
Impact of Peptide Structure on Colonic Stability and Tissue Permeability
 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Peptide Synthesis
2.2.1. Cyclisation
2.2.2. N-Terminal Acetylation
2.2.3. Peptide Purification and Analysis
2.3. Peptide Stability in a Human Colon Model
2.4. Peptide Tissue Permeability
2.5. Data Analysis and Statistics
3. Results and Discussion
3.1. Synthesis of Oxytocin-Based Peptides
3.2. Insertion of Three D-Amino Acids to Oxytocin Confers Significant Stabilisation
3.3. Chloroacetyl Cyclisation Shows Superior Colonic Stabilisation over Native Oxytocin
3.4. Site of D-Amino Acid Substitution May Be more Important Than Number of Substitutions
3.5. Number of D-Amino Acid Substitutions in Linear Analogues Positively Correlates with Colonic Stability
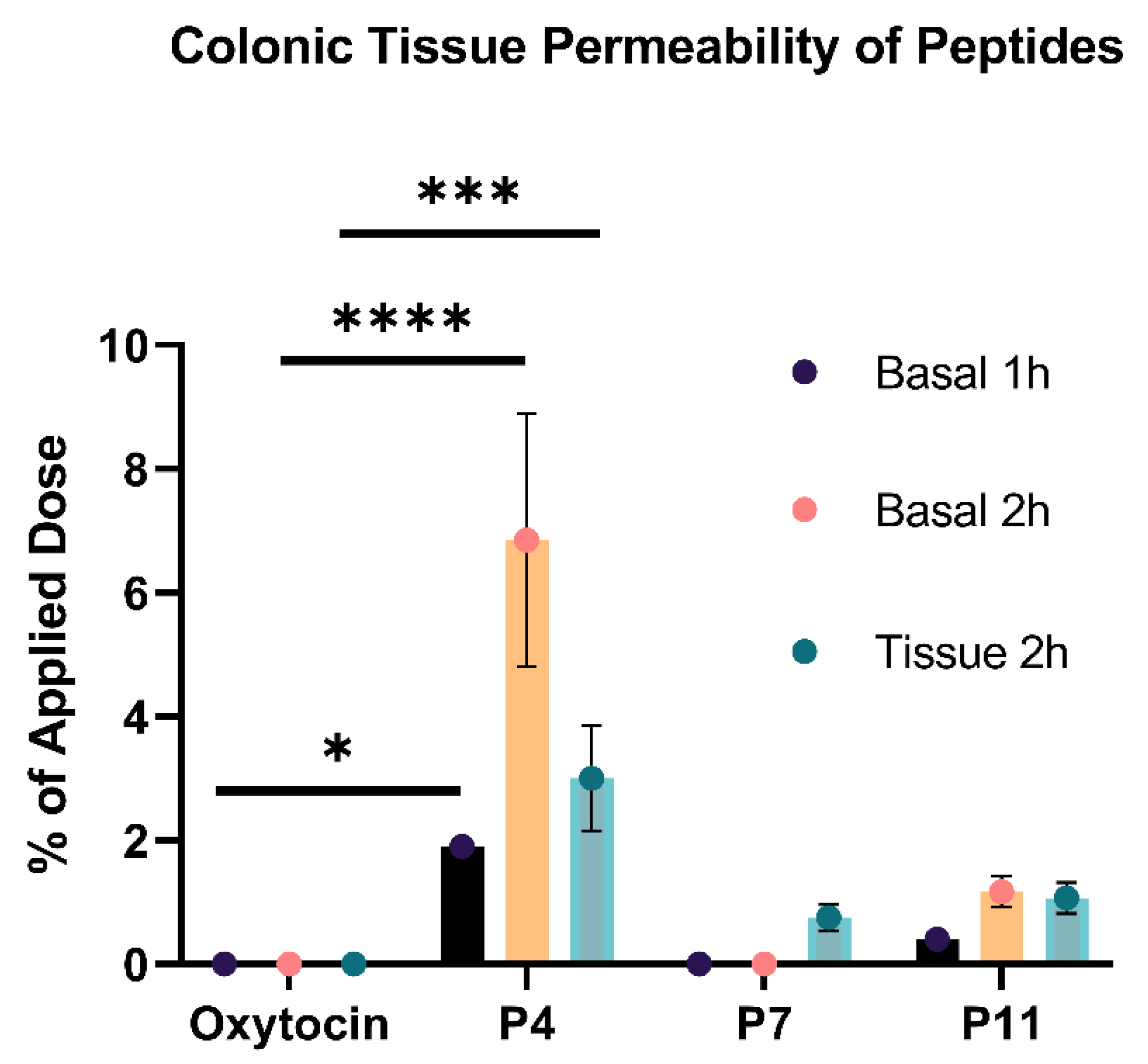
3.6. Number of D-Amino Acid Insertions Positively Correlates with Colonic Tissue Permeability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Vass, P.; Démuth, B.; Hirsch, E.; Nagy, B.; Andersen, S.K.; Vigh, T.; Verreck, G.; Csontos, I.; Nagy, Z.K.; Marosi, G. Drying technology strategies for colon-targeted oral delivery of biopharmaceuticals. J. Control. Release 2019, 296, 162–178. [Google Scholar] [CrossRef] [PubMed]
- Räder, A.F.B.; Weinmüller, M.; Reichart, F.; Schumacher-Klinger, A.; Merzbach, S.; Gilon, C.; Hoffman, A.; Kessler, H. Orally Active Peptides: Is There a Magic Bullet? Angew. Chem. 2018, 57, 14414–14438. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. Advances in oral peptide therapeutics. Nat. Rev. Drug Discov. 2019, 19, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.M.; Rauch, M.; Gilmore, M.S. The Commensal Microbiology of the Gastrointestinal Tract. Adv. Exp. Med. Biol. 2008, 635, 15–28. [Google Scholar] [CrossRef]
- Soenen, S.; Rayner, C.K.; Jones, K.L.; Horowitz, M. The ageing gastrointestinal tract. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 12–18. [Google Scholar] [CrossRef]
- Han, Y.; Gao, Z.; Chen, L.; Kang, L.; Huang, W.; Jin, M.; Wang, Q.; Bae, Y.H. Multifunctional oral delivery systems for enhanced bioavailability of therapeutic peptides/proteins. Acta Pharm. Sin. B 2019, 9, 902–922. [Google Scholar] [CrossRef]
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E.W. Antibacterial peptides for therapeutic use: Obstacles and realistic outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472. [Google Scholar] [CrossRef]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef]
- Mahato, R.I.; Narang, A.S.; Thoma, L.; Miller, D.D. Emerging trends in oral delivery of peptide and protein drugs. Crit. Rev. Ther. Drug Carr. Syst. 2003, 20, 153–214. [Google Scholar] [CrossRef]
- Paone, P.; Cani, P.D. Mucus barrier, mucins and gut microbiota: The expected slimy partners? Gut 2020, 69, 2232–2243. [Google Scholar] [CrossRef] [PubMed]
- Lundquist, P.; Artursson, P. Oral absorption of peptides and nanoparticles across the human intestine: Opportunities, limitations and studies in human tissues. Adv. Drug Deliv. Rev. 2016, 106, 256–276. [Google Scholar] [CrossRef] [PubMed]
- Taherali, F.; Varum, F.; Basit, A.W. A slippery slope: On the origin, role and physiology of mucus. Adv. Drug Deliv. Rev. 2018, 124, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Carino, G.P.; Mathiowitz, E. Oral insulin delivery. Adv. Drug Deliv. Rev. 1999, 35, 249–257. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Gokarn, Y.; Mitragotri, S. Non-invasive delivery strategies for biologics. Nat. Rev. Drug Discov. 2018, 18, 19–40. [Google Scholar] [CrossRef]
- Aguirre, T.; Teijeiro-Osorio, D.; Rosa, M.; Coulter, I.; Alonso, M.; Brayden, D. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv. Drug Deliv. Rev. 2016, 106, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Maher, S.; Mrsny, R.J.; Brayden, D.J. Intestinal permeation enhancers for oral peptide delivery. Adv. Drug Deliv. Rev. 2016, 106, 277–319. [Google Scholar] [CrossRef]
- Aungst, B.J.; Saitoh, H.; Burcham, D.L.; Huang, S.-M.; Mousa, S.A.; Hussain, M.A. Enhancement of the intestinal absorption of peptides and nonpeptides. J. Control. Release 1996, 41, 19–31. [Google Scholar] [CrossRef]
- Fralick, M.; Schneeweiss, S.; Wallis, C.J.D.; Jung, E.H.; Kesselheim, A.S. Desmopressin and the risk of hyponatremia: A population-based cohort study. PLoS Med. 2019, 16, e1002930. [Google Scholar] [CrossRef] [Green Version]
- Menon, C.; Berry, E.; Ockelford, P. Beneficial effect of D.D.A.V.P. on bleeding-time in von willebrand’s disease. Lancet 1978, 312, 743–744. [Google Scholar] [CrossRef]
- Emc. Neoral Soft Gelatin Capsules—Summary of Product Characteristics (SmPC). Available online: https://www.medicines.org.uk/emc/product/1034/smpc#INDICATIONS (accessed on 14 July 2022).
- Gursoy, R.N.; Benita, S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed. Pharmacother. 2004, 58, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Biron, E.; Chatterjee, J.; Ovadia, O.; Langenegger, D.; Brueggen, J.; Hoyer, D.; Schmid, H.A.; Jelinek, R.; Gilon, C.; Hoffman, A.; et al. Improving Oral Bioavailability of Peptides by Multiple N-Methylation: Somatostatin Analogues. Angew. Chem. Int. Ed. 2008, 47, 2595–2599. [Google Scholar] [CrossRef] [PubMed]
- Haviv, F.; Fitzpatrick, T.D.; Swenson, R.E.; Nichols, C.J.; Mort, N.A.; Bush, E.N.; Diaz, G.; Bammert, G.; Nguyen, A. Effect of N-Methyl Substitution of the Peptide Bonds in Luteinizing Hormone-Releasing Hormone Agonists. J. Med. Chem. 1993, 36, 363–369. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Approves First Oral GLP-1 Treatment for Type 2 Diabetes. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-oral-glp-1-treatment-type-2-diabetes (accessed on 27 May 2022).
- GlobeNewsWire. Chiasma Announces FDA Approval of MYCAPSSA® (Octreotide). Available online: https://www.globenewswire.com/news-release/2020/06/26/2054210/0/en/Chiasma-Announces-FDA-Approval-of-MYCAPSSA-Octreotide-Capsules-the-First-and-Only-Oral-Somatostatin-Analog.html (accessed on 27 May 2022).
- Eldor, R.; Arbit, E.; Corcos, A.; Kidron, M. Glucose-Reducing Effect of the ORMD-0801 Oral Insulin Preparation in Patients with Uncontrolled Type 1 Diabetes: A Pilot Study. PLoS ONE 2013, 8, e59524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biermasz, N.R. New medical therapies on the horizon: Oral octreotide. Pituitary 2017, 20, 149–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Sangfuang, N.; McCoubrey, L.E.; Yadav, V.; Elbadawi, M.; Orlu, M.; Gaisford, S.; Basit, A.W. Advancing oral delivery of biologics: Machine learning predicts peptide stability in the gastrointestinal tract. Int. J. Pharm. 2023, 634, 122643. [Google Scholar] [CrossRef] [PubMed]
- Esparza, K.; Jayawardena, D.; Onyuksel, H. Phospholipid Micelles for Peptide Drug Delivery. Methods Mol. Biol. 2019, 2000, 43–57. [Google Scholar] [CrossRef]
- Plaza-Oliver, M.; Santander-Ortega, M.J.; Lozano, M.V. Current approaches in lipid-based nanocarriers for oral drug delivery. Drug Deliv. Transl. Res. 2021, 11, 471. [Google Scholar] [CrossRef]
- Hess, S.; Ovadia, O.; Shalev, D.E.; Senderovich, H.; Qadri, B.; Yehezkel, T.; Salitra, Y.; Sheynis, T.; Jelinek, R.; Gilon, C.; et al. Effect of structural and conformation modifications, including backbone cyclization, of hydrophilic hexapeptides on their intestinal permeability and enzymatic stability. J. Med. Chem. 2007, 50, 6201–6211. [Google Scholar] [CrossRef]
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Samanen, J.; Ali, F.; Romoff, T.; Calvo, R.; Sorenson, E.; Vasko, J.; Storer, B.; Berry, D.; Bennett, D.; Strohsacker, M.; et al. Development of a Small RGD Peptide Fibrinogen Receptor Antagonist with Potent Antiaggregatory Activity in Vitro. J. Med. Chem. 1991, 34, 3114–3125. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Xu, B. Inspiration from the mirror: D-amino acid containing peptides in biomedical approaches. Biomol. Concepts 2016, 7, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Maxian, T.; Gerlitz, L.; Riedl, S.; Rinner, B.; Zweytick, D. Effect of l- to d-amino acid substitution on stability and activity of antitumor peptide rdp215 against human melanoma and glioblastoma. Int. J. Mol. Sci. 2021, 22, 8469. [Google Scholar] [CrossRef]
- Awad, A.; Madla, C.M.; McCoubrey, L.E.; Ferraro, F.; Gavins, F.K.; Buanz, A.; Gaisford, S.; Orlu, M.; Siepmann, F.; Siepmann, J.; et al. Clinical translation of advanced colonic drug delivery technologies. Adv. Drug Deliv. Rev. 2022, 181, 114076. [Google Scholar] [CrossRef]
- Varum, F.; Freire, A.C.; Bravo, R.; Basit, A.W. OPTICORETM, an innovative and accurate colonic targeting technology. Int. J. Pharm. 2020, 583, 119372. [Google Scholar] [CrossRef]
- Bak, A.; Ashford, M.; Brayden, D.J. Local delivery of macromolecules to treat diseases associated with the colon. Adv. Drug Deliv. Rev. 2018, 136–137, 2–27. [Google Scholar] [CrossRef] [PubMed]
- McCoubrey, L.E.; Favaron, A.; Awad, A.; Orlu, M.; Gaisford, S.; Basit, A.W. Colonic drug delivery: Formulating the next generation of colon-targeted therapeutics. J. Control. Release 2023, 353, 1107–1126. [Google Scholar] [CrossRef]
- Durán-Lobato, M.; Niu, Z.; Alonso, M.J. Oral Delivery of Biologics for Precision Medicine. Adv. Mater. 2020, 32, 1901935. [Google Scholar] [CrossRef]
- Wang, J.; Yadav, V.; Smart, A.L.; Tajiri, S.; Basit, A.W. Stability of peptide drugs in the colon. Eur. J. Pharm. Sci. 2015, 78, 31–36. [Google Scholar] [CrossRef]
- Wang, J.; Yadav, V.; Parry, A.; Tajiri, S.; Basit, A.W. Toward Oral Delivery of Biopharmaceuticals: An Assessment of the Gastrointestinal Stability of 17 Peptide Drugs. Mol. Pharm. 2015, 12, 966–973. [Google Scholar] [CrossRef] [Green Version]
- Timmerman, P.; Beld, J.; Puijk, W.C.; Meloen, R.H. Rapid and Quantitative Cyclization of Multiple Peptide Loops onto Synthetic Scaffolds for Structural Mimicry of Protein Surfaces. ChemBioChem 2005, 6, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Lung, F.-D.T.; King, C.R.; Roller, P.P. Development of non-phosphorylated cyclic thioether peptide binding to the Grb2-SH2 domain. Lett. Pept. Sci. 1999, 6, 45–49. [Google Scholar] [CrossRef]
- Varland, S.; Osberg, C.; Arnesen, T. N-terminal modifications of cellular proteins: The enzymes involved, their substrate specificities and biological effects. Proteomics 2015, 15, 2385–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ree, R.; Varland, S.; Arnesen, T. Spotlight on protein N-terminal acetylation. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jörnvall, H. Acetylation of protein N-terminal amino groups structural observations on α-amino acetylated proteins. J. Theor. Biol. 1975, 55, 1–12. [Google Scholar] [CrossRef]
- Aksnes, H.; Hole, K.; Arnesen, T. Molecular, Cellular, and Physiological Significance of N-Terminal Acetylation. Int. Rev. Cell Mol. Biol. 2015, 316, 267–305. [Google Scholar] [CrossRef]
- Yadav, V.; Gaisford, S.; Merchant, H.A.; Basit, A.W. Colonic bacterial metabolism of corticosteroids. Int. J. Pharm. 2013, 457, 268–274. [Google Scholar] [CrossRef]
- Basit, A.W.; Newton, J.M.; Lacey, L.F. Susceptibility of the H2-receptor antagonists cimetidine, famotidine and nizatidine, to metabolism by the gastrointestinal microflora. Int. J. Pharm. 2002, 237, 23–33. [Google Scholar] [CrossRef]
- Yadav, V.; Mai, Y.; McCoubrey, L.E.; Wada, Y.; Tomioka, M.; Kawata, S.; Charde, S.; Basit, A.W. 5-Aminolevulinic Acid as a Novel Therapeutic for Inflammatory Bowel Disease. Biomedicines 2021, 9, 578. [Google Scholar] [CrossRef]
- Vertzoni, M.; Kersten, E.; van der Mey, D.; Muenster, U.; Reppas, C. Evaluating the clinical importance of bacterial degradation of therapeutic agents in the lower intestine of adults using adult fecal material. Eur. J. Pharm. Sci. 2018, 125, 142–150. [Google Scholar] [CrossRef]
- Mai, Y.; Murdan, S.; Awadi, M.; Basit, A. Establishing an in vitro permeation model to predict the in vivo sex-related influence of PEG 400 on oral drug absorption. Int. J. Pharm. 2018, 542, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Riniker, S.; Landrum, G.A. Similarity maps—A visualization strategy for molecular fingerprints and machine-learning methods. J. Cheminformatics 2013, 5, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Burbach, J.P. N-acetyl-vasopressin- and N-acetyl-oxytocin-like substances: Isolation and characterization in the rat neurointermediate pituitary and presence in the brain. J. Neuroendocrinol. 1989, 1, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Tugyi, R.; Uray, K.; Iván, D.; Fellinger, E.; Perkins, A.; Hudecz, F. Partial D-Amino Acid Substitution: Improved Enzymatic Stability and Preserved Ab Recognition of a MUC2 Epitope Peptide. Proc. Natl. Acad. Sci. USA 2005, 102, 413–418. [Google Scholar] [CrossRef]
- Lu, J.; Xu, H.; Xia, J.; Ma, J.; Xu, J.; Li, Y.; Feng, J. D- and Unnatural Amino Acid Substituted Antimicrobial Peptides With Improved Proteolytic Resistance and Their Proteolytic Degradation Characteristics. Front. Microbiol. 2020, 11, 563030. [Google Scholar] [CrossRef] [PubMed]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminformatics 2015, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Gimpl, G.; Reitz, J.; Brauer, S.; Trossen, C. Oxytocin receptors: Ligand binding, signalling and cholesterol dependence. Prog. Brain Res. 2008, 170, 193–204. [Google Scholar] [CrossRef]
- Moberg, K.U.; Handlin, L.; Kendall-Tackett, K.; Petersson, M. Oxytocin is a principal hormone that exerts part of its effects by active fragments. Med. Hypotheses 2019, 133, 109394. [Google Scholar] [CrossRef]
- Nielsen, D.S.; Shepherd, N.E.; Xu, W.; Lucke, A.J.; Stoermer, M.J.; Fairlie, D.P. Orally Absorbed Cyclic Peptides. Chem. Rev. 2017, 117, 8094–8128. [Google Scholar] [CrossRef] [Green Version]
- Góngora-Benítez, M.; Tulla-Puche, J.; Albericio, F. Multifaceted roles of disulfide bonds. peptides as therapeutics. Chem. Rev. 2014, 114, 901–926. [Google Scholar] [CrossRef]
- Li, X.; Du, X.; Li, J.; Gao, Y.; Pan, Y.; Shi, J.; Zhou, N.; Xu, B. Introducing d -amino acid or simple glycoside into small peptides to enable supramolecular hydrogelators to resist proteolysis. Langmuir 2012, 28, 13512–13517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, V.; House, A.; Matiz, S.; McCoubrey, L.E.; Bettano, K.A.; Bhave, L.; Wang, M.; Fan, P.; Zhou, S.; Woodhouse, J.D.; et al. Ileocolonic-Targeted JAK Inhibitor: A Safer and More Effective Treatment for Inflammatory Bowel Disease. Pharmaceutics 2022, 14, 2385. [Google Scholar] [CrossRef] [PubMed]
- Witek, J.; Wang, S.; Schroeder, B.; Lingwood, R.; Dounas, A.; Roth, H.J.; Fouché, M.; Blatter, M.; Lemke, O.; Keller, B.; et al. Rationalization of the Membrane Permeability Differences in a Series of Analogue Cyclic Decapeptides. J. Chem. Inf. Model. 2018, 59, 294–308. [Google Scholar] [CrossRef]
- Linker, S.M.; Schellhaas, C.; Kamenik, A.S.; Veldhuizen, M.M.; Waibl, F.; Roth, H.-J.; Fouché, M.; Rodde, S.; Riniker, S. Lessons for Oral Bioavailability: How Conformationally Flexible Cyclic Peptides Enter and Cross Lipid Membranes. J. Med. Chem. 2023, 66, 2773–2788. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhao, G.; Huang, Y.; Cai, M.; Yan, Q.; Wang, H.; Chen, Y. Enantiomeric Effect of d-Amino Acid Substitution on the Mechanism of Action of α-Helical Membrane-Active Peptides. Int. J. Mol. Sci. 2017, 19, 67. [Google Scholar] [CrossRef] [Green Version]
- Castro, T.G.; Micaêlo, N.M.; Melle-Franco, M. Modeling the secondary structures of the peptaibols antiamoebin I and zervamicin II modified with D-amino acids and proline analogues. J. Mol. Model. 2017, 23, 313. [Google Scholar] [CrossRef]
- Maher, S.; Brayden, D.J.; Casettari, L.; Illum, L. Application of Permeation Enhancers in Oral Delivery of Macromolecules: An Update. Pharmaceutics 2019, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- Maher, S.; Geoghegan, C.; Brayden, D.J. Intestinal permeation enhancers to improve oral bioavailability of macromolecules: Reasons for low efficacy in humans. Expert Opin. Drug Deliv. 2021, 18, 273–300. [Google Scholar] [CrossRef]
- Vaga, S.; Lee, S.; Ji, B.; Andreasson, A.; Talley, N.J.; Agréus, L.; Bidkhori, G.; Kovatcheva-Datchary, P.; Park, J.; Lee, D.; et al. Compositional and functional differences of the mucosal microbiota along the intestine of healthy individuals. Sci. Rep. 2020, 10, 14977. [Google Scholar] [CrossRef]
- Tannergren, C.; Borde, A.; Boreström, C.; Abrahamsson, B.; Lindahl, A. Evaluation of an in vitro faecal degradation method for early assessment of the impact of colonic degradation on colonic absorption in humans. Eur. J. Pharm. Sci. 2014, 57, 200–206. [Google Scholar] [CrossRef]
- Arnold, Y.E.; Thorens, J.; Bernard, S.; Kalia, Y.N. Drug transport across porcine intestine using an ussing chamber system: Regional differences and the effect of P-glycoprotein and CYP3A4 activity on drug absorption. Pharmaceutics 2019, 11, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michiba, K.; Maeda, K.; Kurimori, K.; Enomoto, T.; Shimomura, O.; Takeuchi, T.; Nishiyama, H.; Oda, T.; Kusuhara, H. Characterization of the human intestinal drug transport with ussing chamber system incorporating freshly isolated human jejunum. Drug Metab. Dispos. 2021, 49, 84–93. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
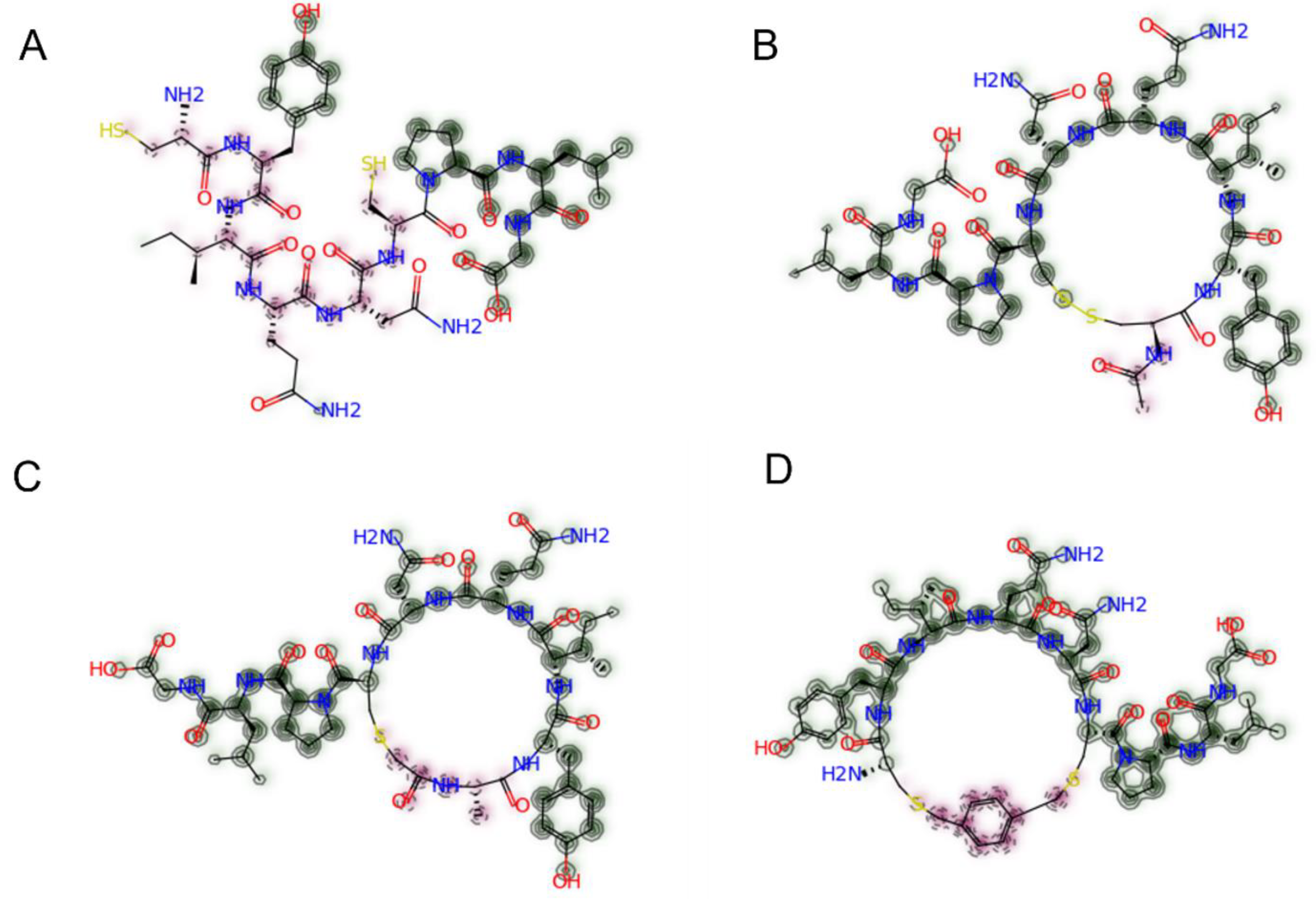
| Peptide | Structure | Amino Acid Sequence | MW (g/mol) | Tanimoto Similarity to Native Oxytocin |
|---|---|---|---|---|
| Oxytocin (Control) | Cyclic (disulfide) |  | 1007.2 | 1.000 |
| P1 | Linear | 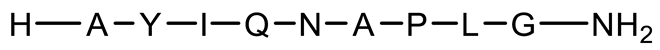 | 946.1 | 0.331 |
| P2 | Cyclic (N-terminal acetylated) |  | 1050.2 | 0.836 |
| P3 | Cyclic (chloroacetyl) | 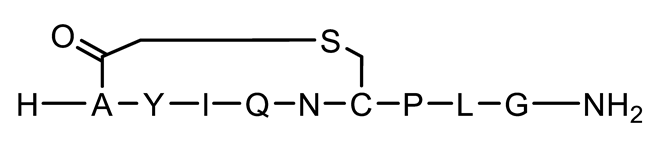 | 1018.2 | 0.777 |
| P4 | Cyclic (disulfide) with 3 D-AAs |  | 1008.2 | 0.645 |
| P5 | Cyclic (chloroacetyl) with 3 D-AAs |  | 1018.2 | 0.609 |
| P6 | Cyclic (thioether) |  | 1112.3 | 0.821 |
| P7 | Cyclic (disulfide) with 1 D-AA | 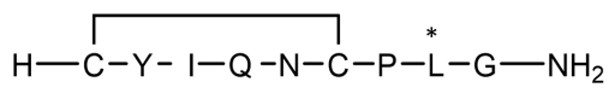 | 1008.2 | 0.852 |
| P8 | Cyclic (disulfide) with 2 D-AAs | 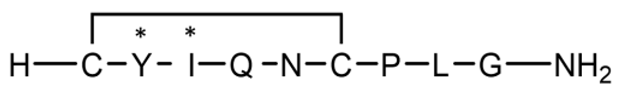 | 1008.2 | 0.753 |
| P9 | Linear with 1 D-AA |  | 946.1 | 0.274 |
| P10 | Linear with 2 D-AAs |  | 946.1 | 0.352 |
| P11 | Linear with 3 D-AAs |  | 946.1 | 0.286 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taherali, F.; Chouhan, N.; Wang, F.; Lavielle, S.; Baran, M.; McCoubrey, L.E.; Basit, A.W.; Yadav, V. Impact of Peptide Structure on Colonic Stability and Tissue Permeability. Pharmaceutics 2023, 15, 1956. https://doi.org/10.3390/pharmaceutics15071956
Taherali F, Chouhan N, Wang F, Lavielle S, Baran M, McCoubrey LE, Basit AW, Yadav V. Impact of Peptide Structure on Colonic Stability and Tissue Permeability. Pharmaceutics. 2023; 15(7):1956. https://doi.org/10.3390/pharmaceutics15071956
Chicago/Turabian StyleTaherali, Farhan, Nerisha Chouhan, Fanjin Wang, Sebastien Lavielle, Maryana Baran, Laura E. McCoubrey, Abdul W. Basit, and Vipul Yadav. 2023. "Impact of Peptide Structure on Colonic Stability and Tissue Permeability" Pharmaceutics 15, no. 7: 1956. https://doi.org/10.3390/pharmaceutics15071956