
Analytical Quality by Design (AQbD) Approach to the Development of In Vitro Release Test for Topical Hydrogel

 , ,
, ,  and
and
Abstract
:1. Introduction
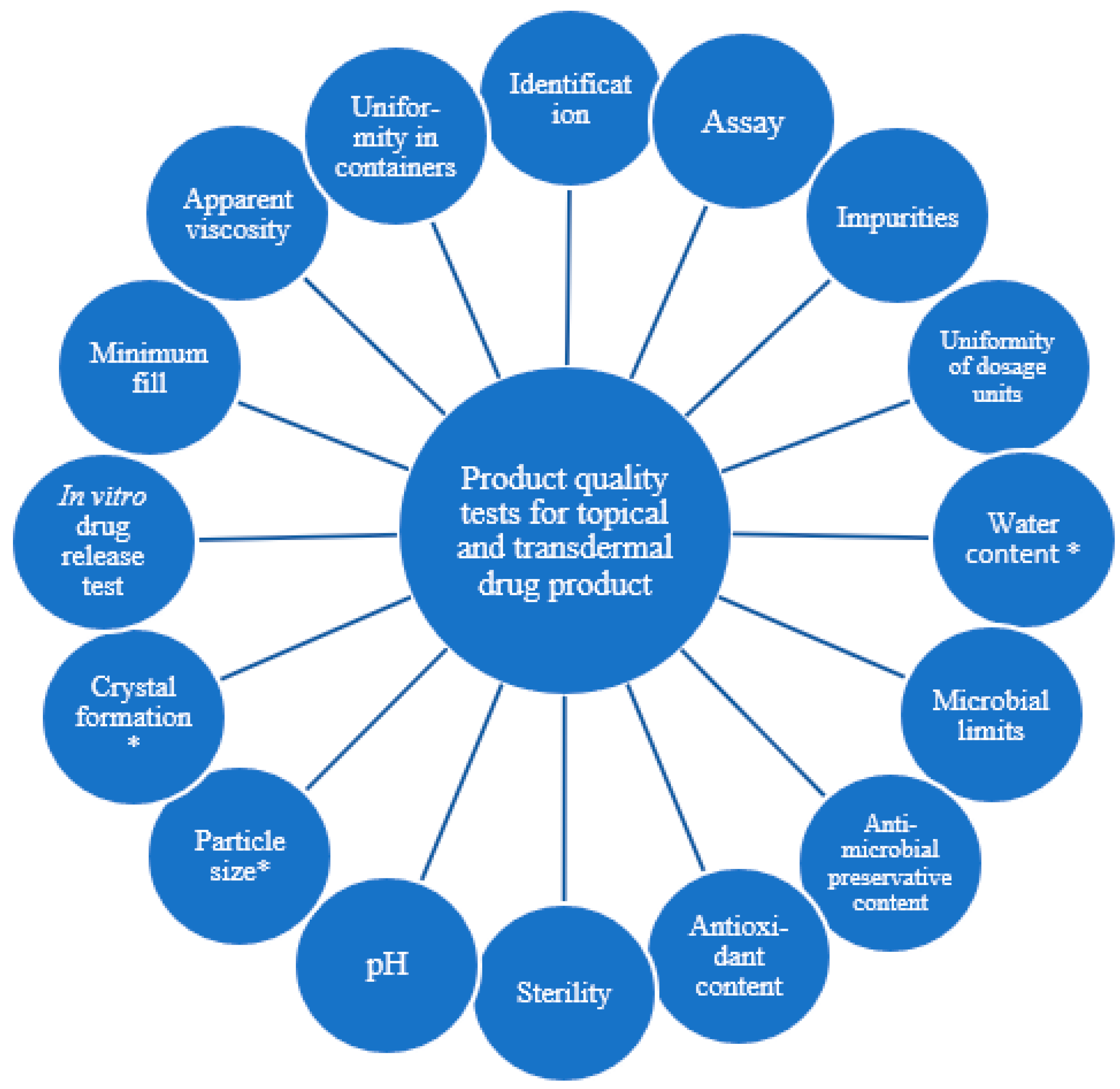
1.1. Product Quality Tests for Topical and Transdermal Drug Product
1.2. Quality by Design Usage in the Development of Topical Semi-Solid Dosages
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. USP Apparatus IV: Flow-Through Cell with a Semi-Solid Adapter
2.2.2. Ultra-High-Performance Liquid Chromatography Analysis
2.2.3. Analytical Quality by Design
2.2.4. Definition of the Analytical Target Profile
2.2.5. Definition of the Critical Method Attributes and Critical Method Parameters
2.2.6. Establishing Failure Mode Effects Analysis (FMEA)
- Low (acceptable) 1 ≤ RPN ≤ 29;
- Medium (to be considered) 30 ≤ RPN ≤ 59;
- High (not acceptable) 60 ≤ RPN ≤ 125.
2.2.7. Design of Experiments (DoE) for IVRT Method
2.2.8. Determination of the Osmolality of Different Media
2.2.9. Performing Membrane Inertness Test
2.2.10. Discriminatory Power of the In Vitro Release Test Method
3. Results
3.1. Definition of ATP and Determination of CMAs
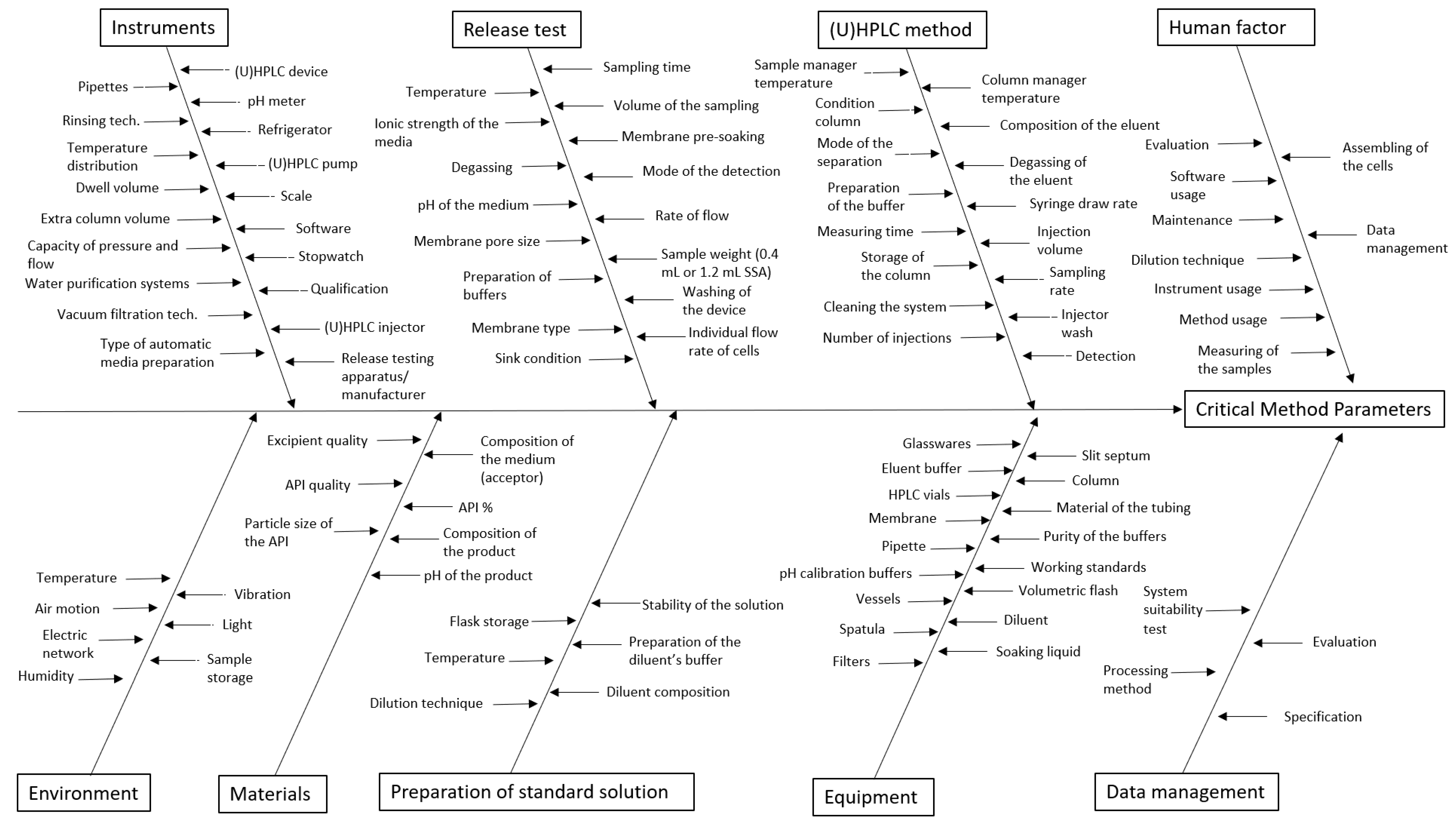
3.2. Identification of the MP using the Ishikawa Diagram
3.3. Initial Risk Assessment using FMEA (Effects of MPs on CMAs)
3.4. Carrying out Preliminary Experiments
3.4.1. In Vitro Release Test Study Design with USP Apparatus IV
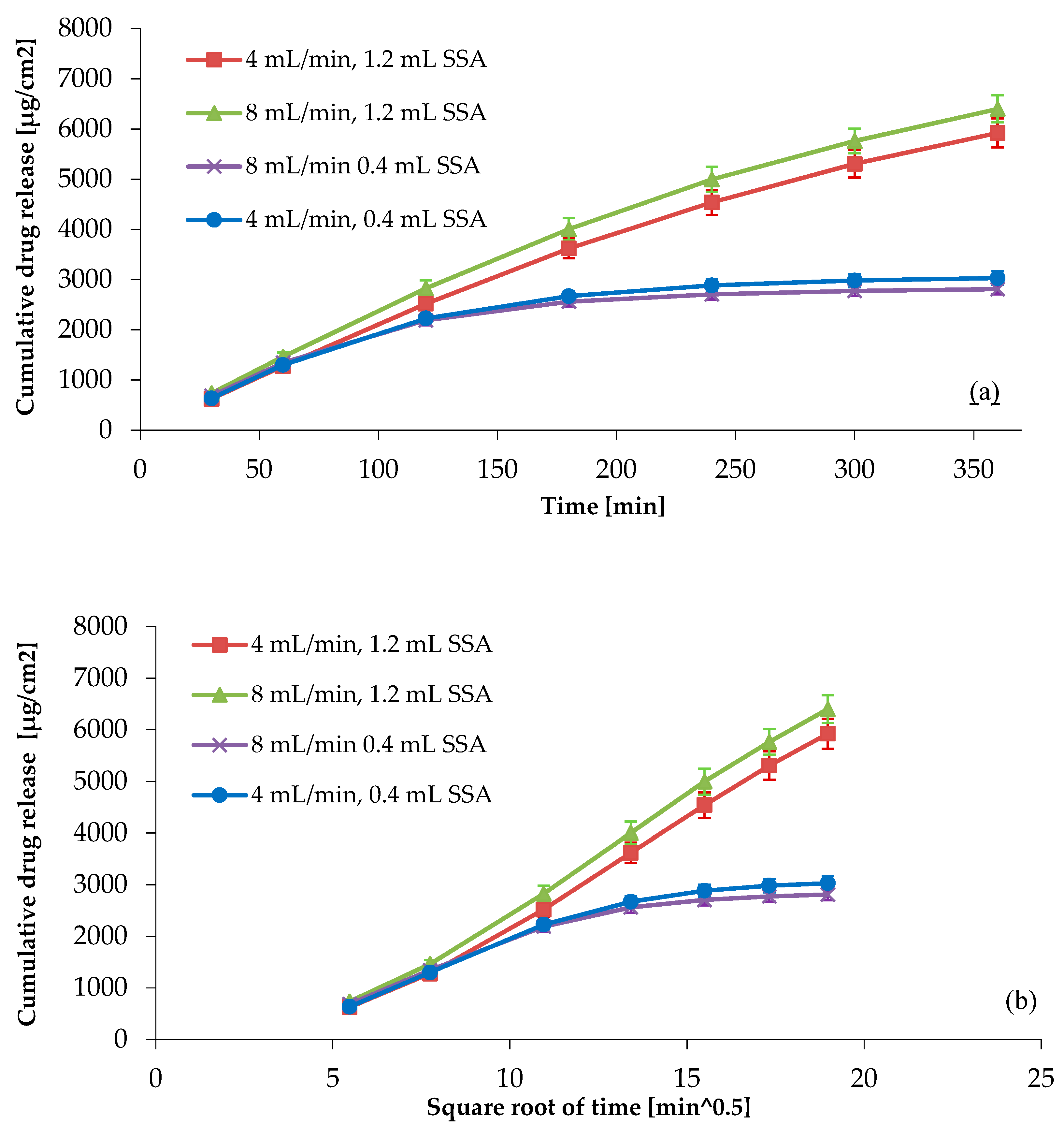
3.4.2. Investigation of the Rate of Flow and Sample Weight with the One-Factor-at-a-Time (OFAT) Method
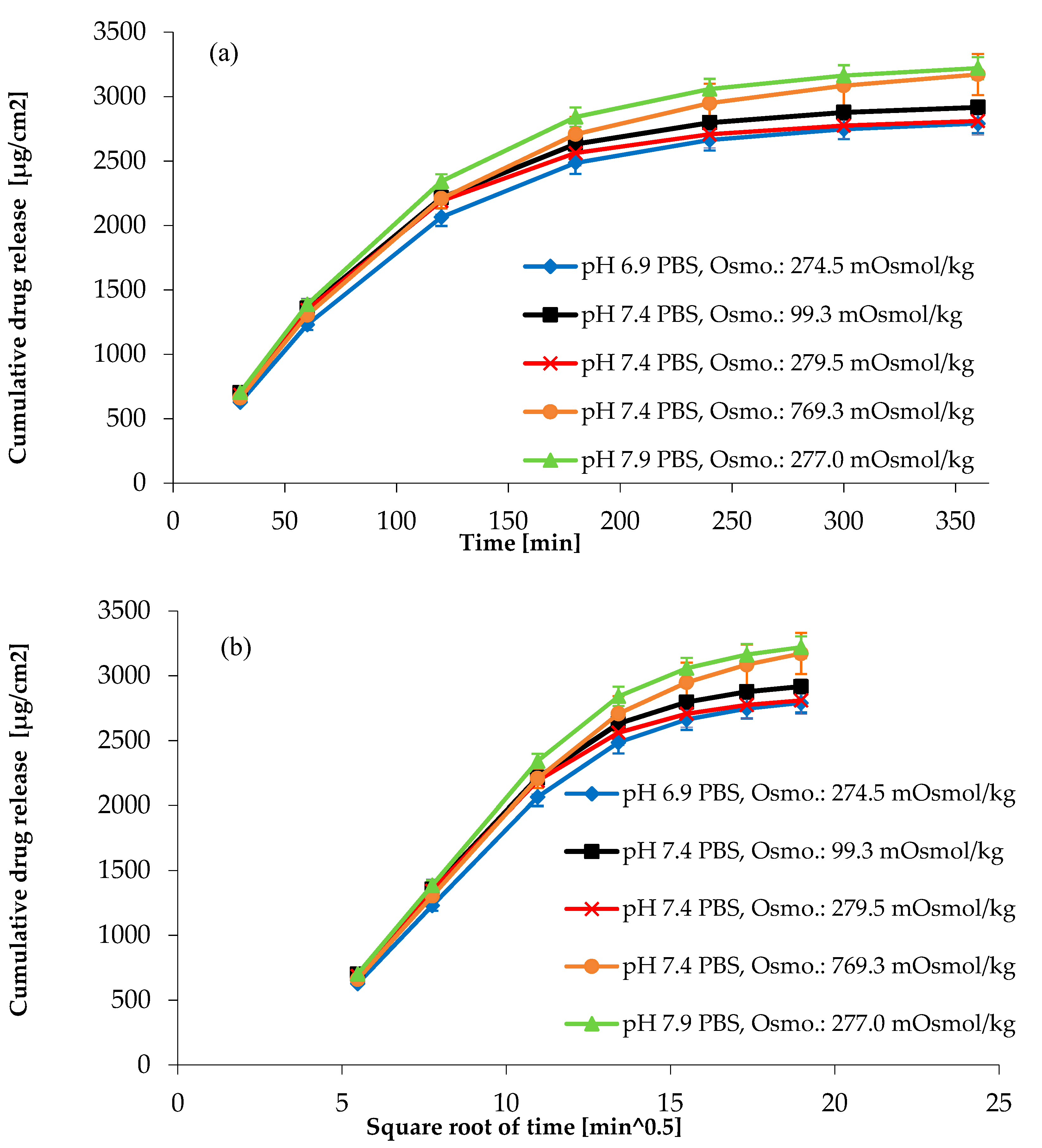
3.4.3. Effect of pH and Osmolality on Drug Release from Topical Hydrogel
3.5. The 23 Full Factorial Design for the IVRT Method
3.6. Updating the FMEA Table
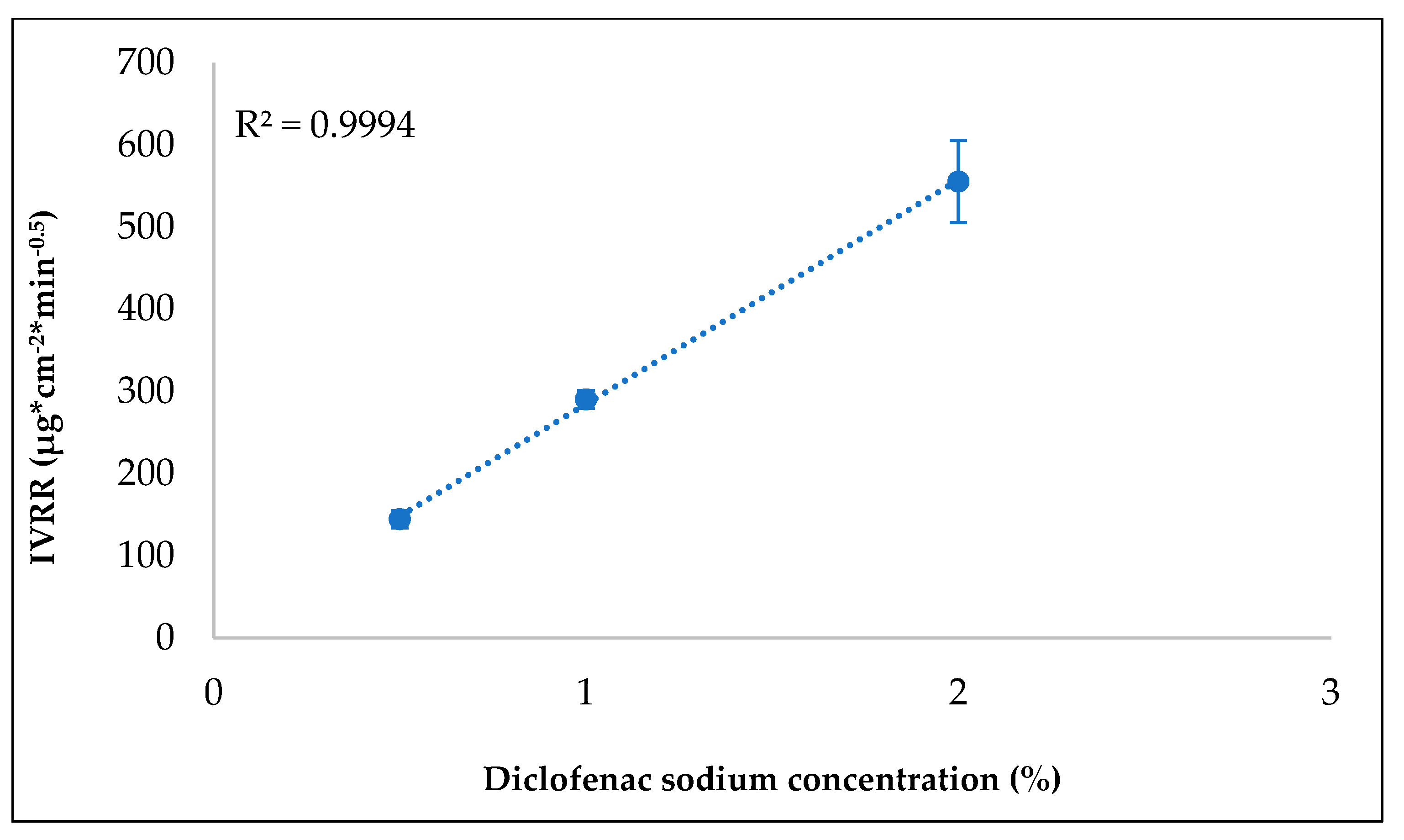
3.7. Investigating Discriminatory Power
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Draft Guideline on Quality and Equivalence of Topical Products EMA/CHMP/QWP/708282/2018; European Medicines Agency: Amsterdam, The Netherlands, 2018.
- Praeparationes Semi Solidae Ad Usum Dermicum. In European Pharmacopoeia; Council of Europe: Strasbourg, France, 2021.
- USP. Chapter <3> Topical and Transdermal Drug Products—Product Quality Tests; USP: Rockvill, MD, USA, 2019. [Google Scholar]
- ICH Q8 (R2) Pharmaceutical Development; European Medicines Agency: London, UK, 2017.
- Kovács, A.; Berkó, S.; Csányi, E.; Csóka, I. Development of Nanostructured Lipid Carriers Containing Salicyclic Acid for Dermal Use Based on the Quality by Design Method. Eur. J. Pharm. Sci. 2017, 99, 246–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, C.T.; Shah, V.P.; Derdzinski, K.; Ewing, G.; Flynn, G.; Maibach, H.; Marques, M.; Rytting, H.; Shaw, S.; Thakker, K.; et al. Topical and Transdermal Drug Products. Dissolution Technol. 2010, 17, 12–25. [Google Scholar] [CrossRef]
- FDA. Guidance for Industry: Nonsterile Semisolid Dosage Forms; Food and Drug Administration: Rockvill, MD, USA, 1997; p. 40.
- USP. Chapter <1724> Semisolid Drug Products—Performance Tests; USP: Rockvill, MD, USA, 2014. [Google Scholar]
- European Medicine Agency. Guideline of the Investigation of Bioequivalence; European Medicine Agency: London, UK, 2010; p. 27.
- USP. Chapter <724> Drug Release; USP: Rockvill, MD, USA, 2019. [Google Scholar]
- JP. Chapter 6.13 Release Test for Preparations for Cutaneous Application. In Japanese Pharmacopoeia, 17th ed.; The Ministry of Health, Labour and Welfare: Tokyo, Japan, 2016. [Google Scholar]
- Chang, R.-K.; Raw, A.; Lionberger, R.; Yu, L. Generic Development of Topical Dermatologic Products, Part II: Quality by Design for Topical Semisolid Products. AAPS J. 2013, 15, 674–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, L.R.; Morgado, J.; Barnett, K.; Harrington, B.; Wang, J.; Harwood, J.; Fortin, D. Analytical Quality by Design (AQbD) in Pharmaceutical Development. Am. Pharm. Rev. 2013, 144191. [Google Scholar]
- Kan, S.; Lu, J.; Liu, J.; Wang, J.; Zhao, Y. A Quality by Design (QbD) Case Study on Enteric-Coated Pellets: Screening of Critical Variables and Establishment of Design Space at Laboratory Scale. Asian J. Pharm. Sci. 2014, 9, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Iurian, S.; Turdean, L.; Tomuta, I. Risk Assessment and Experimental Design in the Development of a Prolonged Release Drug Delivery System with Paliperidone. DDDT 2017, 11, 733–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ICH Q9 on Quality Risk Management. Steps 5; European Medicines Agency: London, UK, 2006.
- Tome, T.; Žigart, N.; Časar, Z.; Obreza, A. Development and Optimization of Liquid Chromatography Analytical Methods by Using AQbD Principles: Overview and Recent Advances. Org. Process Res. Dev. 2019, 23, 1784–1802. [Google Scholar] [CrossRef] [Green Version]
- Raman, N.V.V.S.S.; Mallu, U.R.; Bapatu, H.R. Analytical Quality by Design Approach to Test Method Development and Validation in Drug Substance Manufacturing. J. Chem. 2015, 2015, e435129. [Google Scholar] [CrossRef]
- Borman, P.; Chatfield, M.; Nethercote, P.; Thomson, D.; Truman, K. The Application of Quality by Design to Analytical Methods. Pharm. Technol. 2007, 31. [Google Scholar]
- Hanna-Brown, M.; Ermer, J.; Katzenbach, S.; Rignall, A.; Gervais, A.; Hoffmann, J.; Wypych, J.; McLaughlin, K.; Borman, P.; Grosche, O.; et al. Analytical Procedure Lifecycle Management: Current Status and Opportunities. Pharm. Technol. 2018, 42, 18–23. [Google Scholar]
- USP. Chapter <711> Dissolution; USP: Rockvill, MD, USA, 2019. [Google Scholar]
- PhEur. Chapter 2.9.3 Dissolution Test for Solid Dosage Form. In European Pharmacopoeia, 10th ed.; Council of Europe: Strasbourg, France, 2021. [Google Scholar]
- Miranda, M.; Pais, A.A.C.C.; Cardoso, C.; Vitorino, C. AQbD as a Platform for IVRT Method Development—A Regulatory Oriented Approach. Int. J. Pharm. 2019, 572, 118695. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.; Borman, P.; Campa, C.; Chatfield, M.; Godfrey, M.; Hamilton, P.; Hoyer, W.; Norelli, F.; Orr, R.; Schofield, T. Using the Analytical Target Profile to Drive the Analytical Method Lifecycle. Anal Chem. 2019, 91, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- USP ⟨1220⟩. Analytical Procedure Lifecycle; USP: Rockvill, MD, USA, 2022. [Google Scholar]
- Borman, P.; Campa, C.; Delpierre, G.; Hook, E.; Jackson, P.; Kelley, W.; Protz, M.; Vandeputte, O. Selection of Analytical Technology and Development of Analytical Procedures Using the Analytical Target Profile. Anal. Chem. 2022, 94, 559–570. [Google Scholar] [CrossRef] [PubMed]
- ICH Q14: Analytical Procedure Development and Revision of Q2(R1) Analytical Validation—Final Concept Paper—ECA Academy. Available online: https://www.gmp-compliance.org/guidelines/gmp-guideline/ich-q14-analytical-procedure-development-and-revision-of-q2r1-analytical-validation-final-concept-paper (accessed on 23 February 2022).
- Hanna-Brown, M.; Borman, P.; Bale, S.; Szucs, R.; Roberts, J.; Jones, C. Development of Chromatographic Methods Using QbD Principles. Sep. Sci. 2010, 2, 12–20. [Google Scholar]
- Barnett, K.; McGregor, P.L.; Martin, G.P.; Blond, D.J.; Weitzel, J.; Ermer, J.; Walfish, S.; Nethercote, P.; Gratzl, G.S.; Kovacs, E.; et al. Analytical Target Profile: Structure and Application throughout the Analytical Lifecycle. In Pharmacopeial Forum; USP: Rockvill, MD, USA, 2016; Volume 42. [Google Scholar]
- Ford, J.L. Design and Evaluation of Hydroxypropyl Methylcellulose Matrix Tablets for Oral Controlled Release: A Historical Perspective. In Hydrophilic Matrix Tablets for Oral Controlled Release; Timmins, P., Pygall, S.R., Melia, C.D., Eds.; AAPS Advances in the Pharmaceutical Sciences Series; Springer: New York, NY, USA, 2014; pp. 17–51. ISBN 978-1-4939-1519-4. [Google Scholar]
- Kochling, J.; Wu, W.; Hua, Y.; Guan, Q.; Castaneda-Merced, J. A Platform Analytical Quality by Design (AQbD) Approach for Multiple UHPLC-UV and UHPLC-MS Methods Development for Protein Analysis. J. Pharm. Biomed. Anal. 2016, 125, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, R.; Kona, R.; Dandu, R.; Xie, W.; Claycamp, G.; Hoag, S.W. Quality by Design I: Application of Failure Mode Effect Analysis (FMEA) and Plackett-Burman Design of Experiments in the Identification of “Main Factors” in the Formulation and Process Design Space for Roller-Compacted Ciprofloxacin Hydrochloride Immediate-Release Tablets. AAPS PharmSciTech 2012, 13, 1243–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, J.; Bezerra, M.; Markl, D.; Berghaus, A.; Borman, P.; Schlindwein, W. Development and Validation of an In-Line API Quantification Method Using AQbD Principles Based on UV-Vis Spectroscopy to Monitor and Optimise Continuous Hot Melt Extrusion Process. Pharmaceutics 2020, 12, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA. Draft Guidance on Acyclovir; Food and Drug Administration: Rockville, MD, USA, 2012.
- Garbacz, G.; Weitschies, W. Investigation of Dissolution Behavior of Diclofenac Sodium Extended Release Formulations under Standard and Biorelevant Test Conditions. Drug Dev. Ind. Pharm. 2010, 36, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Lapidus, H.; Lordi, N.G. Some Factors Affecting the Release of a Water-Soluble Drug from a Compressed Hydrophilic Matrix. J. Pharm. Sci. 1966, 55, 840–843. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Ranking | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| Occurrence (O) | nearly impossible | randomly occurring | 50% chance of occurring | likely to occur | certain to occur |
| Severity (S) | no effects | insignificant effect | moderate effect | strong effect | severe effect |
| Detectability (D) | excellent | good | moderate | poor | undetectable |
| ATP Element | Target |
|---|---|
| Target sample (product name) | Diclofenac sodium 1% topical gel |
| API name | Diclofenac sodium |
| Dosage strength | 1% (10 mg/g) |
| Dosage forms | Hydrogel |
| Route of administration | Topical |
| Matrix | Propylene glycol (50%), HPMC (1.5%), purified water (47.5%) |
| Packaging | Plastic tube |
| Regulatory specification | ICH, EMA (European Medicines Agency), FDA (Food and Drug Administration) |
| Release/in vitro release test | The release tests should be sensitive to relevant changes in the ingredients and process parameters. They should have adequate release efficiency, release profiles, and reproducibility. They should meet regulatory requirements [1]. Precision RSD ≤ 10% (6 parallel). |
| Analytical measurements | Analytical measurements: the procedure must be able to accurately quantify diclofenac sodium in IVRT samples over the range of 25–200% of the nominal concentration with an accuracy of 2.0% |
| CMA Parameters | Target | Justification |
|---|---|---|
| Release efficiency in 6 h | Q (6 h) ≥ 70% | IVRT is a fundamental tool used to identifyformulation factors that influence the release of the API, an effective method to monitor lot-to-lot changes and stability during development. A draft guideline on the quality and equivalence of topical products described this criterion [1]. |
| Characterize the release profile | 6 time points should be obtained in the linear portion of the drug release profile | |
| RSD% of the released API amount of the 6 parallel samples at given sampling points | RSD ≤ 10% (6 parallel) | RSD values below 10% are considered to be an indication of the good reproducibility of the IVRT method. |
| Accuracy | Between 98 and 102% | In the case of UHPLC measurements, the weak point of the true value determination is accuracy. |
| System suitability test of the chromatography system | USP plate count: N ≥ 3000 | There is a need for a chromatography system in which the API can properly separate from the matrix components. The plate count has a fundamental impact on the extent of measurement error through the peak’s capability of being integrated. Therefore, the chromatography method should be suitable within the purpose to detect the API in IVRT samples at 25% of the nominal concentration. |
| Method Parameter | Critical Method Attributes | Cause of the Deviation | Effect of the Deviation | F (Occurrence) | S (Severity) | D (Perceptibility) | RPN | Action/Strategy of Risk Decrease |
|---|---|---|---|---|---|---|---|---|
| Release test | ||||||||
| Ionic strength of the medium | min. 70% (Q)—6 h | The gelling agent is HPMC | Release might change | 4 | 5 | 4 | 80 | We need to investigate the effect of the ionic strength of the medium (pH 7.4 PBS ± NaCl). |
| Ionic strength of the medium | 6 time points should be obtained in the linear portion of the drug release profile | The gelling agent is HPMC | Release might change | 4 | 5 | 4 | 80 | We need to investigate the effect of the ionic strength of the medium (pH 7.4 PBS ± NaCl). |
| pH of the medium | min. 70% (Q)—6 h | Changing the pH of the medium | RSD might be increasing; outliers below 70% | 3 | 5 | 4 | 60 | Controlled parameter: prescription is needed to make the medium pH 7.4 ± 0.5. Investigation of the effect of pH change is needed. |
| Membrane type | min. 70% (Q)—6 h | Different membrane and manufacturer | The membrane should be inert and not be rate-limiting to active substance release | 4 | 5 | 3 | 60 | We need to investigate the inertness of the membrane in pH 7.4 PBS medium. |
| Rate of flow | min. 70% (Q)—6 h | The increase in the rate of flow, maintaining the concentration gradient, results in faster drug release | Release kinetic might change; increase or decrease in RSD | 5 | 5 | 3 | 75 | We need to investigate the effect of the flow rate changing (4 mL/min to 8 mL/min). |
| Rate of flow | 6 time points should be obtained in the linear portion of the drug release profile | Quicker flowing causes quicker release | Release kinetic might change | 5 | 5 | 3 | 75 | We need to investigate the effect of the flow rate changing (4 mL/min to 8 mL/ min). |
| Sample weight (0.4 mL or 1.2 mL SSA) | min. 70% (Q)—6 h | Different size of SSA | Sample weight increasing, leading to release kinetic change/release rate change | 5 | 5 | 3 | 75 | We need to investigate the effect of the sample weight (0.4 mL or 1.2 mL SSA). |
| Sample weight (0.4 mL or 1.2 mL SSA) | 6 time points should be obtained in the linear portion of the drug release profile | Different size of SSA | Sample weight increasingleading to release kinetic change/release rate change | 5 | 5 | 3 | 75 | We need to investigate the effect of the sample weight (0.4 mL or 1.2 mL SSA). |
| Individual flow rate of cells | min. 70% (Q)—6 h | The release of API might be changing cell by cell | RSD might be increasing; outliers above 70% | 3 | 5 | 5 | 75 | Measuring the flow rate cell by cell of the release and calculating the release with the measured flow rate. Conducting training about how to assemble the cells. Annual maintenance. |
| Individual flow rate of cells | 6 time points should be obtained in the linear portion of the drug release profile | The release of API might be changing cell by cell | RSD might be increasing; fluctuating release curve is caused by RSD% | 3 | 5 | 5 | 75 | Measuring the flow rate cell by cell of the dissolution and calculating the dissolution with the measured flow rate. |
| Individual flow rate of cells | RSDConc ≤ 10% (6 vessels) | The release of API might be changing cell by cell | Fluctuating release curve is caused by RSD% | 3 | 5 | 5 | 75 | Conducting training about how to assemble the cells. Annual maintenance. |
| API% | min. 70% (Q)—6 h | Sink conditions must be ensured in the receptor medium | Limited drug solubility effects can play a major role in the control of API release | 5 | 5 | 3 | 75 | What is the hydrogel diclofenac sodium’s maximum dosage that we are going to use? |
| API% | 6 time points should be obtained in the linear portion of the drug release profile | The method’s requirement is to detect different IVRRs according to the strength of the formulations | The IVRT method might not be sensitive | 4 | 5 | 3 | 60 | We need to investigate the discriminatory ability of the IVRT method (different formulation strengths: 0.5, 1, and 2%). |
| Composition of the product | min. 70% (Q)—6 h | Gelling agent type | Release might change | 4 | 5 | 3 | 60 | We need to prescribe that the matrix is fixed. |
| Media | Osmolality | Flow Rate | Semi-Solid Adapter | Computed Released Amount at the End of the Experiment at 6 h | IVRR | Lag Time | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | RSD | Mean | SD | RSD | Mean | SD | RSD | ||||
| mOsmol/kg | mL/min | mL | % | % | % | µg × cm−2 × min0.5 | µg × cm−2 × min0.5 | % | min | min | % | |
| pH 7.4 PBS | 279.5 | 4 | 1.2 | 75.5 | 3.5 | 4.6 | 420.2 | 21.6 | 5.2 | 22.9 | 1.3 | 5.5 |
| pH 7.4 PBS | 279.5 | 8 | 0.4 | 100.6 | 3.6 | 3.6 | 273.8 | 10.2 | 3.7 | 8.6 | 1.2 | 14.0 |
| pH 7.4 PBS | 279.5 | 4 | 0.4 | 99.5 | 4.6 | 4.7 | 278.5 | 10.5 | 3.8 | 11.7 | 0.7 | 6.0 |
| pH 7.4 PBS | 279.5 | 8 | 1.2 | 81.2 | 3.5 | 4.3 | 446.7 | 18.2 | 4.1 | 20.1 | 1.5 | 7.3 |
| pH 7.4 PBS + NaCl | 769.3 | 8 | 0.4 | 94.4 | 2.2 | 2.3 | 274.6 | 9.5 | 3.5 | 9.7 | 0.6 | 6.3 |
| pH 7.4 PBS–NaCl | 99.3 | 8 | 0.4 | 91.3 | 1.8 | 1.9 | 275.6 | 4.5 | 1.6 | 8.3 | 0.5 | 5.7 |
| pH 6.9 PBS | 274.5 | 8 | 0.4 | 86.5 | 2.5 | 2.9 | 262.1 | 8.6 | 3.3 | 9.4 | 0.8 | 8.2 |
| pH 7.9 PBS | 277.0 | 8 | 0.4 | 99.5 | 3.0 | 3.0 | 299.1 | 8.9 | 3.0 | 9.7 | 0.7 | 7.0 |
| Experiment | Flow Rate (mL/min) | Volume of SSA (mL) | pH |
|---|---|---|---|
| 1 | 4.00 | 0.40 | 7.40 |
| 2 | 8.00 | 0.40 | 7.40 |
| 3 | 4.00 | 1.20 | 7.40 |
| 4 | 8.00 | 1.20 | 7.40 |
| 5 | 4.00 | 0.40 | 7.90 |
| 6 | 8.00 | 0.40 | 7.90 |
| 7 | 4.00 | 1.20 | 7.90 |
| 8 | 8.00 | 1.20 | 7.90 |
| Factor | Effect | t(32) | p | Coefficient | Standard Error Coefficient |
|---|---|---|---|---|---|
| Mean/intercept | 365.9818 | 137.8254 | 0.0000 | 365.9818 | 2.6554 |
| (1) A: Flow rate (mL/min) | 4.2335 | 0.7971 | 0.4312 | 2.1168 | 2.6554 |
| (2) B: Volume of SSA (mL) | 158.0885 | 29.7673 | 0.0000 | 79.0443 | 2.6554 |
| (3) C: pH | 23.9005 | 4.5004 | 0.0001 | 11.9503 | 2.6554 |
| 1 by 2 | 6.8665 | 1.2929 | 0.2053 | 3.4333 | 2.6554 |
| 1 by 3 | −3.5875 | −0.6755 | 0.5042 | −1.7938 | 2.6554 |
| 2 by 3 | 0.5395 | 0.1016 | 0.9197 | 0.2698 | 2.6554 |
| 1 × 2 × 3 | −7.2765 | −1.3701 | 0.1802 | −3.6383 | 2.6554 |
| Factor | Effect | t(32) | p | Coefficient | Standard Error Coefficient |
|---|---|---|---|---|---|
| Mean/intercept | 88.2920 | 150.1317 | 0.0000 | 88.2920 | 0.5881 |
| (1) A: Flow rate (mL/min) | 1.8480 | 1.5712 | 0.1260 | 0.9240 | 0.5881 |
| (2) B: Volume of SSA (mL) | −22.4020 | −19.0462 | 0.0000 | −11.2010 | 0.5881 |
| (3) C: pH | −1.5510 | −1.3187 | 0.1966 | −0.7755 | 0.5881 |
| 1 by 2 | 0.9680 | 0.8230 | 0.4166 | 0.4840 | 0.5881 |
| 1 by 3 | −1.3130 | −1.1163 | 0.2726 | −0.6565 | 0.5881 |
| 2 by 3 | −0.9070 | −0.7711 | 0.4463 | −0.4535 | 0.5881 |
| 1 × 2 × 3 | −1.1150 | −0.9480 | 0.3502 | −0.5575 | 0.5881 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szoleczky, R.; Budai-Szűcs, M.; Csányi, E.; Berkó, S.; Tonka-Nagy, P.; Csóka, I.; Kovács, A. Analytical Quality by Design (AQbD) Approach to the Development of In Vitro Release Test for Topical Hydrogel. Pharmaceutics 2022, 14, 707. https://doi.org/10.3390/pharmaceutics14040707
Szoleczky R, Budai-Szűcs M, Csányi E, Berkó S, Tonka-Nagy P, Csóka I, Kovács A. Analytical Quality by Design (AQbD) Approach to the Development of In Vitro Release Test for Topical Hydrogel. Pharmaceutics. 2022; 14(4):707. https://doi.org/10.3390/pharmaceutics14040707
Chicago/Turabian StyleSzoleczky, Réka, Mária Budai-Szűcs, Erzsébet Csányi, Szilvia Berkó, Péter Tonka-Nagy, Ildikó Csóka, and Anita Kovács. 2022. "Analytical Quality by Design (AQbD) Approach to the Development of In Vitro Release Test for Topical Hydrogel" Pharmaceutics 14, no. 4: 707. https://doi.org/10.3390/pharmaceutics14040707
APA StyleSzoleczky, R., Budai-Szűcs, M., Csányi, E., Berkó, S., Tonka-Nagy, P., Csóka, I., & Kovács, A. (2022). Analytical Quality by Design (AQbD) Approach to the Development of In Vitro Release Test for Topical Hydrogel. Pharmaceutics, 14(4), 707. https://doi.org/10.3390/pharmaceutics14040707