

Utility of Physiologically Based Pharmacokinetic Modeling to Investigate the Impact of Physiological Changes of Pregnancy and Cancer on Oncology Drug Pharmacokinetics
 ,
,
Abstract
:1. Introduction
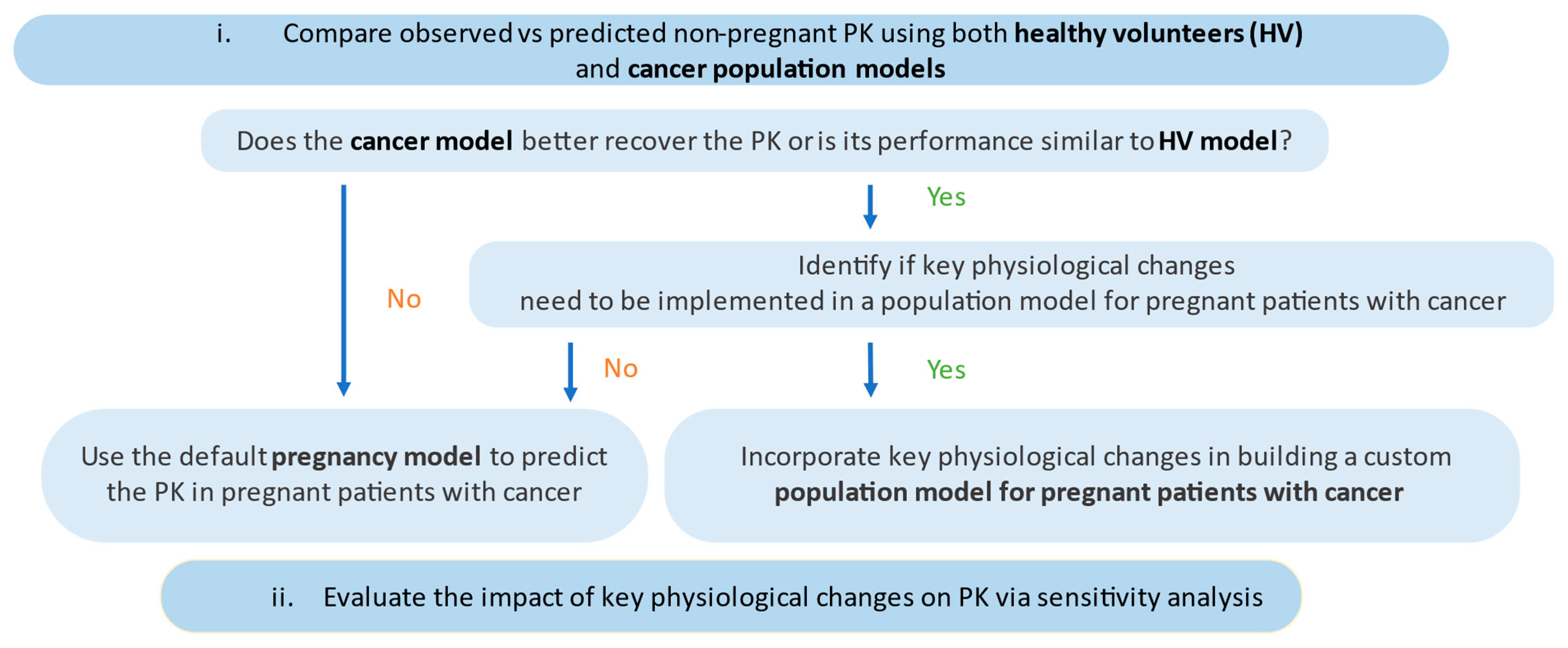
2. Materials and Methods
2.1. Compound Selection and Clinical Data Extraction
2.2. PBPK Model Development and Application
2.3. Drug Model Development and Verification
2.4. Physiological Changes Implemented in Cancer and Pregnancy Population Models
2.4.1. Cancer Population Model
2.4.2. Pregnancy Population Model
2.5. Development and Application of a Population Model for Pregnant Patients with Cancer
2.6. Predictive Performance
2.7. Software
3. Results
3.1. Model Evaluation
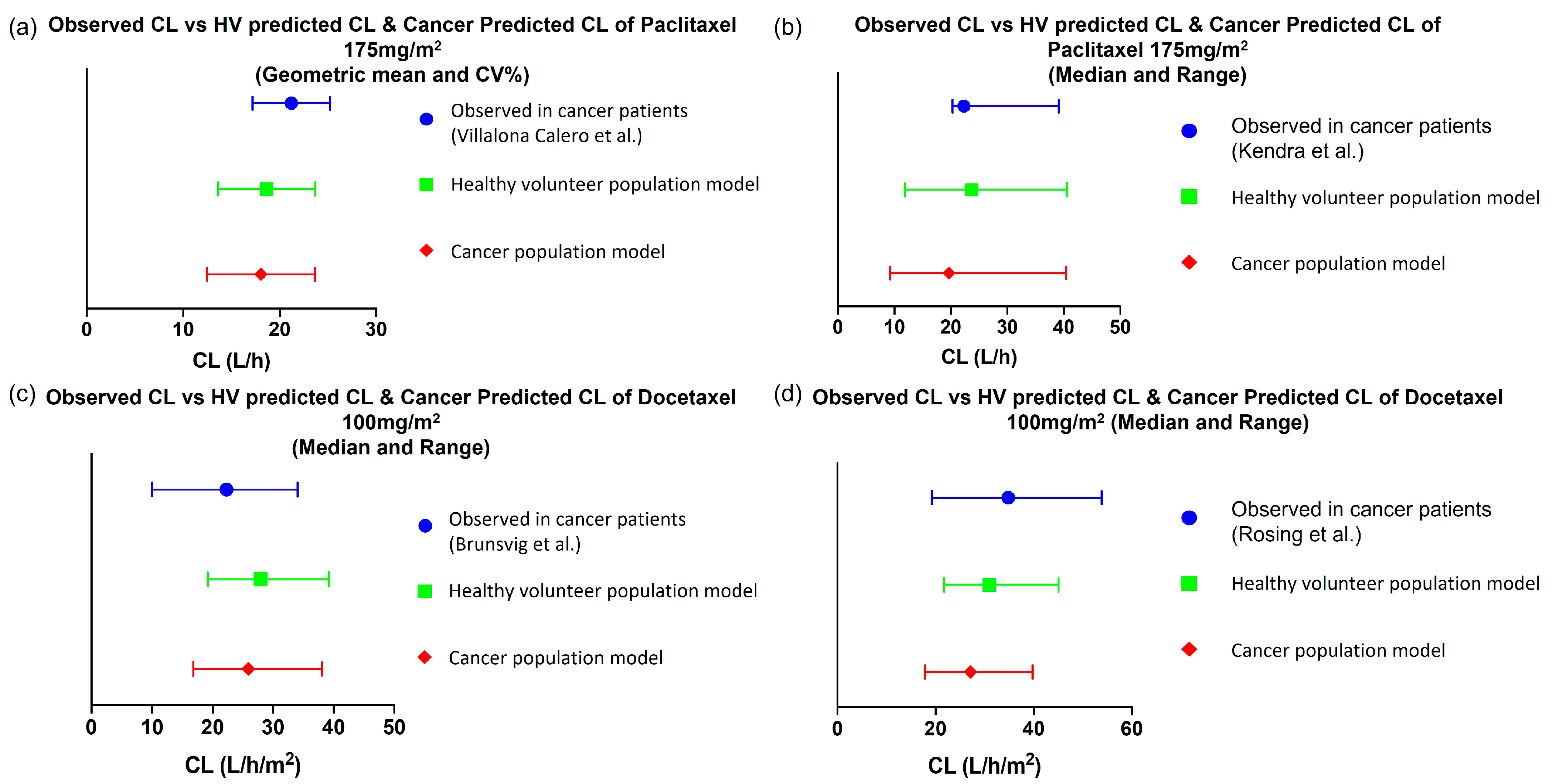
3.1.1. Predictive PK Performance of Drug Models in Non-Pregnant Population
Paclitaxel
Docetaxel
Acalabrutinib
3.1.2. Evaluating the Effect of Physiological Changes in Cancer on the PK of Paclitaxel and Docetaxel
3.2. Evaluating the Effect of Physiological Changes in Pregnancy on the PK of Paclitaxel and Docetaxel
3.3. Model Application
4. Discussion
5. Clinical Implications
6. Limitations
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, Y.Y.; Roberts, C.L.; Dobbins, T.; Stavrou, E.; Black, K.; Morris, J.; Young, J. Incidence and outcomes of pregnancy-associated cancer in Australia, 1994–2008: A population-based linkage study. BJOG 2012, 119, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Eibye, S.; Kjaer, S.K.; Mellemkjaer, L. Incidence of pregnancy-associated cancer in Denmark, 1977–2006. Obstet. Gynecol. 2013, 122, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, A.; Cairncross, Z.F.; Friedenreich, C.M.; Ray, J.G.; Nelson, G.; Fell, D.B.; Lisonkova, S.; Bhatti, P.; McMorris, C.; Sikdar, K.C.; et al. Incidence of Pregnancy-Associated Cancer in Two Canadian Provinces: A Population-Based Study. Int. J. Environ. Res. Public Health 2021, 18, 3100. [Google Scholar] [CrossRef] [PubMed]
- Cottreau, C.M.; Dashevsky, I.; Andrade, S.E.; Li, D.K.; Nekhlyudov, L.; Raebel, M.A.; Ritzwoller, D.P.; Partridge, A.H.; Pawloski, P.A.; Toh, S. Pregnancy-Associated Cancer: A U.S. Population-Based Study. J. Womens Health 2019, 28, 250–257. [Google Scholar] [CrossRef]
- McCormick, A.; Peterson, E. Cancer in Pregnancy. Obstet. Gynecol. Clin. N. Am. 2018, 45, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Coppola, P.; Kerwash, E.; Cole, S. Physiologically Based Pharmacokinetics Model in Pregnancy: A Regulatory Perspective on Model Evaluation. Front. Pediatr. 2021, 9, 687978. [Google Scholar] [CrossRef] [PubMed]
- Pariente, G.; Leibson, T.; Carls, A.; Adams-Webber, T.; Ito, S.; Koren, G. Pregnancy-Associated Changes in Pharmacokinetics: A Systematic Review. PLoS Med. 2016, 13, e1002160. [Google Scholar] [CrossRef]
- Abduljalil, K.; Furness, P.; Johnson, T.N.; Rostami-Hodjegan, A.; Soltani, H. Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: A database for parameters required in physiologically based pharmacokinetic modelling. Clin. Pharmacokinet. 2012, 51, 365–396. [Google Scholar] [CrossRef]
- Dallmann, A.; Ince, I.; Meyer, M.; Willmann, S.; Eissing, T.; Hempel, G. Gestation-Specific Changes in the Anatomy and Physiology of Healthy Pregnant Women: An Extended Repository of Model Parameters for Physiologically Based Pharmacokinetic Modeling in Pregnancy. Clin. Pharmacokinet. 2017, 56, 1303–1330. [Google Scholar] [CrossRef]
- Ezuruike, U.; Blenkinsop, A.; Pansari, A.; Abduljalil, K. Quantification of Fetal Renal Function Using Fetal Urine Production Rate and Its Reflection on the Amniotic and Fetal Creatinine Levels During Pregnancy. Front. Pediatr. 2022, 10, 841495. [Google Scholar] [CrossRef]
- Abduljalil, K.; Pansari, A.; Jamei, M. Prediction of maternal pharmacokinetics using physiologically based pharmacokinetic models: Assessing the impact of the longitudinal changes in the activity of CYP1A2, CYP2D6 and CYP3A4 enzymes during pregnancy. J. Pharmacokinet. Pharmacodyn. 2020, 47, 361–383. [Google Scholar] [CrossRef]
- Coutant, D.E.; Kulanthaivel, P.; Turner, P.K.; Bell, R.L.; Baldwin, J.; Wijayawardana, S.R.; Pitou, C.; Hall, S.D. Understanding Disease-Drug Interactions in Cancer Patients: Implications for Dosing within the Therapeutic Window. Clin. Pharmacol. Ther. 2015, 98, 76–86. [Google Scholar] [CrossRef]
- Cheeti, S.; Budha, N.R.; Rajan, S.; Dresser, M.J.; Jin, J.Y. A physiologically based pharmacokinetic (PBPK) approach to evaluate pharmacokinetics in patients with cancer. Biopharm. Drug Dispos. 2013, 34, 141–154. [Google Scholar] [CrossRef]
- Thai, H.T.; Mazuir, F.; Cartot-Cotton, S.; Veyrat-Follet, C. Optimizing pharmacokinetic bridging studies in paediatric oncology using physiologically-based pharmacokinetic modelling: Application to docetaxel. Br. J. Clin. Pharmacol. 2015, 80, 534–547. [Google Scholar] [CrossRef]
- Kremer, J.M.; Wilting, J.; Janssen, L.H. Drug binding to human alpha-1-acid glycoprotein in health and disease. Pharmacol. Rev. 1988, 40, 1–47. [Google Scholar]
- Schwenger, E.; Reddy, V.P.; Moorthy, G.; Sharma, P.; Tomkinson, H.; Masson, E.; Vishwanathan, K. Harnessing Meta-analysis to Refine an Oncology Patient Population for Physiology-Based Pharmacokinetic Modeling of Drugs. Clin. Pharmacol. Ther. 2018, 103, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Vasilogianni, A.M.; Al-Majdoub, Z.M.; Achour, B.; Peters, S.A.; Rostami-Hodjegan, A.; Barber, J. Proteomics of colorectal cancer liver metastasis: A quantitative focus on drug elimination and pharmacodynamics effects. Br. J. Clin. Pharmacol. 2022, 88, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Launay-Vacher, V.; Janus, N.; Deray, G. Renal insufficiency and cancer treatments. ESMO Open 2016, 1, e000091. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.M.; Scott, J.M.; McPartlin, J.M.; Fernandez-Ballart, J.D. The pregnancy-related decrease in fasting plasma homocysteine is not explained by folic acid supplementation, hemodilution, or a decrease in albumin in a longitudinal study. Am. J. Clin. Nutr. 2002, 76, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Stock, B.; Patterson, R.J.; Levy, G. Serum protein binding of drugs during and after pregnancy in humans. Clin. Pharmacol. Ther. 1980, 28, 253–261. [Google Scholar] [CrossRef]
- Feghali, M.; Venkataramanan, R.; Caritis, S. Pharmacokinetics of drugs in pregnancy. Semin. Perinatol. 2015, 39, 512–519. [Google Scholar] [CrossRef]
- Choi, S.Y.; Koh, K.H.; Jeong, H. Isoform-specific regulation of cytochromes P450 expression by estradiol and progesterone. Drug Metab. Dispos. 2013, 41, 263–269. [Google Scholar] [CrossRef]
- Khatri, R.; Kulick, N.; Rementer, R.J.B.; Fallon, J.K.; Sykes, C.; Schauer, A.P.; Malinen, M.M.; Mosedale, M.; Watkins, P.B.; Kashuba, A.D.M.; et al. Pregnancy-Related Hormones Increase Nifedipine Metabolism in Human Hepatocytes by Inducing CYP3A4 Expression. J. Pharm. Sci. 2021, 110, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Kendra, K.L.; Plummer, R.; Salgia, R.; O’Brien, M.E.; Paul, E.M.; Suttle, A.B.; Compton, N.; Xu, C.F.; Ottesen, L.H.; Villalona-Calero, M.A. A multicenter phase I study of pazopanib in combination with paclitaxel in first-line treatment of patients with advanced solid tumors. Mol. Cancer Ther. 2015, 14, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Dallmann, A.; Ince, I.; Coboeken, K.; Eissing, T.; Hempel, G. A Physiologically Based Pharmacokinetic Model for Pregnant Women to Predict the Pharmacokinetics of Drugs Metabolized Via Several Enzymatic Pathways. Clin. Pharmacokinet. 2018, 57, 749–768. [Google Scholar] [CrossRef]
- Fundation, N.K. Estimated Glomerular Filtration Rate (eGFR). Available online: https://www.kidney.org/atoz/content/gfr (accessed on 21 June 2023).
- US Food and Drug Administration: CALQUENCE (acalabrutinib) Tablets: Updated 8/2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/216387Orig2s000Correctedlbl.pdf (accessed on 30 October 2023).
- Mendes, M.S.; Hatley, O.; Gill, K.L.; Yeo, K.R.; Ke, A.B. A physiologically based pharmacokinetic-pharmacodynamic modelling approach to predict incidence of neutropenia as a result of drug-drug interactions of paclitaxel in cancer patients. Eur. J. Pharm. Sci. 2020, 150, 105355. [Google Scholar] [CrossRef]
- Zhou, D.; Podoll, T.; Xu, Y.; Moorthy, G.; Vishwanathan, K.; Ware, J.; Slatter, J.G.; Al-Huniti, N. Evaluation of the Drug-Drug Interaction Potential of Acalabrutinib and Its Active Metabolite, ACP-5862, Using a Physiologically-Based Pharmacokinetic Modeling Approach. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Shou, M.; Martinet, M.; Korzekwa, K.R.; Krausz, K.W.; Gonzalez, F.J.; Gelboin, H.V. Role of human cytochrome P450 3A4 and 3A5 in the metabolism of taxotere and its derivatives: Enzyme specificity, interindividual distribution and metabolic contribution in human liver. Pharmacogenetics 1998, 8, 391–401. [Google Scholar] [CrossRef]
- van Zuylen, L.; Verweij, J.; Nooter, K.; Brouwer, E.; Stoter, G.; Sparreboom, A. Role of intestinal P-glycoprotein in the plasma and fecal disposition of docetaxel in humans. Clin. Cancer Res. 2000, 6, 2598–2603. [Google Scholar]
- Calquence (Acalabrutinib) Multi-Discipline Review. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/210259Orig1s000MultidisciplineR.pdf (accessed on 5 July 2023).
- Wright, J.G.; Boddy, A.V.; Highley, M.; Fenwick, J.; McGill, A.; Calvert, A.H. Estimation of glomerular filtration rate in cancer patients. Br. J. Cancer 2001, 84, 452–459. [Google Scholar] [CrossRef]
- Wagner, C.; Zhao, P.; Pan, Y.; Hsu, V.; Grillo, J.; Huang, S.M.; Sinha, V. Application of Physiologically Based Pharmacokinetic (PBPK) Modeling to Support Dose Selection: Report of an FDA Public Workshop on PBPK. CPT Pharmacomet. Syst. Pharmacol. 2015, 4, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Villalona-Calero, M.A.; Weiss, G.R.; Burris, H.A.; Kraynak, M.; Rodrigues, G.; Drengler, R.L.; Eckhardt, S.G.; Reigner, B.; Moczygemba, J.; Burger, H.U.; et al. Phase I and pharmacokinetic study of the oral fluoropyrimidine capecitabine in combination with paclitaxel in patients with advanced solid malignancies. J. Clin. Oncol. 1999, 17, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, E.; Verweij, J.; De Bruijn, P.; Loos, W.J.; Pillay, M.; Buijs, D.; Sparreboom, A. Measurement of fraction unbound paclitaxel in human plasma. Drug Metab. Dispos. 2000, 28, 1141–1145. [Google Scholar] [PubMed]
- Brunsvig, P.F.; Andersen, A.; Aamdal, S.; Kristensen, V.; Olsen, H. Pharmacokinetic analysis of two different docetaxel dose levels in patients with non-small cell lung cancer treated with docetaxel as monotherapy or with concurrent radiotherapy. BMC Cancer 2007, 7, 197. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Harrington, B.; O’Brien, S.; Jones, J.A.; Schuh, A.; Devereux, S.; Chaves, J.; Wierda, W.G.; Awan, F.T.; Brown, J.R.; et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Rosing, H.; Lustig, V.; van Warmerdam, L.J.; Huizing, M.T.; ten Bokkel Huinink, W.W.; Schellens, J.H.; Rodenhuis, S.; Bult, A.; Beijnen, J.H. Pharmacokinetics and metabolism of docetaxel administered as a 1-h intravenous infusion. Cancer Chemother. Pharmacol. 2000, 45, 213–218. [Google Scholar] [CrossRef]
- van Hasselt, J.G.C.; van Calsteren, K.; Heyns, L.; Han, S.; Mhallem Gziri, M.; Schellens, J.H.M.; Beijnen, J.H.; Huitema, A.D.R.; Amant, F. Optimizing anticancer drug treatment in pregnant cancer patients: Pharmacokinetic analysis of gestation-induced changes for doxorubicin, epirubicin, docetaxel and paclitaxel. Ann. Oncol. 2014, 25, 2059–2065. [Google Scholar] [CrossRef]
- Janssen, J.M.; Van Calsteren, K.; Dorlo, T.P.C.; Halaska, M.J.; Fruscio, R.; Ottevanger, P.; Schroder, C.P.; Boere, I.; Witteveen, P.O.; Painter, R.C.; et al. Population Pharmacokinetics of Docetaxel, Paclitaxel, Doxorubicin and Epirubicin in Pregnant Women with Cancer: A Study from the International Network of Cancer, Infertility and Pregnancy (INCIP). Clin. Pharmacokinet. 2021, 60, 775–784. [Google Scholar] [CrossRef]
- Calquence (Acalabrutinib) NDA 210259 S-006/S-007 Multi-Discipline Review. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/210259Orig1s006,%20s007.pdf (accessed on 15 August 2023).
- Leong, R.; Grimstein, M.; DeMaria, P.; Norsworthy, K.J.; Fletcher, E.P.; Shord, S. Landscape and Regulatory Perspective on Oncology Drugs in Pregnancy. J. Clin. Pharmacol. 2023, 63 (Suppl. S1), S170–S175. [Google Scholar] [CrossRef]
- Lu, D.; Joshi, A.; Li, H.; Zhang, N.; Ren, M.M.; Gao, Y.; Wada, R.; Jin, J.Y. Model-based meta-analysis for quantifying Paclitaxel dose response in cancer patients. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, e115. [Google Scholar] [CrossRef]
- Stroh, M.; Green, M.; Cha, E.; Zhang, N.; Wada, R.; Jin, J. Meta-analysis of published efficacy and safety data for docetaxel in second-line treatment of patients with advanced non-small-cell lung cancer. Cancer Chemother. Pharmacol. 2016, 77, 485–494. [Google Scholar] [CrossRef]
- Harvey, V.; Mouridsen, H.; Semiglazov, V.; Jakobsen, E.; Voznyi, E.; Robinson, B.A.; Groult, V.; Murawsky, M.; Cold, S. Phase III trial comparing three doses of docetaxel for second-line treatment of advanced breast cancer. J. Clin. Oncol. 2006, 24, 4963–4970. [Google Scholar] [CrossRef]
- Baker, S.D.; van Schaik, R.H.N.; Rivory, L.P.; Tije, A.J.T.; Dinh, K.; Graveland, W.J.; Schenk, P.W.; Charles, K.A.; Clarke, S.J.; Carducci, M.A.; et al. Factors affecting cytochrome P-450 3A activity in cancer patients. Clin. Cancer Res. 2004, 10, 8341–8350. [Google Scholar] [CrossRef]
- Harvey, R.D.; Morgan, E.T. Cancer, inflammation, and therapy: Effects on cytochrome P450-mediated drug metabolism and implications for novel immunotherapeutic agents. Clin. Pharm. Ther. 2014, 96, 449–457. [Google Scholar] [CrossRef]
- Walle, T.; Walle, U.K.; Kumar, G.N.; Bhalla, K.N. Taxol metabolism and disposition in cancer patients. Drug Metab. Dispos. 1995, 23, 506–512. [Google Scholar]
- Berg, S.L.; Tolcher, A.; A O’Shaughnessy, J.; Denicoff, A.M.; Noone, M.; Ognibene, F.P.; Cowan, K.H.; Balis, F.M. Effect of R-verapamil on the pharmacokinetics of paclitaxel in women with breast cancer. J. Clin. Oncol. 1995, 13, 2039–2042. [Google Scholar] [CrossRef]
- Engels, F.K.; Tije, A.J.; Baker, S.D.; Lee, C.K.K.; Loos, W.J.; Vulto, A.G.; Verweij, J.; Sparreboom, A. Effect of cytochrome P450 3A4 inhibition on the pharmacokinetics of docetaxel. Clin. Pharmacol. Ther. 2004, 75, 448–454. [Google Scholar] [CrossRef]
- Urien, S.; Barré, J.; Morin, C.; Paccaly, A.; Montay, G.; Tillement, J.-P. Docetaxel serum protein binding with high affinity to alpha 1-acid glycoprotein. Investig. New Drugs 1996, 14, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Pronk, L.C.; Vasey, P.; Sparreboom, A.; Reigner, B.; Planting, A.S.T.; Gordon, R.J.; Osterwalder, B.; Verweij, J.; Twelves, C. A phase I and pharmacokinetic study of the combination of capecitabine and docetaxel in patients with advanced solid tumours. Br. J. Cancer 2000, 83, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Bruno, R.; Vivier, N.; Vergniol, J.C.; De Phillips, S.L.; Montay, G.; Sheiner, L.B. A population pharmacokinetic model for docetaxel (Taxotere): Model building and validation. J. Pharmacokinet. Biopharm. 1996, 24, 153–172. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.J.; Rivory, L.P. Clinical pharmacokinetics of docetaxel. Clin. Pharmacokinet. 1999, 36, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Oostendorp, R.L.; Huitema, A.; Rosing, H.; Jansen, R.S.; ter Heine, R.; Keessen, M.; Beijnen, J.H.; Schellens, J.H. Coadministration of ritonavir strongly enhances the apparent oral bioavailability of docetaxel in patients with solid tumors. Clin. Cancer Res. 2009, 15, 4228–4233. [Google Scholar] [CrossRef]
- Hamberg, P.; Mathijssen, R.H.J.; De Bruijn, P.; Leonowens, C.; Van Der Biessen, D.; Eskens, F.A.L.M.; Sleijfer, S.; Verweij, J.; De Jonge, M.J.A. Impact of pazopanib on docetaxel exposure: Results of a phase I combination study with two different docetaxel schedules. Cancer Chemother. Pharmacol. 2015, 75, 365–371. [Google Scholar] [CrossRef]
- Engels, F.K.; Mathot, R.A.; Loos, W.J.; van Schaik, R.H.N.; Verweij, J. Influence of high-dose ketoconazole on the pharmacokinetics of docetaxel. Cancer Biol. Ther. 2006, 5, 833–839. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Healthy Subjects | Cancer | Pregnancy | |
|---|---|---|---|
| Albumin (g/L) | 40–50 [12] | 35–41 [12,13,14] | 38.5 (30 GW), 37.6 (34 GW) [8] |
| Alpha-1-acid glycoprotein (AGP) (g/L) | 0.5–1 [15] | 1.34–1.38 [12,13,14] | 0.6 (30–35 GW) [8] |
| CYP3A4 expression/activity | -- | 10–33% decrease [12,16,17], No change [13,24] | 60% increase throughout [25], 75% increase (27 GW), 130% increase (at term) [11], 50–100% increase [21] |
| CYP2C8 expression/activity | -- | No change to minimum reduction [16,17] | 150% increase [22,23] |
| GFR (mL/min/1.73 m2) | 90–120 [26] | Reduced, less than 90 [18] | 160 (26 GW), 156 (36 GW) [8] |
| Physiological Parameter | Cancer (SimCYP V21) | Pregnancy (32 GW) (SimCYP V21) | Modified Pregnancy |
|---|---|---|---|
| Albumin | 15% decrease | 25% decrease | 25% decrease |
| Alpha-1-acid glycoprotein (AGP) | 100% increase | 30% decrease | 30% decrease |
| CYP3A4 abundance | No change | 100% increase | 100% increase |
| CYP2C8 abundance | No change | No change | 150% increase # |
| GFR (mL/min/1.73 m2) | 30–40% decrease | 25% increase | 25% increase |
| Acalabrutinib 100 mg BID (Day 8) | Cmax (ng/mL) | AUC0–24h (ng/mL·h) |
|---|---|---|
| Observed: non-pregnant patients with cancer (Byrd et al. (2016) [38]) | 827 | 1850 |
| Predicted: HV population | 585 | 1702 |
| Predicted: Cancer population | 566 | 1719 |
| Ratio HV/Cancer | 1.03 | 0.99 |
| Predicted: Pregnancy (32 GW) | 251 | 662 |
| Ratio Pregnancy/Cancer | 0.44 | 0.39 |
| Ratio Pregnancy/HV | 0.43 | 0.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Grimstein, M.; Pressly, M.; Fletcher, E.P.; Shord, S.; Leong, R. Utility of Physiologically Based Pharmacokinetic Modeling to Investigate the Impact of Physiological Changes of Pregnancy and Cancer on Oncology Drug Pharmacokinetics. Pharmaceutics 2023, 15, 2727. https://doi.org/10.3390/pharmaceutics15122727
Yang X, Grimstein M, Pressly M, Fletcher EP, Shord S, Leong R. Utility of Physiologically Based Pharmacokinetic Modeling to Investigate the Impact of Physiological Changes of Pregnancy and Cancer on Oncology Drug Pharmacokinetics. Pharmaceutics. 2023; 15(12):2727. https://doi.org/10.3390/pharmaceutics15122727
Chicago/Turabian StyleYang, Xinxin, Manuela Grimstein, Michelle Pressly, Elimika Pfuma Fletcher, Stacy Shord, and Ruby Leong. 2023. "Utility of Physiologically Based Pharmacokinetic Modeling to Investigate the Impact of Physiological Changes of Pregnancy and Cancer on Oncology Drug Pharmacokinetics" Pharmaceutics 15, no. 12: 2727. https://doi.org/10.3390/pharmaceutics15122727