, and Prevention Strategies. Ecohealth 2019, 16, 378–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmar, A.J.; Drozda, A.A.; Blader, I.J. Drug Repurposing Screening Identifies Novel Compounds That Effectively Inhibit Toxoplasma gondii Growth. mSphere 2016, 1, e00042-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunay, I.R.; Gajurel, K.; Dhakal, R.; Liesenfeld, O.; Montoya, J.G. Treatment of Toxoplasmosis: Historical Perspective, Animal Models, and Current Clinical Practice. Clin. Microbiol. Rev. 2018, 31, e00057-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Group, S.S.; Thiebaut, R.; Leproust, S.; Chene, G.; Gilbert, R. Effectiveness of prenatal treatment for congenital toxoplasmosis: A meta-analysis of individual patients’ data. Lancet 2007, 369, 115–122. [Google Scholar] [CrossRef]
- Buxton, D.; Innes, E.A. A commercial vaccine for ovine toxoplasmosis. Parasitology 1995, 110, S11–S16. [Google Scholar] [CrossRef]
- Buxton, D. Toxoplasmosis: The first commercial vaccine. Parasitol. Today 1993, 9, 335–337. [Google Scholar] [CrossRef]
- AMCSF. Risk Profile in Relation to Toxoplasma in the Food Chain. Available online: http://www.innocua.net/web/download-310/acmsfrtaxopasm.pdf (accessed on 2 June 2021).
- Rezaei, F.; Sarvi, S.; Sharif, M.; Hejazi, S.H.; Pagheh, A.S.; Aghayan, S.A.; Daryani, A. A systematic review of Toxoplasma gondii antigens to find the best vaccine candidates for immunization. Microb. Pathog. 2019, 126, 172–184. [Google Scholar] [CrossRef]
- Saadatnia, G.; Golkar, M. A review on human toxoplasmosis. Scand. J. Infect. Dis. 2012, 44, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Hajissa, K.; Zakaria, R.; Suppian, R.; Mohamed, Z. Epitope-based vaccine as a universal vaccination strategy against Toxoplasma gondii infection: A mini-review. J. Adv. Vet. Anim. Res. 2019, 6, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Panas, M.W.; Boothroyd, J.C. Seizing control: How dense granule effector proteins enable Toxoplasma to take charge. Mol. Microbiol. 2021, 115, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Pagheh, A.S.; Sarvi, S.; Sharif, M.; Rezaei, F.; Ahmadpour, E.; Dodangeh, S.; Omidian, Z.; Hassannia, H.; Mehrzadi, S.; Daryani, A. Toxoplasma gondii surface antigen 1 (SAG1) as a potential candidate to develop vaccine against toxoplasmosis: A systematic review. Comp. Immunol. Microbiol. Infect. Dis. 2020, 69, 101414. [Google Scholar] [CrossRef]
- Dlugonska, H. Toxoplasma rhoptries: Unique secretory organelles and source of promising vaccine proteins for immunoprevention of toxoplasmosis. J. Biomed. Biotechnol. 2008, 2008, 632424. [Google Scholar] [CrossRef] [Green Version]
- Dubois, D.J.; Soldati-Favre, D. Biogenesis and secretion of micronemes in Toxoplasma gondii. Cell. Microbiol. 2019, 21, e13018. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.M.; Corvi, M.M.; Diambra, L. Gene target discovery with network analysis in Toxoplasma gondii. Sci. Rep. 2019, 9, 646. [Google Scholar] [CrossRef] [Green Version]
- Verma, R.; Khanna, P. Development of Toxoplasma gondii vaccine: A global challenge. Hum. Vaccines Immunother. 2013, 9, 291–293. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhou, H. Moving towards improved vaccines for Toxoplasma gondii. Expert Opin. Biol. Ther. 2018, 18, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Dubey, J.P.; Lindsay, D.S.; Speer, C.A. Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin. Microbiol. Rev. 1998, 11, 267–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radke, J.R.; Behnke, M.S.; Mackey, A.J.; Radke, J.B.; Roos, D.S.; White, M.W. The transcriptome of Toxoplasma gondii. BMC Biol. 2005, 3, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darkin-Rattray, S.J.; Gurnett, A.M.; Myers, R.W.; Dulski, P.M.; Crumley, T.M.; Allocco, J.J.; Cannova, C.; Meinke, P.T.; Colletti, S.L.; Bednarek, M.A.; et al. Apicidin: A novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. USA 1996, 93, 13143–13147. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.E.; Stilger, K.L.; Elias, E.V.; Naguleswaran, A.; Sullivan, W.J., Jr. A decade of epigenetic research in Toxoplasma gondii. Mol. Biochem. Parasitol. 2010, 173, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Rose, K.L.; Zhang, J.; Beavis, R.C.; Ueberheide, B.; Garcia, B.A.; Chait, B.; Zhao, Y.; Hunt, D.F.; Segal, E.; et al. Proteome-wide prediction of acetylation substrates. Proc. Natl. Acad. Sci. USA 2009, 106, 13785–13790. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Wang, J.; Asher, S.; Hoang, L.; Guardiani, C.; Ivanov, I.; Zheng, Y.G. Histone H4 acetylation differentially modulates arginine methylation by an in Cis mechanism. J. Biol. Chem. 2011, 286, 20323–20334. [Google Scholar] [CrossRef] [Green Version]
- Vaca, H.R.; Celentano, A.M.; Macchiaroli, N.; Kamenetzky, L.; Camicia, F.; Rosenzvit, M.C. Histone deacetylase enzymes as potential drug targets of Neglected Tropical Diseases caused by cestodes. Int. J. Parasitol. Drugs Drug Resist. 2019, 9, 120–132. [Google Scholar] [CrossRef]
- Copeland, R.A.; Solomon, M.E.; Richon, V.M. Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discov. 2009, 8, 724–732. [Google Scholar] [CrossRef]
- Wigle, T.J. Promoting illiteracy in epigenetics: An emerging therapeutic strategy. Curr. Chem. Genom. 2011, 5, 48–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrakovcic, M.; Bohner, L.; Hanisch, M.; Frohlich, L.F. Epigenetic Targeting of Autophagy via HDAC Inhibition in Tumor Cells: Role of p53. Int. J. Mol. Sci. 2018, 19, 3952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strobl, J.S.; Cassell, M.; Mitchell, S.M.; Reilly, C.M.; Lindsay, D.S. Scriptaid and suberoylanilide hydroxamic acid are histone deacetylase inhibitors with potent anti-Toxoplasma gondii activity in vitro. J. Parasitol. 2007, 93, 694–700. [Google Scholar] [CrossRef]
- Saksouk, N.; Bhatti, M.M.; Kieffer, S.; Smith, A.T.; Musset, K.; Garin, J.; Sullivan, W.J., Jr.; Cesbron-Delauw, M.F.; Hakimi, M.A. Histone-modifying complexes regulate gene expression pertinent to the differentiation of the protozoan parasite Toxoplasma gondii. Mol. Cell. Biol. 2005, 25, 10301–10314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duraisingh, M.T.; Voss, T.S.; Marty, A.J.; Duffy, M.F.; Good, R.T.; Thompson, J.K.; Freitas-Junior, L.H.; Scherf, A.; Crabb, B.S.; Cowman, A.F. Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell 2005, 121, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonkin, C.J.; Carret, C.K.; Duraisingh, M.T.; Voss, T.S.; Ralph, S.A.; Hommel, M.; Duffy, M.F.; Silva, L.M.; Scherf, A.; Ivens, A.; et al. Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum. PLoS Biol. 2009, 7, e84. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Chen, S.; Aleem, M.; He, S.; Yang, Y.; Zhou, T.; Liu, J.; Luo, J.; Yan, R.; Xu, L.; et al. Histone deacetylase SIR2 in Toxoplasma gondii modulates functions of murine macrophages in vitro and protects mice against acute toxoplasmosis in vivo. Microb. Pathog. 2021, 154, 104835. [Google Scholar] [CrossRef]
- Freitas-Junior, L.H.; Hernandez-Rivas, R.; Ralph, S.A.; Montiel-Condado, D.; Ruvalcaba-Salazar, O.K.; Rojas-Meza, A.P.; Mancio-Silva, L.; Leal-Silvestre, R.J.; Gontijo, A.M.; Shorte, S.; et al. Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell 2005, 121, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Assolini, J.P.; Concato, V.M.; Goncalves, M.D.; Carloto, A.C.M.; Conchon-Costa, I.; Pavanelli, W.R.; Melanda, F.N.; Costa, I.N. Nanomedicine advances in toxoplasmosis: Diagnostic, treatment, and vaccine applications. Parasitol. Res. 2017, 116, 1603–1615. [Google Scholar] [CrossRef] [PubMed]
- Saade, F.; Petrovsky, N. Technologies for enhanced efficacy of DNA vaccines. Expert Rev. Vaccines 2012, 11, 189–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Meng, Q.; Li, Q.; Liu, J.; Zhou, M.; Jin, Z.; Zhao, K. Chitosan Derivatives and Their Application in Biomedicine. Int. J. Mol. Sci. 2020, 21, 487. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, N.; Shahbazi, M.A.; Araujo, F.; Zhang, H.; Makila, E.M.; Kauppila, J.; Sarmento, B.; Salonen, J.J.; Hirvonen, J.T.; Santos, H.A. Chitosan-modified porous silicon microparticles for enhanced permeability of insulin across intestinal cell monolayers. Biomaterials 2014, 35, 7172–7179. [Google Scholar] [CrossRef] [PubMed]
- Wedmore, I.; McManus, J.G.; Pusateri, A.E.; Holcomb, J.B. A special report on the chitosan-based hemostatic dressing: Experience in current combat operations. J. Trauma 2006, 60, 655–658. [Google Scholar] [CrossRef] [Green Version]
- Illum, L. Chitosan and its use as a pharmaceutical excipient. Pharm. Res. 1998, 15, 1326–1331. [Google Scholar] [CrossRef]
- Jain, R.A. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials 2000, 21, 2475–2490. [Google Scholar] [CrossRef]
- Allahyari, M.; Mohit, E. Peptide/protein vaccine delivery system based on PLGA particles. Hum. Vaccines Immunother. 2016, 12, 806–828. [Google Scholar] [CrossRef] [Green Version]
- Cheraghipour, K.; Masoori, L.; Ezzatkhah, F.; Salimikia, I.; Amiri, S.; Makenali, A.S.; Taherpour, F.; Mahmoudvand, H. Effect of chitosan on Toxoplasma gondii infection: A systematic review. Parasite Epidemiol. Control 2020, 11, e00189. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, G.; Wang, W.; Xie, Q.; Zhang, M.; Yuan, C.; Hassan, I.A.; Liu, X.; Xu, L.; Yan, R.; et al. Pathogenicity of two Toxoplasma gondii strains in chickens of different ages infected via intraperitoneal injection. Avian Pathol. 2014, 43, 91–95. [Google Scholar] [CrossRef] [Green Version]
- McCall, R.L.; Sirianni, R.W. PLGA nanoparticles formed by single- or double-emulsion with vitamin E-TPGS. J. Vis. Exp. 2013, 51015. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Yan, W.; Xu, Z.; Ni, H. Formation mechanism of monodisperse, low molecular weight chitosan nanoparticles by ionic gelation technique. Colloids Surf. B Biointerfaces 2012, 90, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Skop, N.B.; Calderon, F.; Levison, S.W.; Gandhi, C.D.; Cho, C.H. Heparin crosslinked chitosan microspheres for the delivery of neural stem cells and growth factors for central nervous system repair. Acta Biomater. 2013, 9, 6834–6843. [Google Scholar] [CrossRef] [PubMed]
- Dadimoghaddam, Y.; Daryani, A.; Sharif, M.; Ahmadpour, E.; Hossienikhah, Z. Tissue tropism and parasite burden of Toxoplasma gondii RH strain in experimentally infected mice. Asian Pac. J. Trop. Med. 2014, 7, 521–524. [Google Scholar] [CrossRef]
- Zheng, B.; Ding, J.Z.; Lou, D.; Tong, Q.B.; Zhuo, X.H.; Ding, H.J.; Kong, Q.M.; Lu, S.H. The Virulence-Related MYR1 Protein of Toxoplasma gondii as a Novel DNA Vaccine Against Toxoplasmosis in Mice. Front. Microbiol. 2019, 10, 734. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Ding, W.; Aleem, M.T.; Su, J.; Liu, J.; Luo, J.; Yan, R.; Xu, L.; Song, X.; Li, X. Toxoplasma gondii Proteasome Subunit Alpha Type 1 with Chitosan: A Promising Alternative to Traditional Adjuvant. Pharmaceutics 2021, 13, 752. [Google Scholar] [CrossRef]
- Homan, W.L.; Vercammen, M.; De Braekeleer, J.; Verschueren, H. Identification of a 200- to 300-fold repetitive 529 bp DNA fragment in Toxoplasma gondii, and its use for diagnostic and quantitative PCR. Int. J. Parasitol. 2000, 30, 69–75. [Google Scholar] [CrossRef]
- Peek, L.J.; Middaugh, C.R.; Berkland, C. Nanotechnology in vaccine delivery. Adv. Drug Deliv. Rev. 2008, 60, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Gratton, S.E.; Ropp, P.A.; Pohlhaus, P.D.; Luft, J.C.; Madden, V.J.; Napier, M.E.; DeSimone, J.M. The effect of particle design on cellular internalization pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 11613–11618. [Google Scholar] [CrossRef] [Green Version]
- Leya, T.; Ahmad, I.; Sharma, R.; Tripathi, G.; Kurcheti, P.P.; Rajendran, K.V.; Bedekar, M.K. Bicistronic DNA vaccine macromolecule complexed with poly lactic-co-glycolic acid-chitosan nanoparticles enhanced the mucosal immunity of Labeo rohita against Edwardsiella tarda infection. Int. J. Biol. Macromol. 2020, 156, 928–937. [Google Scholar] [CrossRef]
- Zheng, F.; Liu, H.; Sun, X.; Zhang, Y.; Zhang, B.; Teng, Z.; Hou, Y.; Wang, B. Development of oral DNA vaccine based on chitosan nanoparticles for the immunization against reddish body iridovirus in turbots (Scophthalmus maximus). Aquaculture 2016, 452, 263–271. [Google Scholar] [CrossRef]
- Yoo, J.; Won, Y.Y. Phenomenology of the Initial Burst Release of Drugs from PLGA Microparticles. ACS Biomater. Sci. Eng. 2020, 6, 6053–6062. [Google Scholar] [CrossRef]
- Koppolu, B.P.; Smith, S.G.; Ravindranathan, S.; Jayanthi, S.; Kumar, T.K.S.; Zaharoff, D.A. Controlling chitosan-based encapsulation for protein and vaccine delivery. Biomaterials 2014, 35, 4382–4389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlosser, P.M.; Bale, A.S.; Gibbons, C.F.; Wilkins, A.; Cooper, G.S. Human health effects of dichloromethane: Key findings and scientific issues. Environ. Health Perspect. 2015, 123, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Li, J.; Xiao, T.; Huang, X.D.; Wang, L.J.; Huang, B.C.; Yin, K.; Liu, G.Z.; Xu, C.; Wei, Q.K. Protective immunity induced by a DNA vaccine cocktail expressing TgSAG1, TgROP2, and the genetic adjuvant HBsAg against Toxoplasma gondii infection. Microb. Pathog. 2020, 147, 104441. [Google Scholar] [CrossRef]
- Khodadadi, M.; Ghaffarifar, F.; Dalimi, A.; Ahmadpour, E. Immunogenicity of in-silico designed multi-epitope DNA vaccine encoding SAG1, SAG3 and SAG5 of Toxoplasma gondii adjuvanted with CpG-ODN against acute toxoplasmosis in BALB/c mice. Acta Trop. 2021, 216, 105836. [Google Scholar] [CrossRef]
- Foroutan, M.; Barati, M.; Ghaffarifar, F. Enhancing immune responses by a novel multi-epitope ROP8 DNA vaccine plus interleukin-12 plasmid as a genetic adjuvant against acute Toxoplasma gondii infection in BALB/c mice. Microb. Pathog. 2020, 147, 104435. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, Y.; Liang, Y.; Wang, S.; Xie, Q.; Nan, X.; Li, P.; Hong, G.; Liu, Q.; Li, X. Molecular characterization and protective immunity of rhoptry protein 35 (ROP35) of Toxoplasma gondii as a DNA vaccine. Vet. Parasitol. 2018, 260, 12–21. [Google Scholar] [CrossRef]
- Sayles, P.C.; Gibson, G.W.; Johnson, L.L. B cells are essential for vaccination-induced resistance to virulent Toxoplasma gondii. Infect. Immun. 2000, 68, 1026–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, N.; Frickel, E.M.; Mostowy, S. Macrophage-Microbe Interactions: Lessons from the Zebrafish Model. Front. Immunol. 2017, 8, 1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germann, T.; Bongartz, M.; Dlugonska, H.; Hess, H.; Schmitt, E.; Kolbe, L.; Kölsch, E.; Podlaski, F.J.; Gately, M.K.; Rüde, E. Interleukin-12 profoundly up-regulates the synthesis of antigen-specific complement-fixing IgG2a, IgG2b and IgG3 antibody subclasses in vivo. Eur. J. Immunol. 1995, 25, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Pifer, R.; Yarovinsky, F. Innate responses to Toxoplasma gondii in mice and humans. Trends Parasitol. 2011, 27, 388–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innes, E.A.; Bartley, P.M.; Buxton, D.; Katzer, F. Ovine toxoplasmosis. Parasitology 2009, 136, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Sonaimuthu, P.; Ching, X.T.; Fong, M.Y.; Kalyanasundaram, R.; Lau, Y.L. Induction of Protective Immunity against Toxoplasmosis in BALB/c Mice Vaccinated with Toxoplasma gondii Rhoptry-1. Front. Microbiol. 2016, 7, 808. [Google Scholar] [CrossRef]
- Ching, X.T.; Fong, M.Y.; Lau, Y.L. Evaluation of Immunoprotection Conferred by the Subunit Vaccines of GRA2 and GRA5 against Acute Toxoplasmosis in BALB/c Mice. Front. Microbiol. 2016, 7, 609. [Google Scholar] [CrossRef]
- Suzuki, Y.; Sa, Q.; Gehman, M.; Ochiai, E. Interferon-gamma- and perforin-mediated immune responses for resistance against Toxoplasma gondii in the brain. Expert Rev. Mol. Med. 2011, 13, e31. [Google Scholar] [CrossRef] [Green Version]
- Dupont, C.D.; Christian, D.A.; Hunter, C.A. Immune response and immunopathology during toxoplasmosis. Semin. Immunopathol. 2012, 34, 793–813. [Google Scholar] [CrossRef] [Green Version]
- Kelly, M.N.; Kolls, J.K.; Happel, K.; Schwartzman, J.D.; Schwarzenberger, P.; Combe, C.; Moretto, M.; Khan, I.A. Interleukin-17/interleukin-17 receptor-mediated signaling is important for generation of an optimal polymorphonuclear response against Toxoplasma gondii infection. Infect. Immun. 2005, 73, 617–621. [Google Scholar] [CrossRef] [Green Version]
- Guiton, R.; Vasseur, V.; Charron, S.; Arias, M.T.; Van Langendonck, N.; Buzoni-Gatel, D.; Ryffel, B.; Dimier-Poisson, I. Interleukin 17 receptor signaling is deleterious during Toxoplasma gondii infection in susceptible BL6 mice. J. Infect. Dis. 2010, 202, 427–435. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Gao, H.; Qian, Y. Th17 differentiation and their pro-inflammation function. Adv. Exp. Med. Biol. 2014, 841, 99–151. [Google Scholar] [CrossRef]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Howard, C.J.; Charleston, B.; Stephens, S.A.; Sopp, P.; Hope, J.C. The role of dendritic cells in shaping the immune response. Anim. Health Res. Rev. 2004, 5, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I. Dendritic cells: Master regulators of the immune response. Cancer Immunol. Res. 2013, 1, 145–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walseng, E.; Furuta, K.; Goldszmid, R.S.; Weih, K.A.; Sher, A.; Roche, P.A. Dendritic cell activation prevents MHC class II ubiquitination and promotes MHC class II survival regardless of the activation stimulus. J. Biol. Chem. 2010, 285, 41749–41754. [Google Scholar] [CrossRef] [Green Version]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Baravalle, G.; Park, H.; McSweeney, M.; Ohmura-Hoshino, M.; Matsuki, Y.; Ishido, S.; Shin, J.S. Ubiquitination of CD86 is a key mechanism in regulating antigen presentation by dendritic cells. J. Immunol. 2011, 187, 2966–2973. [Google Scholar] [CrossRef] [Green Version]
- Grosche, L.; Knippertz, I.; Konig, C.; Royzman, D.; Wild, A.B.; Zinser, E.; Sticht, H.; Muller, Y.A.; Steinkasserer, A.; Lechmann, M. The CD83 Molecule—An Important Immune Checkpoint. Front. Immunol. 2020, 11, 721. [Google Scholar] [CrossRef]
- Trombetta, E.S.; Mellman, I. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 2005, 23, 975–1028. [Google Scholar] [CrossRef]
- Fooksman, D.R. Organizing MHC Class II Presentation. Front. Immunol. 2014, 5, 158. [Google Scholar] [CrossRef] [Green Version]
- Fredriksen, A.B.; Sandlie, I.; Bogen, B. DNA vaccines increase immunogenicity of idiotypic tumor antigen by targeting novel fusion proteins to antigen-presenting cells. Mol. Ther. 2006, 13, 776–785. [Google Scholar] [CrossRef]
- Grodeland, G.; Mjaaland, S.; Roux, K.H.; Fredriksen, A.B.; Bogen, B. DNA vaccine that targets hemagglutinin to MHC class II molecules rapidly induces antibody-mediated protection against influenza. J. Immunol. 2013, 191, 3221–3231. [Google Scholar] [CrossRef]
- Lyons, R.E.; Anthony, J.P.; Ferguson, D.J.; Byrne, N.; Alexander, J.; Roberts, F.; Roberts, C.W. Immunological studies of chronic ocular toxoplasmosis: Up-regulation of major histocompatibility complex class I and transforming growth factor beta and a protective role for interleukin-6. Infect. Immun. 2001, 69, 2589–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colbert, J.D.; Cruz, F.M.; Rock, K.L. Cross-presentation of exogenous antigens on MHC I molecules. Curr. Opin. Immunol. 2020, 64, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int. Rev. Immunol. 2009, 28, 239–260. [Google Scholar] [CrossRef]
- Dimeloe, S.; Burgener, A.V.; Grahlert, J.; Hess, C. T-cell metabolism governing activation, proliferation and differentiation; a modular view. Immunology 2017, 150, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Qin, T.; Huang, Y.; Zheng, S.; Bo, R.; Liu, Z.; Xing, J.; Hu, Y.; Liu, J.; Wang, D. Exploring the immunopotentiation of Chinese yam polysaccharide poly(lactic-co-glycolic acid) nanoparticles in an ovalbumin vaccine formulation in vivo. Drug Deliv. 2017, 24, 1099–1111. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The multifaceted role of CD4+ T cells in CD8+ T cell memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Treatment (Each Mouse) | Time for Immunization | Time for Infection | Infection Dose (Each Mouse) |
|---|---|---|---|---|
| Blank | Equal volume of PBS | Week zero and two | Week four | 1 × 103 T. gondii for each mouse |
| Control | 100 μg pVAX1 plasmids | |||
| CS | Equal amount of chitosan nanospheres loading PBS | |||
| PLGA | Equal amount of PLGA nanospheres loading PBS | |||
| pVAX1-SIR2 | 100 μg pVAX1-SIR2 plasmids | |||
| pVAX1-SIR2/CS | PLGA nanospheres loading 100 μg pVAX1-SIR2 plasmids | |||
| pVAX1-SIR2/PLGA | Chitosan nanospheres loading 100 μg pVAX1-SIR2 plasmids |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Z.; Lu, Y.; Cao, W.; Aleem, M.T.; Liu, J.; Luo, J.; Yan, R.; Xu, L.; Song, X.; Li, X. Nano DNA Vaccine Encoding Toxoplasma gondii Histone Deacetylase SIR2 Enhanced Protective Immunity in Mice. Pharmaceutics 2021, 13, 1582. https://doi.org/10.3390/pharmaceutics13101582
Yu Z, Lu Y, Cao W, Aleem MT, Liu J, Luo J, Yan R, Xu L, Song X, Li X. Nano DNA Vaccine Encoding Toxoplasma gondii Histone Deacetylase SIR2 Enhanced Protective Immunity in Mice. Pharmaceutics. 2021; 13(10):1582. https://doi.org/10.3390/pharmaceutics13101582
Chicago/Turabian StyleYu, Zhengqing, Yujia Lu, Wandi Cao, Muhammad Tahir Aleem, Junlong Liu, Jianxun Luo, Ruofeng Yan, Lixin Xu, Xiaokai Song, and Xiangrui Li. 2021. "Nano DNA Vaccine Encoding Toxoplasma gondii Histone Deacetylase SIR2 Enhanced Protective Immunity in Mice" Pharmaceutics 13, no. 10: 1582. https://doi.org/10.3390/pharmaceutics13101582