
Drug Repurposing for the Identification of Compounds with Anti-SARS-CoV-2 Capability via Multiple Targets

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
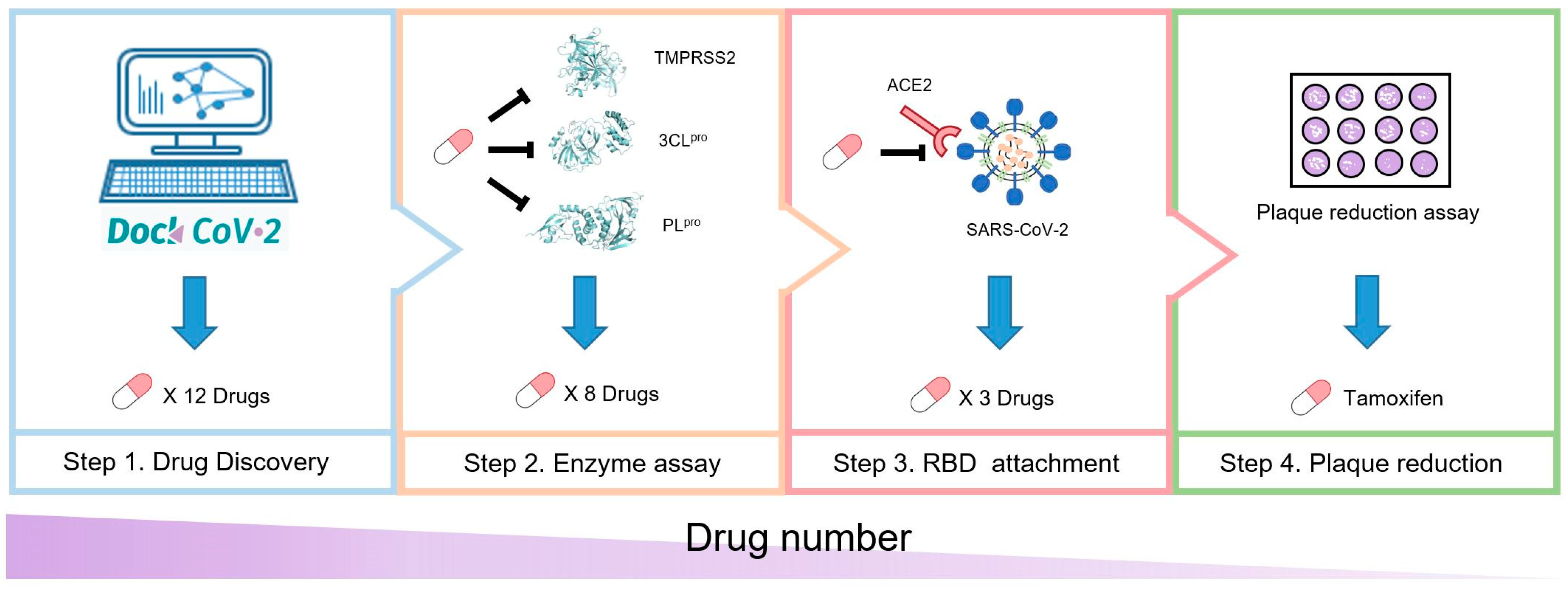
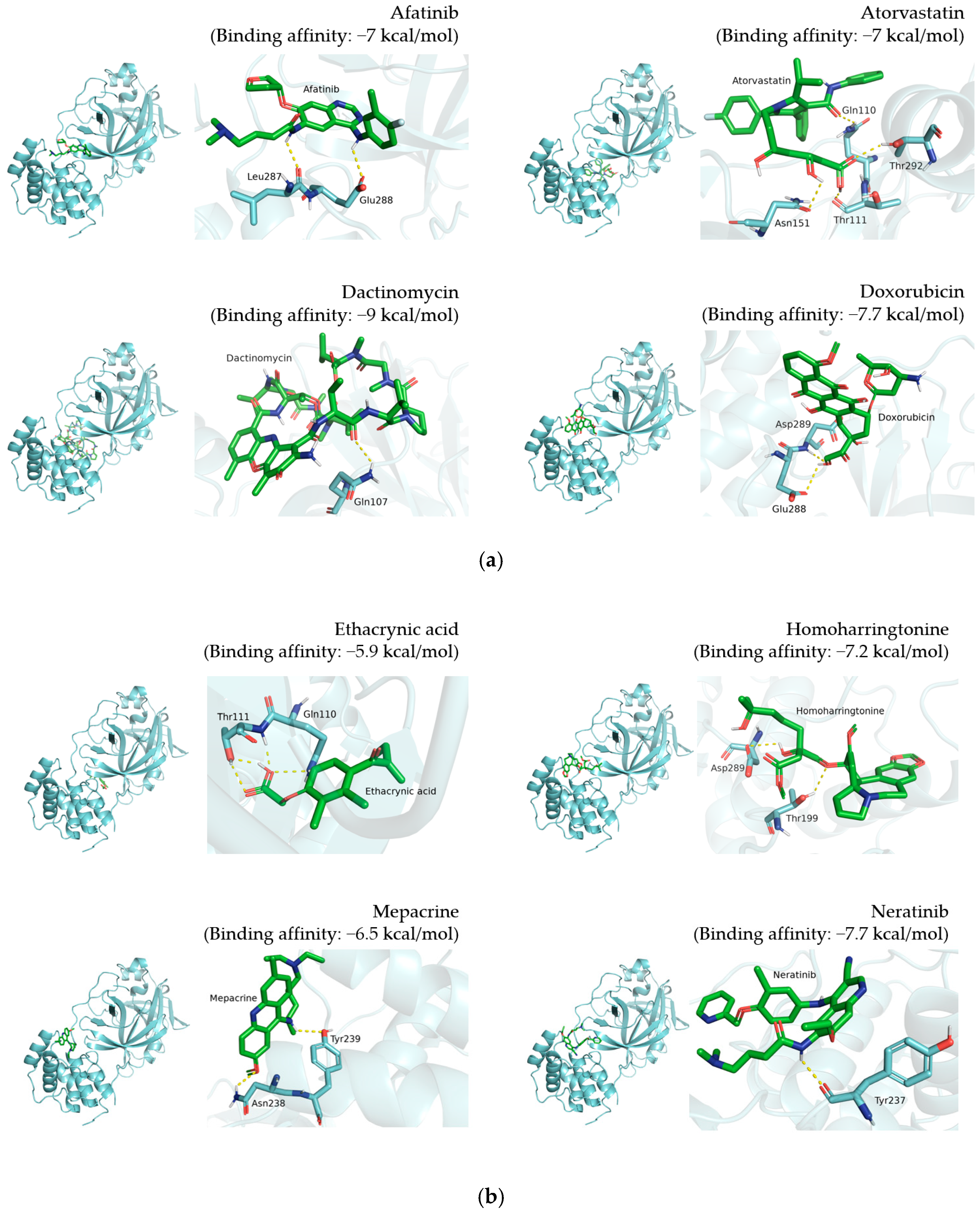
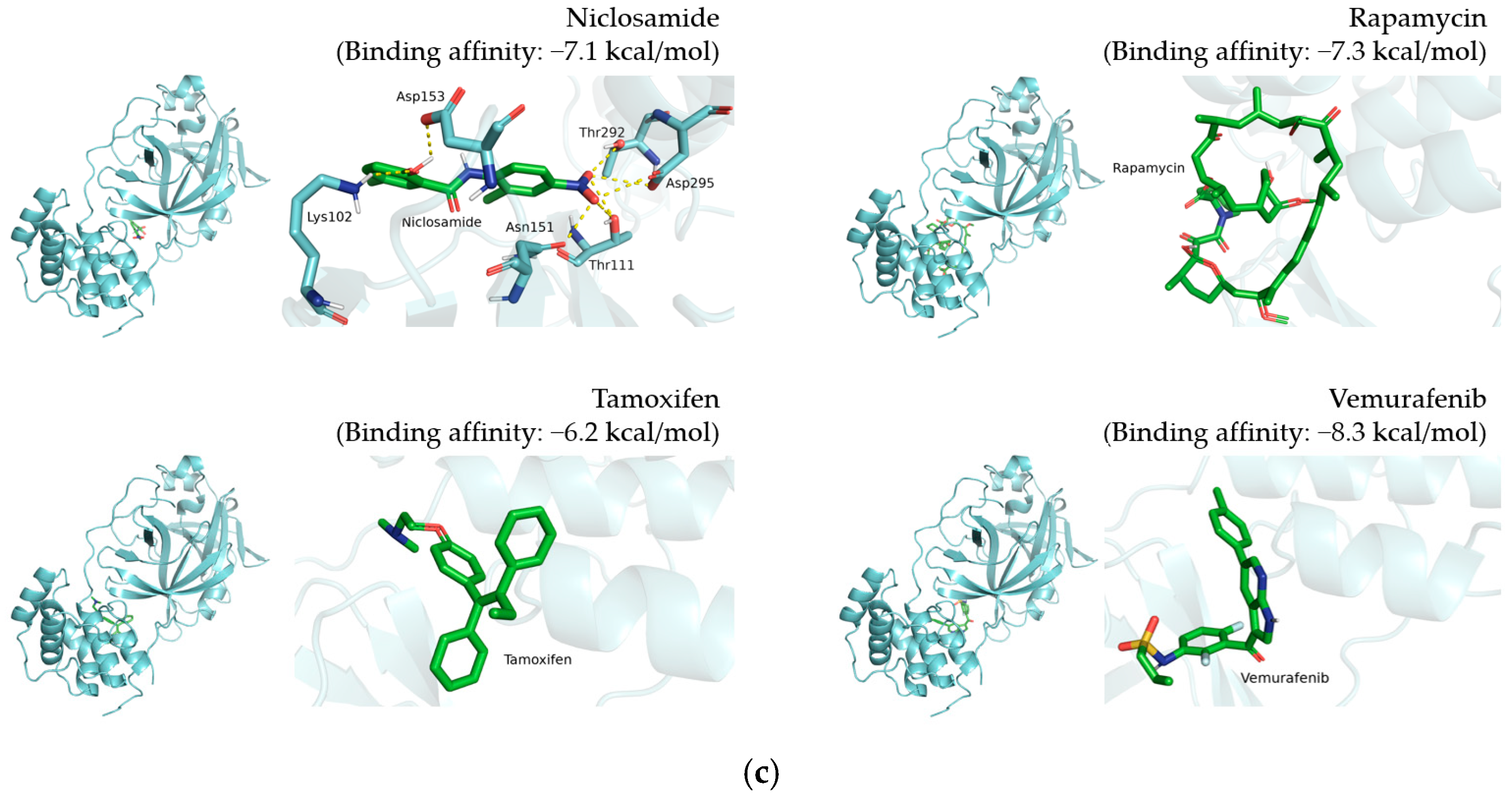
2.2.1. Analysis and Visualization of Docking Results of 12 Candidate Compounds from DockCoV2
2.2.2. Expression and Purification of SARS-CoV-2 3CLpro
2.2.3. Expression and Purification of SARS-CoV-2 PLpro
2.2.4. Inhibition Assay of 3CLpro and PLpro
2.2.5. Expression, Purification, and Inhibition Assay of Human TMPRSS2
2.2.6. RBD–ACE2 Attachment Assay
2.2.7. Virus Preparation
2.2.8. Plaque Reduction Assay
3. Results
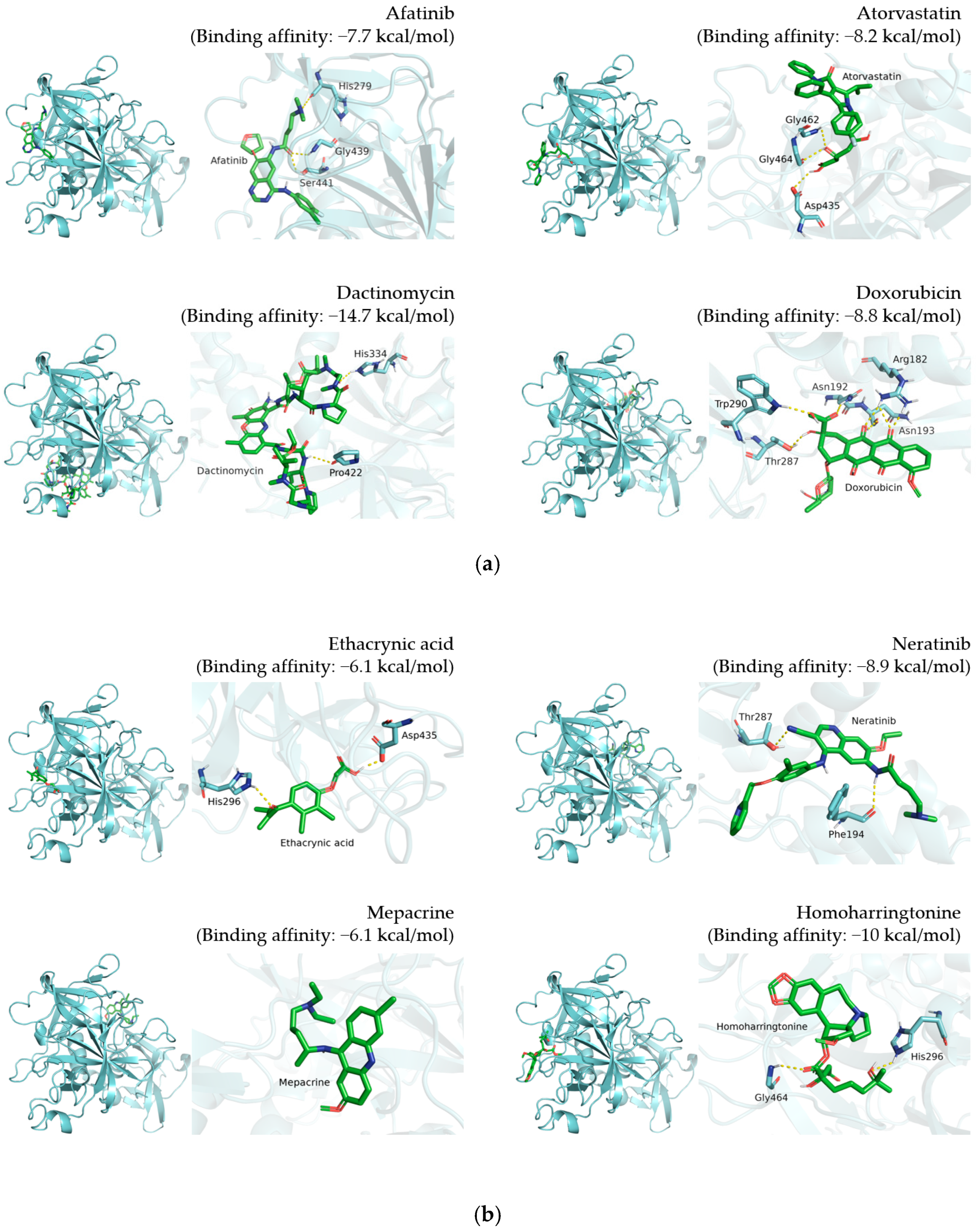
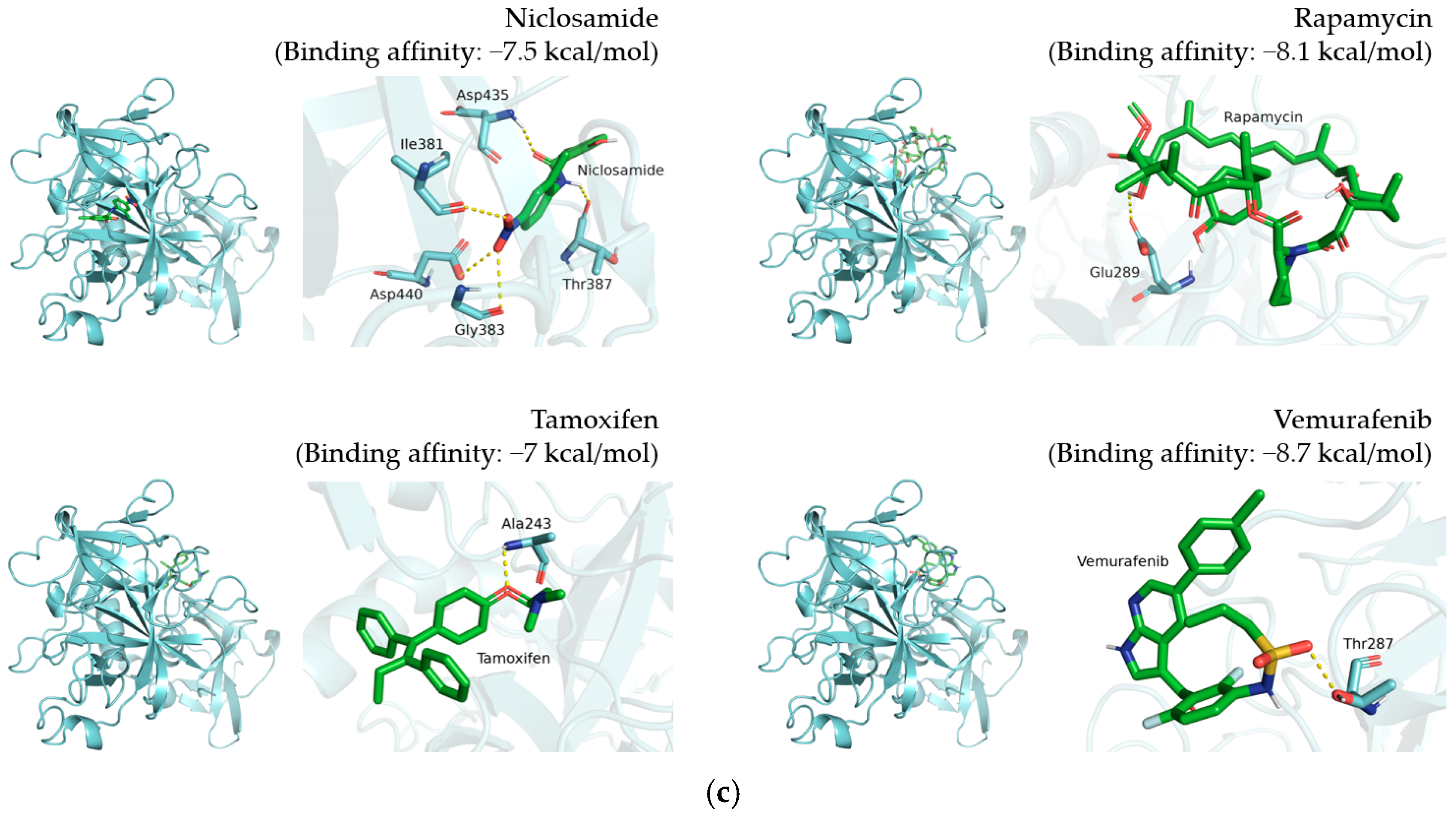
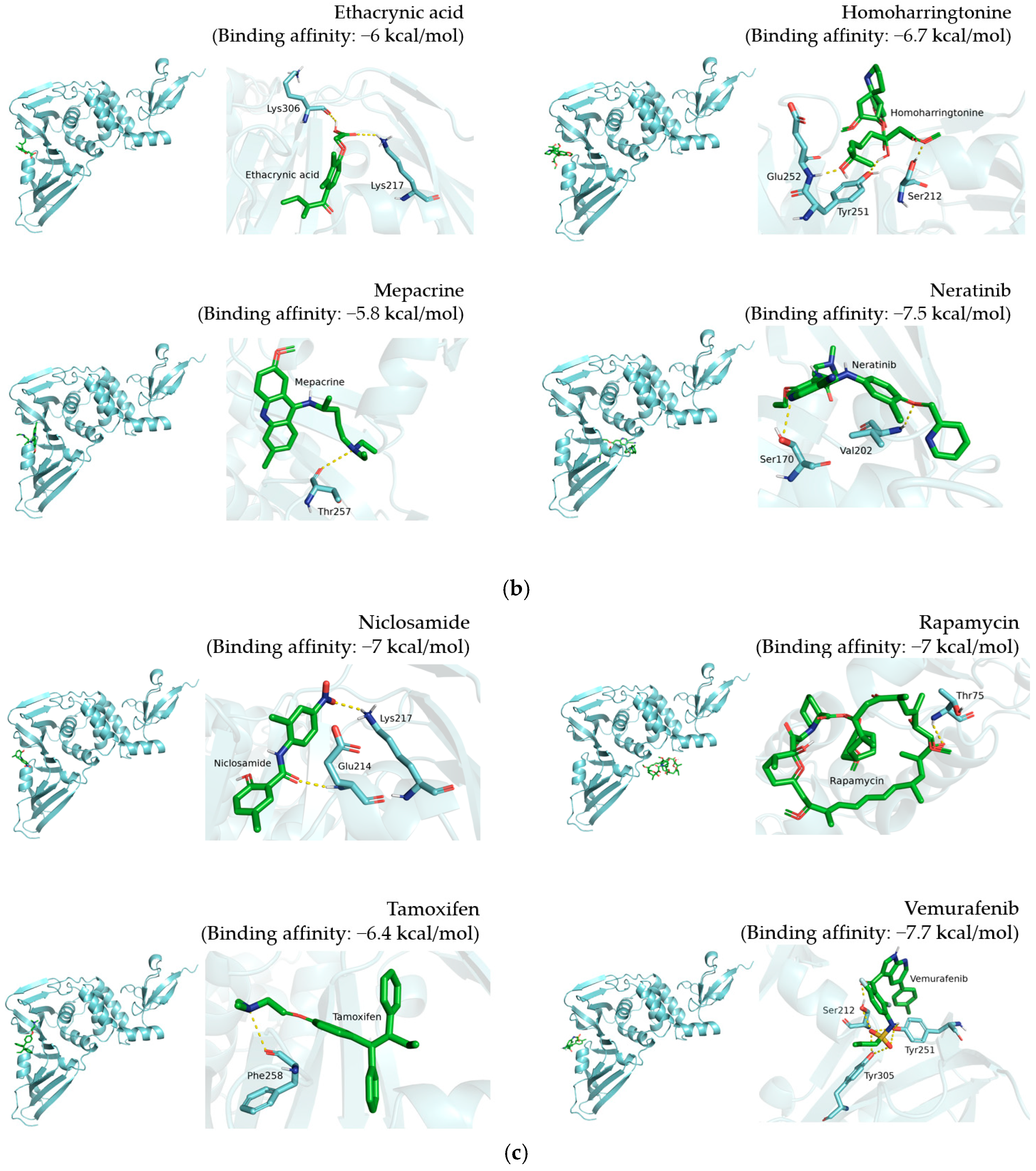
3.1. Selection of 12 Candidate Compounds and Identification of the Corresponding Interactions with TMPRSS2 Residues
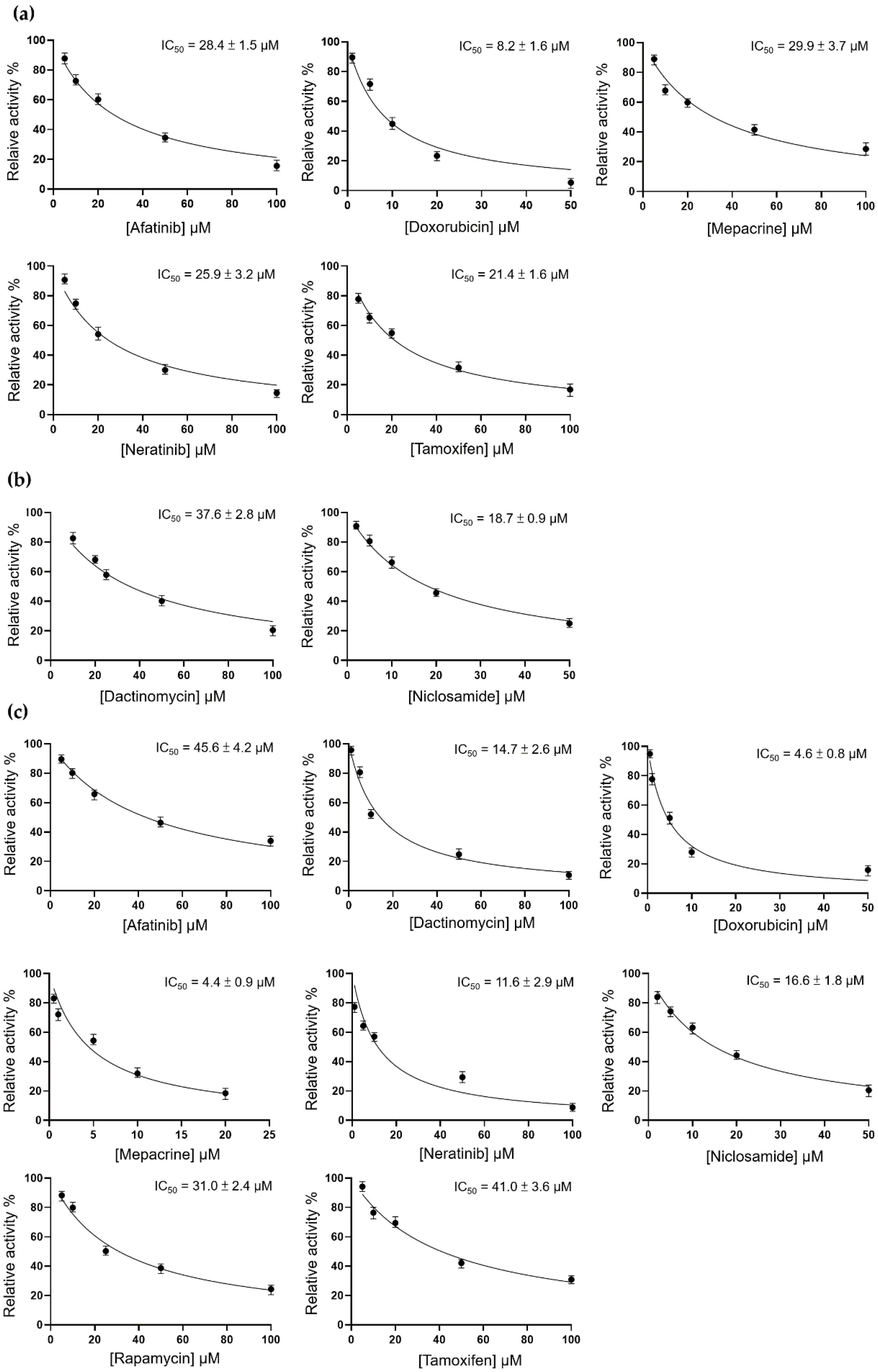
3.2. Inhibition of TMPRSS2, 3CLpro, and PLpro by 12 Candidate Compounds
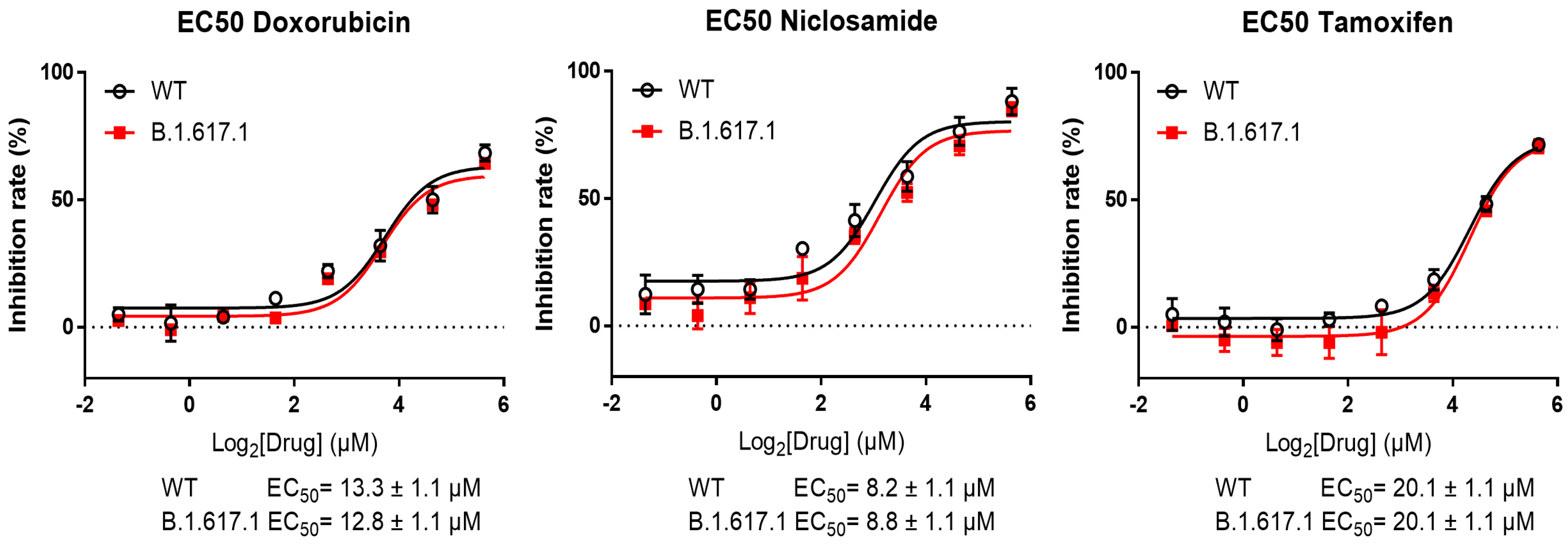
3.3. Inhibition of the Spike RBD for Wild-Type and B.1.617.1-Variant SARS-CoV-2 by Three Candidate Compounds
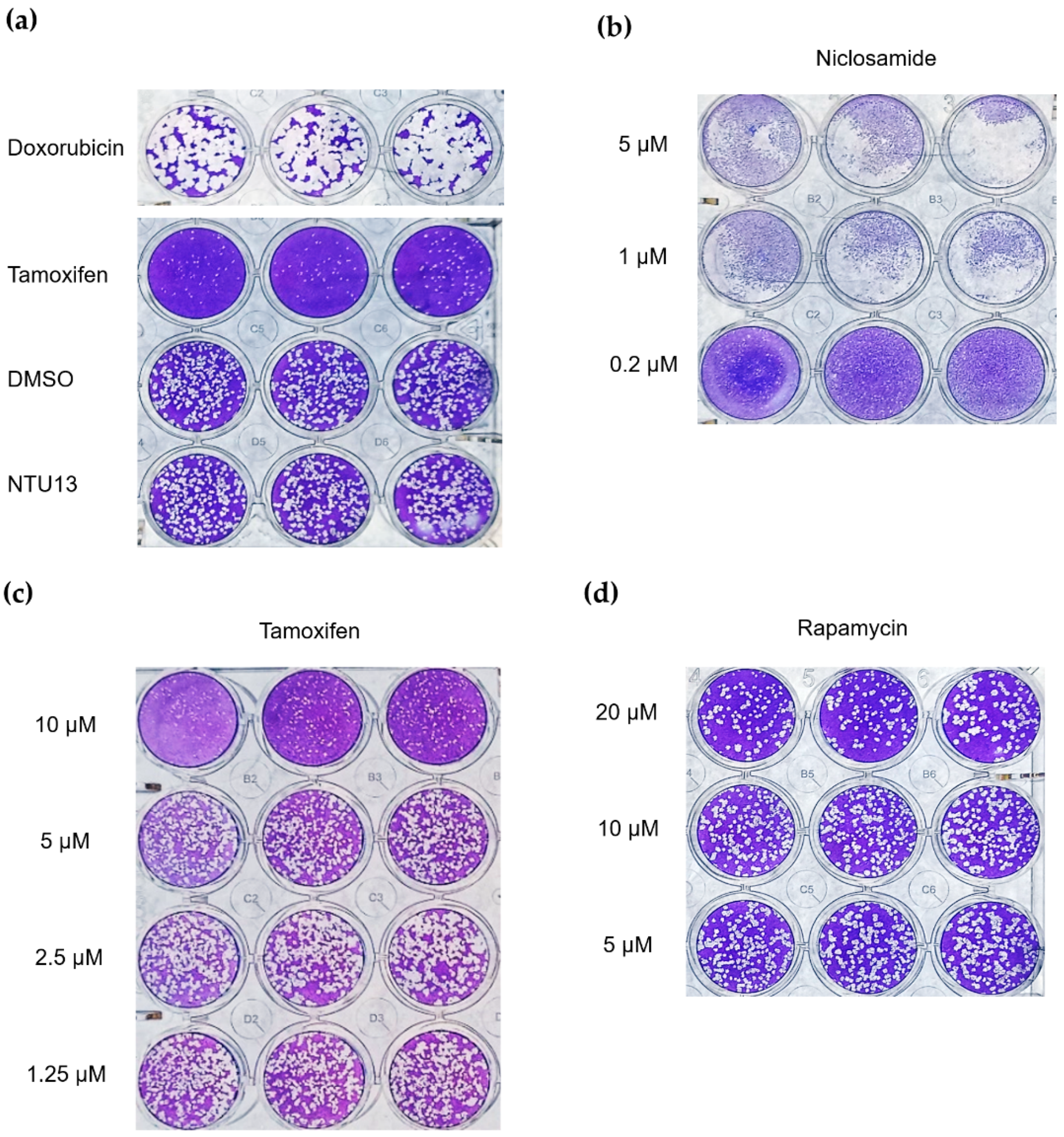
3.4. Verification with Plaque Reduction Assay for Three Candidate Compounds
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, E.J.; Rouphael, N.G.; Widge, A.T.; Jackson, L.A.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Safety and Immunogenicity of SARS-CoV-2 mRNA-1273 Vaccine in Older Adults. N. Engl. J. Med. 2020, 383, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Florindo, H.F.; Kleiner, R.; Vaskovich-Koubi, D.; Acúrcio, R.C.; Carreira, B.; Yeini, E.; Tiram, G.; Liubomirski, Y.; Satchi-Fainaro, R. Immune-mediated approaches against COVID-19. Nat. Nanotechnol. 2020, 15, 630–645. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Harcourt, B.H.; Jukneliene, D.; Kanjanahaluethai, A.; Bechill, J.; Severson, K.M.; Smith, C.M.; Rota, P.A.; Baker, S.C. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 2004, 78, 13600–13612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, B.; Goyal, D. Targeting the dimerization of the main protease of coronaviruses: A potential broad-spectrum therapeutic strategy. ACS Comb. Sci. 2020, 22, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Sharma, A.; Kumar, S.; Singh, G.; Barnwal, R.P. SARS-CoV-2: Insights into its structural intricacies and functional aspects for drug and vaccine development. Int. J. Biol. Macromol. 2021, 179, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Benítez-Cardoza, C.G.; Vique-Sánchez, J.L. Potential inhibitors of the interaction between ACE2 and SARS-CoV-2 (RBD), to develop a drug. Life Sci. 2020, 256, 117970. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Li, Y.; Han, L.; Liu, Z.; Wang, R. Comparative Assessment of Scoring Functions on an Updated Benchmark: 2. Evaluation Methods and General Results. J. Chem. Inf. Model. 2014, 54, 1717–1736. [Google Scholar] [CrossRef] [PubMed]
- Masoudi-Sobhanzadeh, Y.; Salemi, A.; Pourseif, M.M.; Jafari, B.; Omidi, Y.; Masoudi-Nejad, A. Structure-based drug repurposing against COVID-19 and emerging infectious diseases: Methods, resources and discoveries. Brief. Bioinform. 2021, 22, bbab113. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Chen, L.; Lan, R.; Shen, R.; Li, P. Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int. J. Antimicrob. Agents 2020, 56, 106012. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.F.; Chang, Y.C.; Hsiao, Y.; Lee, K.H.; Hsiao, Y.C.; Lin, Y.H.; Tu, Y.C.E.; Huang, H.C.; Chen, C.Y.; Juan, H.F. DockCoV2: A drug database against SARS-CoV-2. Nucleic Acids Res. 2021, 49, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Chen, Y.; Wang, Y.C.; Wang, W.J.; Yang, C.S.; Tsai, C.L.; Hou, M.H.; Chen, H.F.; Shen, Y.C.; Hung, M.C. Tannic acid suppresses SARS-CoV-2 as a dual inhibitor of the viral main protease and the cellular TMPRSS2 protease. Am. J. Cancer Res. 2020, 10, 4538–4546. [Google Scholar] [PubMed]
- The PyMOL Molecular Graphics System; Version 2.0; Schrödinger LLC.: New York, NY, USA, 2002.
- Kuo, C.J.; Chao, T.L.; Kao, H.C.; Tsai, Y.M.; Liu, Y.K.; Wang, L.H.; Hsieh, M.C.; Chang, S.Y.; Liang, P.H. Kinetic Characterization and Inhibitor Screening for the Proteases Leading to Identification of Drugs against SARS-CoV-2. Antimicrob. Agents Chemother. 2021, 65, e02577-20. [Google Scholar] [CrossRef]
- Bhattacharjee, M.J.; Lin, J.J.; Chang, C.Y.; Chiou, Y.T.; Li, T.N.; Tai, C.W.; Shiu, T.F.; Chen, C.A.; Chou, C.Y.; Chakraborty, P.; et al. Identifying primate ACE2 variants that confer resistance to SARS-CoV-2. Mol. Biol. Evol. 2021, 38, 2715–2731. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.T.; Chao, T.L.; Kao, H.C.; Pang, Y.H.; Lee, W.H.; Hsieh, C.H.; Chang, S.Y.; Huang, H.C.; Juan, H.F. Enhancement of the IFN-β-induced host signature informs repurposed drugs for COVID-19. Heliyon 2020, 6, e05646. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hu, L.; Huang, X.; Wang, C.; Zhang, Z.; Wang, Y.; Zhang, D.; Ye, W. Potential of coronavirus 3C-like protease inhibitors for the development of new anti-SARS-CoV-2 drugs: Insights from structures of protease and inhibitors. Int. J. Antimicrob. Agents 2020, 56, 106055. [Google Scholar] [CrossRef]
- Rajput, A.; Thakur, A.; Mukhopadhyay, A.; Kamboj, S.; Rastogi, A.; Gautam, S.; Jassal, H.; Kumar, M. Prediction of repurposed drugs for Coronaviruses using artificial intelligence and machine learning. Comput. Struct. Biotechnol. J. 2021, 19, 3133–3148. [Google Scholar] [CrossRef] [PubMed]
- Konno, S.; Kobayashi, K.; Senda, M.; Funai, Y.; Seki, Y.; Tamai, I.; Schäkel, L.; Sakata, K.; Pillaiyar, T.; Taguchi, A.; et al. 3CL Protease Inhibitors with an Electrophilic Arylketone Moiety as Anti-SARS-CoV-2 Agents. J. Med. Chem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L. 3CL Protease Inhibitor of Non-Peptide SARS Coronavirus and Use Thereof. China Patent CN 1965833, 23 May 2007. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamri, M.A.; Tahir, U.L.; Qamar, M.; Mirza, M.U.; Alqahtani, S.M.; Froeyen, M.; Chen, L.L. Discovery of human coronaviruses pan-papain-like protease inhibitors using computational approaches. J. Pharm. Anal. 2020, 10, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Pitsillou, E.; Liang, J.; Ververis, K.; Hung, A.; Karagiannis, T.C. Interaction of small molecules with the SARS-CoV-2 papa-in-like protease: In silico studies and in vitro validation of protease activity inhibition using an enzymatic inhibition assay. J. Mol. Graph. Model. 2021, 104, 107851. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Dhanjal, J.K.; Bhargava, P.; Kaul, A.; Wang, J.; Zhang, H.; Kaul, S.C.; Wadhwa, R.; Sundar, D. Withanone and Withaferin-A are predicted to interact with transmembrane protease serine 2 (TMPRSS2) and block entry of SARS-CoV-2 into cells. J. Biomol. Struct. Dyn. 2020, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Grottesi, A.; Bešker, N.; Emerson, A.; Manelfi, C.; Beccari, A.R.; Frigerio, F.; Lindahl, E.; Cerchia, C.; Talarico, C. Computational Studies of SARS-CoV-2 3CLpro: Insights from MD Simulations. Int. J. Mol. Sci. 2020, 21, 5346. [Google Scholar] [CrossRef]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B 2021, 11, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Báez-Santos, Y.M.; St John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.; Le Bas, A.; Ruza, R.R.; Duyvesteyn, H.M.E.; Mikolajek, H.; Malinauskas, T.; Tan, T.K.; Rijal, P.; Dumoux, M.; Ward, P.N.; et al. Neutralizing nanobodies bind SARS-CoV-2 spike RBD and block interaction with ACE2. Nat. Struct. Mol. Biol. 2020, 27, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Bojadzic, D.; Alcazar, O.; Chen, J.; Chuang, S.T.; Condor Capcha, J.M.; Shehadeh, L.A.; Buchwald, P. Small-Molecule Inhibitors of the Coronavirus Spike: ACE2 Protein–Protein Interaction as Blockers of Viral Attachment and Entry for SARS-CoV-2. ACS Infect. Dis. 2021, 7, 1519–1534. [Google Scholar] [CrossRef]
- Baird, J.K. Resistance to chloroquine unhinges vivax malaria therapeutics. Antimicrob. Agents Chemother. 2011, 55, 1827–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Mukherjee, U. A comprehensive review of immunosuppression used for liver transplantation. J. Transplant. 2009, 2009, 701464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skeel, R.T. Handbook of Cancer Chemotherapy, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2003; pp. 2–831. [Google Scholar]
- Barrett, P.N.; Terpening, S.J.; Snow, D.; Cobb, R.R.; Kistner, O. Vero cell technology for rapid development of inactivated whole virus vaccines for emerging viral diseases. Expert Rev. Vaccines 2017, 16, 883–894. [Google Scholar] [CrossRef]
- Rhim, J.S.; Schell, K.; Creasy, B.; Case, W. Biological Characteristics and Viral Susceptibility of an African Green Monkey Kidney Cell Line (Vero). Proc. Soc. Exp. Biol. Med. 1969, 132, 670–678. [Google Scholar] [CrossRef]
- Desmyter, J.; Melnick, J.L.; Rawls, W.E. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J. Virol. 1968, 2, 955–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spruth, M.; Kistner, O.; Savidis-Dacho, H.; Hitter, E.; Crowe, B.; Gerencer, M.; Brühl, P.; Grillberger, L.; Reiter, M.; Tauer, C.; et al. A double-inactivated whole virus candidate SARS coronavirus vaccine stimulates neutralising and protective antibody responses. Vaccine 2006, 24, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hou, Y.; Huang, J.; Pan, W.; Ma, Q.; Shi, Y.; Li, C.; Zhao, J.; Jia, Z.; Jiang, H.; et al. Lianhuaqingwen exerts anti-viral and anti-inflammatory activity against novel coronavirus (SARS-CoV-2). Pharmacol. Res. 2020, 156, 104761. [Google Scholar]
- Liu, J.; Liu, Y.; Xia, H.; Zou, J.; Weaver, S.C.; Swanson, K.A.; Cai, H.; Cutler, M.; Cooper, D.; Muik, A.; et al. BNT162b2-elicited neutralization of B.1.617 and other SARS-CoV-2 variants. Nature 2021, 596, 273–275. [Google Scholar] [CrossRef]
- Marín-Palma, D.; Tabares-Guevara, J.H.; Zapata-Cardona, M.I.; Flórez-Álvarez, L.; Yepes, L.M.; Rugeles, M.T.; Zapata-Builes, W.; Hernandez, J.C.; Taborda, N.A. Curcumin Inhibits In Vitro SARS-CoV-2 Infection in Vero E6 Cells through Multiple Antiviral Mechanisms. Molecules 2021, 26, 6900. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulos, A.A.; Karakasiliotis, I.; Kotzampasi, D.M.; Dimitriou, M.; Sourvinos, G.; Kampa, M.; Pirintsos, S.; Castanas, E.; Daskalakis, V. Natural Polyphenols Inhibit the Dimerization of the SARS-CoV-2 Main Protease: The Case of Fortunellin and Its Structural Analogs. Molecules 2021, 26, 6068. [Google Scholar] [CrossRef] [PubMed]
- Alaaeldin, R.; Mustafa, M.; Abuo-Rahma, G.E.A.; Fathy, M. In vitro inhibition and molecular docking of a new ciprofloxacin-chalcone against SARS-CoV-2 main protease. Fundam. Clin. Pharmacol. 2021, 7, 1–11. [Google Scholar]
- Georgakis, G.V.; Younes, A. From Rapa Nui to Rapamycin: Targeting PI3K/Akt/MTOR for Cancer Therapy. Expert Rev. Anticancer Ther. 2006, 6, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Seto, B. Rapamycin and mTOR: A serendipitous discovery and implications for breast cancer. Clin. Transl Med. 2012, 1, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Wang, X.; Proud, C.G. mTOR Inhibitors in Cancer Therapy. F1000Res 2016, 5, 2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Sage, V.; Cinti, A.; Amorim, R.; Mouland, A.J. Adapting the stress response: Viral subversion of the mTOR signaling pathway. Viruses 2016, 8, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Li, R.; Liu, S. Immunoregulation with mTOR Inhibitors to Prevent COVID-19 Severity: A Novel Intervention Strategy beyond Vaccines and Specific Antiviral Medicines. J. Med. Virol. 2020, 92, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H. Antiviral Potential of ERK/MAPK and PI3K/AKT/MTOR Signaling Modulation for Middle East Respiratory Syndrome Coronavirus Infection as Identified by Temporal Kinome Analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yang, F.; Zou, S.; Qu, L. Rapamycin: A Bacteria-Derived Immunosuppressant That Has Anti-Atherosclerotic Effects and Its Clinical Application. Front. Pharmacol. 2019, 9, 1520. [Google Scholar] [CrossRef] [PubMed]
- Pindiprolu, S.H.; Pindiprolu, S.K.S.S. CD133 receptor mediated delivery of STAT3 inhibitor for simultaneous elimination of cancer cells and cancer stem cells in oral squamous cell carcinoma. Med. Hypotheses 2019, 129, 109241. [Google Scholar] [CrossRef]
- Xu, J.; Shi, P.Y.; Li, H.; Zhou, J. Broad Spectrum Antiviral Agent Niclosamide and Its Therapeutic Potential. ACS Infect. Dis. 2020, 6, 909–915. [Google Scholar] [CrossRef]
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S.Y.; Park, S.; Shum, D.; Kim, S. Identification of Antiviral Drug Candidates against SARS-CoV-2 from FDA-Approved Drugs. Antimicrob. Agents Chemother. 2020, 64, e00819-20. [Google Scholar] [CrossRef] [PubMed]
- Jurgeit, A.; McDowell, R.; Moese, S.; Meldrum, E.; Schwendener, R.; Greber, U.F. Niclosamide is a proton carrier and targets acidic endosomes with broad antiviral effects. PLoS Pathog. 2012, 8, e1002976. [Google Scholar] [CrossRef] [Green Version]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mosbauer, K.; Zellner, A.; et al. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat. Commun. 2019, 10, 5770. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Weiss, R.B. The anthracyclines: Will we ever find a better doxorubicin? Semin. Oncol. 1992, 19, 670–686. [Google Scholar]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef]
- Jamal, Q.M.S.; Ahmad, V.; Alharbi, A.H.; Ansari, M.A.; Alzohairy, M.A.; Almatroudi, A.; Alghamdi, S.; Alomary, M.N.; AlYahya, S.; Shesha, N.T.; et al. Therapeutic development by repurposing drugs targeting SARS-CoV-2 spike protein interactions by simulation studies. Saudi J. Biol. Sci. 2021, 28, 4560–4568. [Google Scholar] [CrossRef] [PubMed]
- Al-Motawa, M.S.; Abbas, H.; Wijten, P.; de la Fuente, A.; Xue, M.; Rabbani, N.; Thornalley, P.J. Vulnerabilities of the SARS-CoV-2 Virus to Proteotoxicity-Opportunity for Repurposed Chemotherapy of COVID-19 Infection. Front. Pharmacol. 2020, 11, 585408. [Google Scholar] [CrossRef] [PubMed]
- Maximov, P.Y.; McDaniel, R.E.; Jordan, V.C. Tamoxifen: Pioneering Medicine in Breast Cancer; Springer: Basel, Switzerland, 2013. [Google Scholar]
- World Health Organization. World Health Organization Model List of Essential Medicines: 21st List 2019; World Health Organization: Geneva, Switzerland, 2019; Volume 55. [Google Scholar]
- Ingvarsson, S. Breast cancer: Introduction. Semin. Cancer Biol. 2001, 11, 323–326. [Google Scholar] [CrossRef] [Green Version]
- Mandeville, R.; Ghali, S.S.; Chausseau, J.P. In vitro stimulation of human NK activity by estrogen antagonist (tamoxifen). Eur. J. Cancer Clin. Oncol. 1984, 20, 983–985. [Google Scholar] [CrossRef]
- Baral, E.; Nagy, E.; Krepart, G.V.; Lotocki, R.J.; Unruh, H.W.; Berczi, I. Antiestrogens sensitize human ovarian and lung carcinomas for lysis by autologous killer cells. Anticancer Res. 2000, 20, 2027–2031. [Google Scholar]
- Montoya, M.C.; Krysan, D.J. Repurposing estrogen receptor antagonists for the treatment of infectious disease. mBio 2018, 9, e02272-18. [Google Scholar] [CrossRef] [Green Version]
- O’Brian, C.A.; Liskamp, R.M.; Solomon, D.H.; Weinstein, I.B. Inhibition of protein kinase C by tamoxifen. Cancer Res. 1985, 45, 2462–2465. [Google Scholar] [PubMed]
- Bekele, R.T.; Venkatraman, G.; Liu, R.Z.; Tang, X.; Mi, S.; Benesch, M.G.; Mackey, J.R.; Godbout, R.; Curtis, J.M.; McMullen, T.P.; et al. Oxidative stress contributes to the tamoxifen-induced killing of breast cancer cells: Implications for tamoxifen therapy and resistance. Sci. Rep. 2016, 6, 21164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangerini, R.; Argellati, F.; Pfeffer, U.; Boccardo, F. Effects of bicalutamide and 4OH-tamoxifen on androgen-regulated gene expression in the LNCaP cell line. Anticancer Res. 2012, 32, 5323–5329. [Google Scholar] [PubMed]
- Karamouzis, M.V.; Papavassiliou, K.A.; Adamopoulos, C.; Papavassiliou, A.G. Targeting androgen/estrogen receptors crosstalk in cancer. Trends Cancer 2016, 2, 35–48. [Google Scholar] [CrossRef]
- Kim, H.; Datta, A.; Talwar, S.; Saleem, S.N.; Mondal, D.; Abdel-Mageed, A.B. Estradiol-ERβ2 signaling axis confers growth and migration of CRPC cells through TMPRSS2-ETV5 gene fusion. Oncotarget 2016, 8, 62820–62833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daurio, N.A.; Tuttle, S.W.; Worth, A.J.; Song, E.Y.; Davis, J.M.; Snyder, N.W.; Blair, I.A.; Koumenis, C. AMPK activation and metabolic reprogramming by tamoxifen through estrogen receptor-independent mechanisms suggests new uses for this therapeutic modality in cancer treatment. Cancer Res. 2016, 76, 3295–3306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Center for Biotechnology Information. PubChem Database. Tamoxifen, CID = 2733526. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Tamoxifen (accessed on 15 November 2021).
- Mesange, F.; Delarue, F.; Puel, J.; Bayard, F.; Faye, J.C. Ligands of the antiestrogen-binding site are able to inhibit virion production of human immunodeficiency virus 1-infected lymphocytes. Mol. Pharmacol. 1996, 50, 75–79. [Google Scholar] [PubMed]
- Watashi, K.; Inoue, D.; Hijikata, M.; Goto, K.; Aly, H.H.; Shimotohno, K. Anti-hepatitis C virus activity of tamoxifen reveals the functional association of estrogen receptor with viral RNA polymerase NS5B. J. Biol. Chem. 2007, 282, 32765–32772. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Seifert, M.; Xu, Y.; Hock, B. Comparative study of estrogenic potencies of estradiol, tamoxifen, bisphenol-A and resveratrol with two in vitro bioassays. Environ. Int. 2004, 30, 329–335. [Google Scholar] [CrossRef]
- Jing, G.; Yuan, K.; Turk, A.N.; Jhala, N.C.; Arnoletti, J.P.; Zhang, K.; McDonald, J.M.; Chen, Y. Tamoxifen enhances therapeutic effects of gemcitabine on cholangiocarcinoma tumorigenesis. Lab. Investig. 2011, 91, 896–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francavilla, A.M.D.; Polimeno, L.; Dileo, A.; Barone, M.; Ove, P.; Coetzee, M.; Eagon, P.; Makowka, L.; Ambrosino, G.; Mazzaferro, V.; et al. The effect of estrogen and tamoxifen on hepatocyte proliferation in Vivo and in Vitro. Hepatology 1989, 9, 614–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morello, K.C.; Wurz, G.T.; DeGregorio, M.W. Pharmacokinetics of selective estrogen receptor modulators. Clin. Pharmacokinet. 2003, 42, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Cusack, B.J.; Young, S.P.; Driskell, J.; Olson, R.D. Doxorubicin and doxorubicinol pharmacokinetics and tissue concentrations following bolus injection and continuous infusion of doxorubicin in the rabbit. Cancer Chemother. Pharmacol. 1993, 32, 53–58. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Compound Structure | TMPRSS2 Residues Involved in Hydrogen Interaction |
|---|---|---|
| Afatinib | 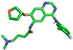 | His279, Gly439, Ser441 |
| Atorvastatin |  | Asp435, Gly462, Gly464 |
| Dactinomycin | 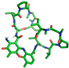 | His334, Pro422 |
| Doxorubicin | 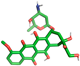 | Arg182, Asn192, Asn193, Thr287, Trp290 |
| Ethacrynic acid |  | His296, Asp435 |
| Homoharringtonine |  | His296, Gly464 |
| Mepacrine |  | None |
| Neratinib |  | Phe194, Thr287 |
| Niclosamide |  | Ile381, Gly383, Thr387, Asp435, Asp440 |
| Rapamycin |  | Glu289 |
| Tamoxifen |  | Ala243 |
| Vemurafenib |  | Thr287 |
| Compound Name | Compound Structure | PLpro Residues Involved in Hydrogen Interaction |
|---|---|---|
| Afatinib |  | Glu214, Thr257, Thr259, Lys306 |
| Atorvastatin |  | Tyr213, Glu214, Thr257 |
| Dactinomycin |  | Thr74, Thr75, Asn128, Gln174, His175 |
| Doxorubicin |  | Glu214, Lys217, Thr259, Lys306, Ser309 |
| Ethacrynic acid |  | Lys217, Lys306 |
| Homoharringtonine | 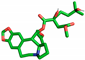 | Ser212, Tyr251, Glu252 |
| Mepacrine | 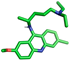 | Thr257 |
| Neratinib | 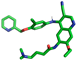 | Ser170, Val202 |
| Niclosamide |  | Glu214, Lys217 |
| Rapamycin | 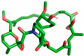 | Thr75 |
| Tamoxifen |  | Phe258 |
| Vemurafenib |  | Ser212, Tyr251, Tyr305 |
| Compound Name | Compound Structure | 3CLpro Residues Involved in Hydrogen Interaction |
|---|---|---|
| Afatinib |  | Leu287, Glu288 |
| Atorvastatin |  | Gln110, Thr111, Asn151, Thr292 |
| Dactinomycin |  | Gln107 |
| Doxorubicin |  | Glu288, Asp289 |
| Ethacrynic acid |  | Gln110, Thr111 |
| Homoharringtonine | 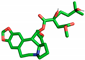 | Thr199, Asp289 |
| Mepacrine | 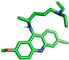 | Asn238, Tyr239 |
| Neratinib |  | Tyr237 |
| Niclosamide |  | Lys102, Thr111, Asn151, Asp153, Thr292, Asp295 |
| Rapamycin | 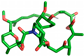 | None |
| Tamoxifen |  | None |
| Vemurafenib | 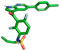 | None |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, P.-C.; Huang, C.-H.; Kuo, C.-J.; Liang, P.-H.; Wang, L.H.-C.; Pan, M.Y.-C.; Chang, S.-Y.; Chao, T.-L.; Ieong, S.-M.; Fang, J.-T.; et al. Drug Repurposing for the Identification of Compounds with Anti-SARS-CoV-2 Capability via Multiple Targets. Pharmaceutics 2022, 14, 176. https://doi.org/10.3390/pharmaceutics14010176
Yu P-C, Huang C-H, Kuo C-J, Liang P-H, Wang LH-C, Pan MY-C, Chang S-Y, Chao T-L, Ieong S-M, Fang J-T, et al. Drug Repurposing for the Identification of Compounds with Anti-SARS-CoV-2 Capability via Multiple Targets. Pharmaceutics. 2022; 14(1):176. https://doi.org/10.3390/pharmaceutics14010176
Chicago/Turabian StyleYu, Pei-Chen, Chen-Hao Huang, Chih-Jung Kuo, Po-Huang Liang, Lily Hui-Ching Wang, Max Yu-Chen Pan, Sui-Yuan Chang, Tai-Ling Chao, Si-Man Ieong, Jun-Tung Fang, and et al. 2022. "Drug Repurposing for the Identification of Compounds with Anti-SARS-CoV-2 Capability via Multiple Targets" Pharmaceutics 14, no. 1: 176. https://doi.org/10.3390/pharmaceutics14010176