
Synthesis & Evaluation of Novel Mannosylated Neoglycolipids for Liposomal Delivery System Applications

 , ,
, , 
Abstract
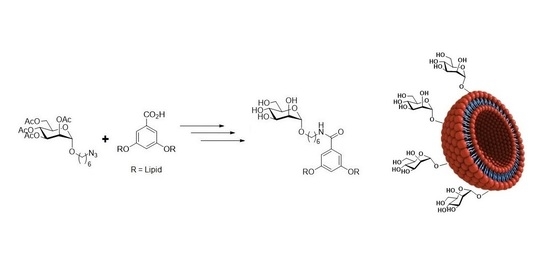
:
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. General Methods
2.1.2. Dynamic Light Scattering
2.1.3. Transmission Electron Microscopy
2.1.4. Chemical Syntheses
General Procedure for methyl 3,5-bis(alkyloxy)benzoates (3-7)-Method A
General Procedure for Hydrolysis of Methyl Ester (8–12)-Method B
General Procedure for the Carbohydrate Synthesis-Method C
General Procedure for Synthesis of Mannolipids (16–20)-Method D
General Procedure for de-O-Acetylation of Compounds (21–25)-Method E
2.2. Biology
2.2.1. Reagents
2.2.2. Human Plasma Collection
2.2.3. HPLC Conditions
2.2.4. Entrapment
2.2.5. Plasma Stability Analysis
2.2.6. Evaluation of Hemolysis
2.2.7. Cell Culture and Nile Red Uptake
2.2.8. Cytokine Analysis
2.2.9. Statistical Analyses
3. Results & Discussion
3.1. Syntheses & Characterization of Neoglycolipids
3.1.1. Synthesis of the Lipid Architecture
3.1.2. Synthesis of Mannosylated Neoglycolipids
3.1.3. Mannosylated Neoglycolipid Self-Assembly Analysis by DLS and TEM
3.1.4. Binding & Agglutination Abilities of Mannoliposomes with Con A
3.2. Biological Evaluation
3.2.1. Cellular Uptake of Mannosylated Liposomes
3.2.2. Preliminary Safety Assessments of Mannosylated Glycoliposomes
3.2.3. Mannosylated Liposomal Entrapment of Dynantin & Protection against Plasma Degradation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sapna Kumari, S.; Goyal, A.; Gürer, E.S.; Yapar, E.A.; Garg, M.; Sood, M.; Sindhu, R.K. Bioactive loaded novel nano-formulations for targeted drug delivery and their therapeutic potential. Pharmaceutics 2022, 14, 1091. [Google Scholar] [CrossRef] [PubMed]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharm. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.I.; Yeh, M.K. Clinical development of liposome-based drugs: Formulation, characterization, and therapeutic efficacy. Int. J. Nanomed. 2012, 7, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Dis. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Boge, L.; Västberg, A.; Umerska, A.; Bysell, H.; Eriksson, J.; Edwards, K.; Millqvist-Fureby, A.; Andersson, M. Freeze-dried and re-hydrated liquid crystalline nanoparticles stabilized with disaccharides for drug-delivery of the plectasin derivative AP114 antimicrobial peptide. J. Colloid Interface Sci. 2018, 522, 126–135. [Google Scholar] [CrossRef]
- Lai, C.H.; Hütter, J.; Hsu, C.W.; Tanaka, H.; Varela-Aramburu, S.; De Cola, L.; Lepenies, B.; Seeberger, P.H. Analysis of Carbohydrate-Carbohydrate Interactions Using Sugar-Functionalized Silicon Nanoparticles for Cell Imaging. Nano Lett. 2016, 16, 807–811. [Google Scholar] [CrossRef]
- Asthana, G.S.; Asthana, A.; Kohli, D.V.; Vyas, S.P. Mannosylated chitosan nanoparticles for delivery of antisense oligonucleotides for macrophage targeting. BioMed Res. Int. 2014, 2014, 526391. [Google Scholar] [CrossRef] [Green Version]
- Carrillo-Conde, B.; Song, E.H.; Chavez-Santoscoy, A.; Phanse, Y.; Ramer-Tait, A.E.; Pohl, N.L.; Wannemuehler, M.J.; Bellaire, B.H.; Narasimhan, B. Mannose-functionalized “pathogen-like” polyanhydride nanoparticles target C-type lectin receptors on dendritic cells. Mol. Pharm. 2011, 8, 1877–1886. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.; Sarmento, B.; Seabra, V. Mannose-functionalized solid lipid nanoparticles are effective in targeting alveolar macrophages. Eur. J. Pharm. Sci. 2018, 114, 103–113. [Google Scholar] [CrossRef]
- Štimac, A.; Cvitaš, J.T.; Frkanec, L.; Vugrek, O.; Frkanec, R. Design and syntheses of mono and multivalent mannosyl-lipoconjugates for targeted liposomal drug delivery. Int. J. Pharm. 2016, 511, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.C.; Chaves, L.L.; Pinheiro, M.; Ferreira, D.; Sarmento, B.; Reis, S. Design and statistical modeling of mannose-decorated dapsone-containing nanoparticles as a strategy of targeting intestinal M-cells. Int. J. Nanomed. 2016, 11, 2601–2617. [Google Scholar] [CrossRef] [Green Version]
- Vieira, A.C.; Chaves, L.L.; Pinheiro, M.; Lima, S.A.C.; Ferreira, D.; Sarmento, B.; Reis, S. Mannosylated solid lipid nanoparticles for the selective delivery of rifampicin to macrophages. Artif. Cells Nanomed. Biotechnol. 2018, 46 (Suppl. S1), 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijagkanalan, W.; Kawakami, S.; Takenaga, M.; Igarashi, R.; Yamashita, F.; Hashida, M. Efficient targeting to alveolar macrophages by intratracheal administration of mannosylated liposomes in rats. J. Control. Release 2008, 125, 121–130. [Google Scholar] [CrossRef]
- Ye, Z.; Zhang, Q.; Wang, S.; Bharate, P.; Varela-Aramburu, S.; Lu, M.; Seeberger, P.H.; Yin, J. Tumour-Targeted Drug Delivery with Mannose-Functionalized Nanoparticles Self-Assembled from Amphiphilic β-Cyclodextrins. Chemistry 2016, 22, 15216–15221. [Google Scholar] [CrossRef]
- Régnier-Vigouroux, A. The mannose receptor in the brain. Int. Rev. Cytol. 2003, 226, 321–342. [Google Scholar] [CrossRef]
- Umezawa, F.; Eto, Y. Liposome targeting to mouse brain: Mannose as a recognition marker. Biochem. Biophys. Res. Commun. 1988, 153, 1038–1044. [Google Scholar] [CrossRef]
- Du, D.; Chang, N.; Sun, S.; Li, M.; Yu, H.; Liu, M.; Liu, X.; Wang, G.; Li, H.; Liu, X.; et al. The role of glucose transporters in the distribution of p-aminophenyl-α-D-mannopyranoside modified liposomes within mice brain. J. Control. Release 2014, 182, 99–110. [Google Scholar] [CrossRef]
- Irache, J.M.; Salman, H.H.; Gamazo, C.; Espuelas, S. Mannose-targeted systems for the delivery of therapeutics. Expert Opin. Drug Deliv. 2008, 5, 703–724. [Google Scholar] [CrossRef]
- Ghosh, S.; Das, N.; Mandal, A.K.; Dungdung, S.R.; Sarkar, S. Mannosylated liposomal cytidine 5' diphosphocholine prevent age related global moderate cerebral ischemia reperfusion induced mitochondrial cytochrome c release in aged rat brain. Neuroscience 2010, 171, 1287–1299. [Google Scholar] [CrossRef]
- Li, X.Y.; Zhao, Y.; Sun, M.G.; Shi, J.F.; Ju, R.J.; Zhang, C.X.; Li, X.T.; Zhao, W.Y.; Mu, L.M.; Zeng, F.; et al. Multifunctional liposomes loaded with paclitaxel and artemether for treatment of invasive brain glioma. Biomaterials 2014, 35, 5591–5604. [Google Scholar] [CrossRef] [PubMed]
- Goyard, D.; Shiao, T.C.; Fraleigh, N.L.; Vu, H.Y.; Lee, H.; Diaz-Mitoma, F.; Le, H.-T.; Roy, R. Expedient synthesis of functional single-component glycoliposomes using thiol-yne chemistry. J. Mater. Chem. B 2016, 4, 4227–4233. [Google Scholar] [CrossRef] [PubMed]
- Lewicky, J.D.; Martel, A.L.; Fraleigh, N.L.; Boraman, A.; Nguyen, T.M.; Schiller, P.W.; Shiao, T.C.; Roy, R.; Le, H.-T. Strengthening peptide-based drug activity with novel glyconanoparticle. PLoS ONE 2018, 3, e0204472. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Nguyen, T.M.; Weltrowska, G.; Berezowska, I.; Lemieux, C.; Chung, N.N.; Schiller, P.W. [2’,6’-Dimethyltyrosine]dynorphin A(1-11)-NH2 analogues lacking an N-terminal amino group: Potent and selective kappa opioid antagonists. J. Med. Chem. 2001, 44, 3048–3053. [Google Scholar] [CrossRef]
- Lewicky, J.D.; Fraleigh, N.L.; Boraman, A.; Martel, A.L.; Nguyen, T.M.; Schiller, P.W.; Shiao, T.C.; Roy, R.; Montaut, S.; Le, H.-T. Mannosylated glycoliposomes for the delivery of a peptide kappa opioid receptor antagonist to the brain. Eur. J. Pharm. Biopharm. 2020, 154, 290–296. [Google Scholar] [CrossRef]
- Lewicky, J.D.; Fraleigh, N.L.; Martel, A.L.; Nguyen, T.M.; Schiller, P.W.; Mousavifar, L.; Roy, R.; Le, A.D.; Funk, D.; Le, H.-T. Improving the Utility of a Dynorphin Peptide Analogue Using Mannosylated Glycoliposomes. Int. J. Mol. Sci. 2021, 22, 7996. [Google Scholar] [CrossRef]
- Helal, M.A.; Habib, E.S.; Chittiboyina, A.G. Selective kappa opioid antagonists for treatment of addiction, are we there yet? Eur. J. Med. Chem. 2017, 141, 632–647. [Google Scholar] [CrossRef]
- Carroll, F.I.; Carlezon, W.A., Jr. Development of κ opioid receptor antagonists. J. Med. Chem. 2013, 56, 2178–2195. [Google Scholar] [CrossRef]
- Mousavifar, L.; Touaibia, M.; Roy, R. Development of Mannopyranoside Therapeutics against Adherent-Invasive Escherichia coli Infections. Acc. Chem. Res. 2018, 51, 2937–2948. [Google Scholar] [CrossRef]
- Percec, V.; Leowanawat, P.; Sun, H.J.; Kulikov, O.; Nusbaum, C.D.; Tran, T.M.; Bertin, A.; Wilson, D.A.; Peterca, M.; Zhang, S.; et al. Modular synthesis of amphiphilic Janus glycodendrimers and their self-assembly into glycodendrimersomes and other complex architectures with bioactivity to biomedically relevant lectins. J. Am. Chem. Soc. 2013, 135, 9055–9077. [Google Scholar] [CrossRef]
- Pagé, D.; Roy, R. Synthesis and biological properties of mannosylated starburst poly(amidoamine) dendrimers. Bioconjugate Chem. 1997, 8, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Moussodia, R.O.; Sun, H.J.; Leowanawat, P.; Muncan, A.; Nusbaum, C.D.; Chelling, K.M.; Heiney, P.A.; Klein, M.L.; André, S.; et al. Mimicking biological membranes with programmable glycan ligands self-assembled from amphiphilic Janus glycodendrimers. Angew. Chem. Int. Ed. Engl. 2014, 53, 10899–10903. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Moussodia, R.O.; Murzeau, C.; Sun, H.J.; Klein, M.L.; Vértesy, S.; André, S.; Roy, R.; Gabius, H.J.; Percec, V. Dissecting molecular aspects of cell interactions using glycodendrimersomes with programmable glycan presentation and engineered human lectins. Angew. Chem. Int. Ed. Engl. 2015, 54, 4036–4040. [Google Scholar] [CrossRef]
- Denmark, S.E.; Kobayashi, T.; Regens, C.S. Total Synthesis of (+)-Papulacandin D. Tetrahedron 2010, 66, 4745–4759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhaware, V.; Shaikh, A.Y.; Kar, M.; Hotha, S.; Sen Gupta, S. Synthesis and self-assembly of amphiphilic homoglycopolypeptide. Langmuir 2013, 29, 5659–5667. [Google Scholar] [CrossRef]
- Li, C.W.; Hon, K.W.; Ghosh, B.; Li, P.H.; Lin, H.Y.; Chan, P.H.; Lin, C.H.; Chen, Y.C.; Mong, K.K. Synthesis of oligomeric mannosides and their structure-binding relationship with concanavalin A. Chem. Asian J 2014, 9, 1786–1796. [Google Scholar] [CrossRef]
- van Ameijde, J.; Liskamp, R.M. Synthesis of novel trivalent amino acid glycoconjugates based on the cyclotriveratrylene (‘CTV’) scaffold. Org. Biomol. Chem. 2003, 1, 2661–2669. [Google Scholar] [CrossRef]
- Guggi, D.; Langoth, N.; Hoffer, M.H.; Wirth, M.; Bernkop-Schnürch, A. Comparative evaluation of cytotoxicity of a glucosamine-TBA conjugate and a chitosan-TBA conjugate. Int. J. Pharm. 2004, 278, 353–360. [Google Scholar] [CrossRef]
- Azefu, Y.; Tamiaki, H.; Sato, R.; Toma, K. Facile synthesis of stable lipid analogues possessing a range of alkyl groups: Application to artificial glycolipids. Bioorg. Med. Chem. 2002, 10, 4013–4022. [Google Scholar] [CrossRef]
- Tamiaki, H.; Azefu, Y.; Shibata, R.; Sato, R.; Toma, K. Oligomethylene spacer length dependent interaction of synthetic galactolipids incorporated in phospholipid layers with ricin. Colloids Surf. B Biointerfaces. 2006, 53, 87–93. [Google Scholar] [CrossRef]
- Wagner, A.; Vorauer-Uhl, K.; Katinger, H. Liposomes produced in a pilot scale: Production, purification and efficiency aspects. Eur. J. Pharm. Biopharm. 2002, 54, 213–219. [Google Scholar] [CrossRef]
- Zhu, T.F.; Szostak, J.W. Preparation of large monodisperse vesicles. PLoS ONE 2009, 4, e5009. [Google Scholar] [CrossRef] [PubMed]
- Nagayasu, A.; Uchiyama, K.; Kiwada, H. The size of liposomes: A factor which affects their targeting efficiency to tumors and therapeutic activity of liposomal antitumor drugs. Adv. Drug Deliv. Rev. 1999, 40, 75–87. [Google Scholar] [CrossRef]
- Davis, M.E.; Chen, Z.G.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug Dis. 2008, 7, 771–782. [Google Scholar] [CrossRef]
- Martel, A.L.; Fraleigh, N.L.; Picard, E.; Lewicky, J.D.; Pawelec, G.; Lee, H.; Ma, G.W.; Mousavifar, L.; Roy, R.; Le, H.-T. Novel immunomodulatory properties of low dose cytarabine entrapped in a mannosylated cationic liposome. Int. J. Pharm. 2021, 606, 120849. [Google Scholar] [CrossRef] [PubMed]
- Illescas, B.M.; Rojo, J.; Delgado, R.; Martín, N. Multivalent Glycosylated Nanostructures To Inhibit Ebola Virus Infection. J. Am. Chem. Soc. 2017, 139, 6018–6025. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Gonçalves, C.; Gallego, T.; François-Heude, M.; Malard, V.; Mateo, V.; Lemoine, F.; Cendret, V.; Djedaini-Pilard, F.; Moreau, V.; et al. Comparative binding and uptake of liposomes decorated with mannose oligosaccharides by cells expressing the mannose receptor or DC-SIGN. Carbohydr. Res. 2020, 487, 107877. [Google Scholar] [CrossRef]
- Agrawal, B.B.; Goldstein, I.J. Physical and chemical characterization of concanavalin A, the hemagglutinin from jack bean (Canavalia ensiformis). Biochim. Biophys. Acta 1967, 133, 376–379. [Google Scholar] [CrossRef] [Green Version]
- Poretz, R.D.; Goldstein, I.J. An examination of the topography of the saccharide binding sites of concanavalin A and of the forces involved in complexation. Biochemistry 1970, 9, 2890–2896. [Google Scholar] [CrossRef]
- Swaminathan, C.P.; Surolia, N.; Surolia, A. Role of Water in the Specific Binding of Mannose and Mannooligosaccharides to Concanavalin A. J. Am. Chem. Soc. 1998, 120, 5153–5159. [Google Scholar] [CrossRef]
- Guerry, D.; Kenna, M.A.; Schrieber, A.D.; Cooper, R.A. Concanavalin A-mediated binding and sphering of human red blood cells by homologous monocytes. J. Exp. Med. 1976, 144, 1695–1700. [Google Scholar] [CrossRef]
- Pashov, A.; MacLeod, S.; Saha, R.; Perry, M.; VanCott, T.C.; Kieber-Emmons, T. Concanavalin A binding to HIV envelope protein is less sensitive to mutations in glycosylation sites than monoclonal antibody 2G12. Glycobiology 2005, 15, 994–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepenies, B.; Lee, J.; Sonkaria, S. Targeting C-type lectin receptors with multivalent carbohydrate ligands. Adv. Drug Deliv. Rev. 2013, 65, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.; Aliu, B.; Jiang, X.; Sharpe, T.; Pang, L.; Hadorn, A.; Rabbani, S.; Ernst, B. Poly-l-lysine glycoconjugates inhibit DC-SIGN-mediated attachment of pandemic viruses. ChemMedChem 2021, 16, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.W.; Ratner, D.M.; Seeberger, P.H.; Hacohen, N. Carbohydrate-mediated targeting of antigen to dendritic cells leads to enhanced presentation of antigen to T cells. Chembiochem 2008, 9, 294–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snipstad, S.; Westrøm, S.; Mørch, Y.; Afadzi, M.; Åslund, A.K.; de Lange Davies, C. Contact-mediated intracellular delivery of hydrophobic drugs from polymeric nanoparticles. Cancer Nanotechnol. 2014, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Go, S.; Sato, C.; Yin, J.; Kannagi, R.; Kitajima, K. Hypoxia-enhanced expression of free deaminoneuraminic acid in human cancer cells. Biochem. Biophys. Res. Commun. 2007, 357, 537–542. [Google Scholar] [CrossRef]
- Dalle Vedove, E.; Costabile, G.; Merkel, O.M. Mannose and Mannose-6-Phosphate Receptor-Targeted Drug Delivery Systems and Their Application in Cancer Therapy. Adv. Healthc. Mater. 2018, 7, e1701398. [Google Scholar] [CrossRef]
- Li, Y.; Wu, H.; Ji, B.; Qian, W.; Xia, S.; Wang, L.; Xu, Y.; Chen, J.; Yang, L.; Mao, H. Targeted Imaging of CD206 Expressing Tumor-Associated M2-like Macrophages Using Mannose-Conjugated Antibiofouling Magnetic Iron Oxide Nanoparticles. ACS Appl. Bio. Mater. 2020, 3, 4335–4347. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, Z.H.; Li, Y.X.; Xu, H.L.; Fang, W.H.; He, F. Targeted Delivery of Nanovaccine to Dendritic Cells via DC-Binding Peptides Induces Potent Antiviral Immunity in vivo. Int. J. Nanomed. 2022, 17, 1593–1608. [Google Scholar] [CrossRef]
- Macri, C.; Dumont, C.; Johnston, A.P.; Mintern, J.D. Targeting dendritic cells: A promising strategy to improve vaccine effectiveness. Clin. Transl. Immunol. 2016, 5, e66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.K.; Wall, K.A. Use of Dendritic Cell Receptors as Targets for Enhancing Anti-Cancer Immune Responses. Cancers 2019, 11, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Guo, R.; Zhang, F. Brain perivascular macrophages: Recent advances and implications in health and diseases. CNS Neurosci. Ther. 2019, 25, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Ohgidani, M.; Kato, T.A.; Haraguchi, Y.; Matsushima, T.; Mizoguchi, Y.; Murakawa-Hirachi, T.; Sagata, N.; Monji, A.; Kanba, S. Microglial CD206 Gene Has Potential as a State Marker of Bipolar Disorder. Front. Immunol. 2017, 7, 676. [Google Scholar] [CrossRef] [Green Version]
- Inglut, C.T.; Sorrin, A.J.; Kuruppu, T.; Vig, S.; Cicalo, J.; Ahmad, H.; Huang, H.C. Immunological and Toxicological Considerations for the Design of Liposomes. Nanomaterials 2020, 10, 190. [Google Scholar] [CrossRef] [Green Version]
- Sia, Z.R.; He, X.; Zhang, A.; Ang, J.C.; Shao, S.; Seffouh, A.; Huang, W.C.; D'Agostino, M.R.; Teimouri Dereshgi, A.; Suryaprakash, S.; et al. A liposome-displayed hemagglutinin vaccine platform protects mice and ferrets from heterologous influenza virus challenge. Proc. Natl. Acad. Sci. USA. 2021, 118, e2025759118. [Google Scholar] [CrossRef]
- Lee, D.H.; Rötger, C.; Appeldoorn, C.C.; Reijerkerk, A.; Gladdines, W.; Gaillard, P.J.; Linker, R.A. Glutathione PEGylated liposomal methylprednisolone (2B3-201) attenuates CNS inflammation and degeneration in murine myelin oligodendrocyte glycoprotein induced experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2014, 274, 96–101. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | Solvent | |
|---|---|---|
| A (%) | B (%) | |
| 0 | 100 | 0 |
| 15 | 40 | 60 |
| 18 | 20 | 80 |
| 26 | 20 | 80 |
| 30 | 100 | 0 |
| 40 | 100 | 0 |
| Compounds | Lipid Tail Size | Diameter (nm) | PDI |
|---|---|---|---|
| 21 | C8 | 252 ± 79 | 0.18 |
| 22 | C10 | n.d. | n.d. |
| 23 | C12 | 254 ± 41 | 0.20 |
| 24 | C14 | 287 ± 27 | 0.04 |
| 25 | C16 | 385 ± 20 | 0.91 |
| Compound | Concentration (μg/mL) | % Hemolysis (±SEM) |
|---|---|---|
| 24 | 100 | 0 |
| 50 | 0 | |
| 25 | 0 | |
| 12.5 | 0 | |
| PBS | - | 0 |
| ACK lysing buffer | - | 100 ± 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mousavifar, L.; Lewicky, J.D.; Taponard, A.; Bagul, R.; Rivat, M.; Abdullayev, S.; Martel, A.L.; Fraleigh, N.L.; Nakamura, A.; Veyrier, F.J.; et al. Synthesis & Evaluation of Novel Mannosylated Neoglycolipids for Liposomal Delivery System Applications. Pharmaceutics 2022, 14, 2300. https://doi.org/10.3390/pharmaceutics14112300
Mousavifar L, Lewicky JD, Taponard A, Bagul R, Rivat M, Abdullayev S, Martel AL, Fraleigh NL, Nakamura A, Veyrier FJ, et al. Synthesis & Evaluation of Novel Mannosylated Neoglycolipids for Liposomal Delivery System Applications. Pharmaceutics. 2022; 14(11):2300. https://doi.org/10.3390/pharmaceutics14112300
Chicago/Turabian StyleMousavifar, Leila, Jordan D. Lewicky, Alexis Taponard, Rahul Bagul, Madleen Rivat, Shuay Abdullayev, Alexandrine L. Martel, Nya L. Fraleigh, Arnaldo Nakamura, Frédéric J. Veyrier, and et al. 2022. "Synthesis & Evaluation of Novel Mannosylated Neoglycolipids for Liposomal Delivery System Applications" Pharmaceutics 14, no. 11: 2300. https://doi.org/10.3390/pharmaceutics14112300