
Inhibition of Protein Aggregation and Endoplasmic Reticulum Stress as a Targeted Therapy for α-Synucleinopathy
 , , , and
, , , and
Abstract
:
1. Introduction
2. The Characteristic Features and Roles of Various Strains of α-Synuclein
2.1. The Structure of SNCA Gene and α-Synuclein Protein
2.2. Other Genes Related to α-Synuclein Pathology
2.3. Post-Translational Modifications
2.4. Physiological Forms of α-Synuclein
2.5. α-Synuclein Oligomers
2.6. Fibrillar Strains of α-Synuclein
3. Endoplasmic Reticulum Stress Induced by α-Synuclein
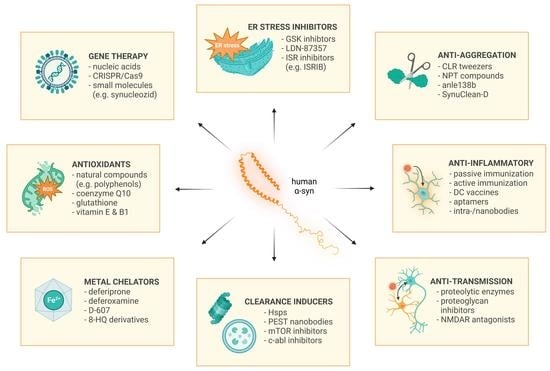
4. Drugs Targeting α-Synuclein
4.1. Gene Therapies
4.2. Natural Compounds
4.3. Monomer Stabilizing Agents
4.4. Increasing the Clearence of α-Synuclein
4.5. α-Synuclein Aggregation Inhibitors
4.6. Endoplasmic Reticulum Stress Inhibition
4.7. Modulators of Mitochondrial Pathways, Oxidative Stress and Metal Metabolism
4.8. Agents Modifying Ubiquitin–Proteasome and Autophagy–Lysosomal System
4.9. Inhibition of α-Synuclein Propagation
4.10. Regulation of Post-Translational Modifications
4.11. Other Approaches
4.12. Passive and Active Immunization
5. Summary and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mhyre, T.R.; Boyd, J.T.; Hamill, R.W.; Maguire-Zeiss, K.A. Parkinson’s Disease. In Protein Aggregation and Fibrillogenesis in Cerebral and Systemic Amyloid Disease; Harris, J.R., Ed.; Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2012; pp. 389–455. ISBN 978-94-007-5416-4. [Google Scholar]
- Ou, Z.; Pan, J.; Tang, S.; Duan, D.; Yu, D.; Nong, H.; Wang, Z. Global Trends in the Incidence, Prevalence, and Years Lived with Disability of Parkinson’s Disease in 204 Countries/Territories From 1990 to 2019. Front. Public Health 2021, 9, 776847. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, Regional, and National Burden of Neurological Disorders, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, Regional, and National Incidence, Prevalence, and Years Lived with Disability for 328 Diseases and Injuries for 195 Countries, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Hamilton, J.L.; Kopil, C.; Beck, J.C.; Tanner, C.M.; Albin, R.L.; Ray Dorsey, E.; Dahodwala, N.; Cintina, I.; Hogan, P.; et al. Current and Projected Future Economic Burden of Parkinson’s Disease in the U.S. NPJ Park. Dis. 2020, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Mechelli, A.; Natale, G.; Volpicelli-Daley, L.; Di Lazzaro, G.; Ghiglieri, V. Alpha-Synuclein in Parkinson’s Disease and Other Synucleinopathies: From Overt Neurodegeneration Back to Early Synaptic Dysfunction. Cell Death Dis. 2023, 14, 176. [Google Scholar] [CrossRef]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and Propagation of α-Synuclein Aggregation in the Nervous System. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef] [Green Version]
- Borghammer, P.; Horsager, J.; Andersen, K.; Van Den Berge, N.; Raunio, A.; Murayama, S.; Parkkinen, L.; Myllykangas, L. Neuropathological Evidence of Body-First vs. Brain-First Lewy Body Disease. Neurobiol. Dis. 2021, 161, 105557. [Google Scholar] [CrossRef]
- Koga, S.; Sekiya, H.; Kondru, N.; Ross, O.A.; Dickson, D.W. Neuropathology and Molecular Diagnosis of Synucleinopathies. Mol. Neurodegener. 2021, 16, 83. [Google Scholar] [CrossRef]
- Irwin, D.J.; Hurtig, H.I. The Contribution of Tau, Amyloid-Beta and Alpha-Synuclein Pathology to Dementia in Lewy Body Disorders. J. Alzheimers Dis. Park. 2018, 8, 444. [Google Scholar] [CrossRef]
- Adamowicz, D.H.; Roy, S.; Salmon, D.P.; Galasko, D.R.; Hansen, L.A.; Masliah, E.; Gage, F.H. Hippocampal α-Synuclein in Dementia with Lewy Bodies Contributes to Memory Impairment and Is Consistent with Spread of Pathology. J. Neurosci. 2017, 37, 1675–1684. [Google Scholar] [CrossRef] [Green Version]
- Asi, Y.T.; Simpson, J.E.; Heath, P.R.; Wharton, S.B.; Lees, A.J.; Revesz, T.; Houlden, H.; Holton, J.L. Alpha-Synuclein MRNA Expression in Oligodendrocytes in MSA. Glia 2014, 62, 964–970. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, U.; Kayed, R. Amyloid β, Tau, and α-Synuclein Aggregates in the Pathogenesis, Prognosis, and Therapeutics for Neurodegenerative Diseases. Prog. Neurobiol. 2022, 214, 102270. [Google Scholar] [CrossRef] [PubMed]
- Van der Perren, A.; Gelders, G.; Fenyi, A.; Bousset, L.; Brito, F.; Peelaerts, W.; Van den Haute, C.; Gentleman, S.; Melki, R.; Baekelandt, V. The Structural Differences between Patient-Derived α-Synuclein Strains Dictate Characteristics of Parkinson’s Disease, Multiple System Atrophy and Dementia with Lewy Bodies. Acta Neuropathol. 2020, 139, 977–1000. [Google Scholar] [CrossRef]
- Ayers, J.I.; Lee, J.; Monteiro, O.; Woerman, A.L.; Lazar, A.A.; Condello, C.; Paras, N.A.; Prusiner, S.B. Different α-Synuclein Prion Strains Cause Dementia with Lewy Bodies and Multiple System Atrophy. Proc. Natl. Acad. Sci. USA 2022, 119, e2113489119. [Google Scholar] [CrossRef]
- Fouke, K.E.; Wegman, M.E.; Weber, S.A.; Brady, E.B.; Román-Vendrell, C.; Morgan, J.R. Synuclein Regulates Synaptic Vesicle Clustering and Docking at a Vertebrate Synapse. Front. Cell Dev. Biol. 2021, 9, 774650. [Google Scholar] [CrossRef]
- Deng, H.; Wang, P.; Jankovic, J. The Genetics of Parkinson Disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhu, R.; Pan, B.; Xu, H.; Olufemi, M.F.; Gathagan, R.J.; Li, Y.; Zhang, L.; Zhang, J.; Xiang, W.; et al. Post-Translational Modifications of Soluble α-Synuclein Regulate the Amplification of Pathological α-Synuclein. Nat. Neurosci. 2023, 26, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Scudamore, O.; Ciossek, T. Increased Oxidative Stress Exacerbates α-Synuclein Aggregation In Vivo. J. Neuropathol. Exp. Neurol. 2018, 77, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berge, N.; Ferreira, N.; Mikkelsen, T.W.; Alstrup, A.K.O.; Tamgüney, G.; Karlsson, P.; Terkelsen, A.J.; Nyengaard, J.R.; Jensen, P.H.; Borghammer, P. Ageing Promotes Pathological Alpha-Synuclein Propagation and Autonomic Dysfunction in Wild-Type Rats. Brain 2021, 144, 1853–1868. [Google Scholar] [CrossRef]
- Alam, P.; Bousset, L.; Melki, R.; Otzen, D.E. α-Synuclein Oligomers and Fibrils: A Spectrum of Species, a Spectrum of Toxicities. J. Neurochem. 2019, 150, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.-J.; Lin, K.-L.; Chen, S.-D.; Liou, C.-W.; Chuang, Y.-C.; Lin, H.-Y.; Lin, T.-K. The Overcrowded Crossroads: Mitochondria, Alpha-Synuclein, and the Endo-Lysosomal System Interaction in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef] [Green Version]
- Trudler, D.; Nazor, K.L.; Eisele, Y.S.; Grabauskas, T.; Dolatabadi, N.; Parker, J.; Sultan, A.; Zhong, Z.; Goodwin, M.S.; Levites, Y.; et al. Soluble α-Synuclein–Antibody Complexes Activate the NLRP3 Inflammasome in HiPSC-Derived Microglia. Proc. Natl. Acad. Sci. USA 2021, 118, e2025847118. [Google Scholar] [CrossRef]
- Liu, M.; Qin, L.; Wang, L.; Tan, J.; Zhang, H.; Tang, J.; Shen, X.; Tan, L.; Wang, C. A-synuclein Induces Apoptosis of Astrocytes by Causing Dysfunction of the Endoplasmic Reticulum-Golgi Compartment. Mol. Med. Rep. 2018, 18, 322–332. [Google Scholar] [CrossRef] [Green Version]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In Vivo Demonstration That α-Synuclein Oligomers Are Toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Minakaki, G.; Menges, S.; Kittel, A.; Emmanouilidou, E.; Schaeffner, I.; Barkovits, K.; Bergmann, A.; Rockenstein, E.; Adame, A.; Marxreiter, F.; et al. Autophagy Inhibition Promotes SNCA/Alpha-Synuclein Release and Transfer via Extracellular Vesicles with a Hybrid Autophagosome-Exosome-like Phenotype. Autophagy 2018, 14, 98–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, J.C. Calling α-Synuclein a Prion Is Scientifically Justifiable. Acta Neuropathol. 2019, 138, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Dehay, B.; Bové, J.; Carballo-Carbajal, I.; Dovero, S.; Pérez-Villalba, A.; Fernagut, P.-O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy Body Extracts from Parkinson Disease Brains Trigger α-Synuclein Pathology and Neurodegeneration in Mice and Monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef]
- Olanow, C.W.; Perl, D.P.; DeMartino, G.N.; McNaught, K.S.P. Lewy-Body Formation Is an Aggresome-Related Process: A Hypothesis. Lancet Neurol. 2004, 3, 496–503. [Google Scholar] [CrossRef]
- Cookson, M.R. α-Synuclein and Neuronal Cell Death. Mol. Neurodegener. 2009, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Brás, I.C.; Outeiro, T.F. Alpha-Synuclein: Mechanisms of Release and Pathology Progression in Synucleinopathies. Cells 2021, 10, 375. [Google Scholar] [CrossRef]
- You, H.; Mariani, L.-L.; Mangone, G.; Le Febvre de Nailly, D.; Charbonnier-Beaupel, F.; Corvol, J.-C. Molecular Basis of Dopamine Replacement Therapy and Its Side Effects in Parkinson’s Disease. Cell Tissue Res. 2018, 373, 111–135. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine Oxidation Mediates Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front. Aging Neurosci. 2020, 11, 373. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Divane, A.; Goedert, M. Assignment of Human α-Synuclein (SNCA) and β-Synuclein (SNCB) Genes to Chromosomes 4q21 and 5q35. Genomics 1995, 27, 379–381. [Google Scholar] [CrossRef]
- Lavedan, C. The Synuclein Family. Genome Res. 1998, 8, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Clough, R.L.; Dermentzaki, G.; Stefanis, L. Functional Dissection of the Alpha-Synuclein Promoter: Transcriptional Regulation by ZSCAN21 and ZNF219. J. Neurochem. 2009, 110, 1479–1490. [Google Scholar] [CrossRef]
- Brenner, S.; Wersinger, C.; Gasser, T. Transcriptional Regulation of the α-Synuclein Gene in Human Brain Tissue. Neurosci. Lett. 2015, 599, 140–145. [Google Scholar] [CrossRef]
- Kantor, B.; Tagliafierro, L.; Gu, J.; Zamora, M.E.; Ilich, E.; Grenier, C.; Huang, Z.Y.; Murphy, S.; Chiba-Falek, O. Downregulation of SNCA Expression by Targeted Editing of DNA Methylation: A Potential Strategy for Precision Therapy in PD. Mol. Ther. 2018, 26, 2638–2649. [Google Scholar] [CrossRef] [Green Version]
- Recasens, A.; Perier, C.; Sue, C.M. Role of MicroRNAs in the Regulation of α-Synuclein Expression: A Systematic Review. Front. Mol. Neurosci. 2016, 9, 128. [Google Scholar] [CrossRef] [Green Version]
- George, J.M. The Synucleins. Genome Biol. 2002, 3, REVIEWS3002. [Google Scholar] [CrossRef] [PubMed]
- Bendor, J.; Logan, T.; Edwards, R.H. The Function of α-Synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of Alpha-Synuclein Secondary Structure upon Binding to Synthetic Membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [Green Version]
- Doherty, C.P.A.; Ulamec, S.M.; Maya-Martinez, R.; Good, S.C.; Makepeace, J.; Khan, G.N.; van Oosten-Hawle, P.; Radford, S.E.; Brockwell, D.J. A Short Motif in the N-Terminal Region of α-Synuclein Is Critical for Both Aggregation and Function. Nat. Struct. Mol. Biol. 2020, 27, 249–259. [Google Scholar] [CrossRef]
- Uéda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular Cloning of CDNA Encoding an Unrecognized Component of Amyloid in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef]
- Giasson, B.I.; Murray, I.V.J.; Trojanowski, J.Q.; Lee, V.M.-Y. A Hydrophobic Stretch of 12 Amino Acid Residues in the Middle of α-Synuclein Is Essential for Filament Assembly*. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-State NMR Structure of a Pathogenic Fibril of Full-Length Human α-Synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- Periquet, M.; Fulga, T.; Myllykangas, L.; Schlossmacher, M.G.; Feany, M.B. Aggregated α-Synuclein Mediates Dopaminergic Neurotoxicity In Vivo. J. Neurosci. 2007, 27, 3338–3346. [Google Scholar] [CrossRef] [Green Version]
- Izawa, Y.; Tateno, H.; Kameda, H.; Hirakawa, K.; Hato, K.; Yagi, H.; Hongo, K.; Mizobata, T.; Kawata, Y. Role of C-Terminal Negative Charges and Tyrosine Residues in Fibril Formation of α-Synuclein. Brain Behav. 2012, 2, 595–605. [Google Scholar] [CrossRef]
- Farzadfard, A.; Pedersen, J.N.; Meisl, G.; Somavarapu, A.K.; Alam, P.; Goksøyr, L.; Nielsen, M.A.; Sander, A.F.; Knowles, T.P.J.; Pedersen, J.S.; et al. The C-Terminal Tail of α-Synuclein Protects against Aggregate Replication but Is Critical for Oligomerization. Commun. Biol. 2022, 5, 123. [Google Scholar] [CrossRef]
- Kim, T.D.; Paik, S.R.; Yang, C.-H. Structural and Functional Implications of C-Terminal Regions of Alpha-Synuclein. Biochemistry 2002, 41, 13782–13790. [Google Scholar] [CrossRef]
- Tanaka, G.; Yamanaka, T.; Furukawa, Y.; Kajimura, N.; Mitsuoka, K.; Nukina, N. Biochemical and Morphological Classification of Disease-Associated Alpha-Synuclein Mutants Aggregates. Biochem. Biophys. Res. Commun. 2019, 508, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Lee, S.-J.; Rochet, J.-C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T. Acceleration of Oligomerization, Not Fibrillization, Is a Shared Property of Both α-Synuclein Mutations Linked to Early-Onset Parkinson’s Disease: Implications for Pathogenesis and Therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Lázaro, D.F.; Rodrigues, E.F.; Langohr, R.; Shahpasandzadeh, H.; Ribeiro, T.; Guerreiro, P.; Gerhardt, E.; Kröhnert, K.; Klucken, J.; Pereira, M.D.; et al. Systematic Comparison of the Effects of Alpha-Synuclein Mutations on Its Oligomerization and Aggregation. PLOS Genet. 2014, 10, e1004741. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, N.J.; Giasson, B.I. The A53E α-Synuclein Pathological Mutation Demonstrates Reduced Aggregation Propensity in Vitro and in Cell Culture. Neurosci. Lett. 2015, 597, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Boyer, D.R.; Li, B.; Sun, C.; Fan, W.; Sawaya, M.R.; Jiang, L.; Eisenberg, D.S. Structures of Fibrils Formed by α-Synuclein Hereditary Disease Mutant H50Q Reveal New Polymorphs. Nat. Struct. Mol. Biol. 2019, 26, 1044–1052. [Google Scholar] [CrossRef]
- Sano, K.; Iwasaki, Y.; Yamashita, Y.; Irie, K.; Hosokawa, M.; Satoh, K.; Mishima, K. Tyrosine 136 Phosphorylation of α-Synuclein Aggregates in the Lewy Body Dementia Brain: Involvement of Serine 129 Phosphorylation by Casein Kinase 2. Acta Neuropathol. Commun. 2021, 9, 182. [Google Scholar] [CrossRef]
- Zhang, C.; Pei, Y.; Zhang, Z.; Xu, L.; Liu, X.; Jiang, L.; Pielak, G.J.; Zhou, X.; Liu, M.; Li, C. C-Terminal Truncation Modulates α-Synuclein’s Cytotoxicity and Aggregation by Promoting the Interactions with Membrane and Chaperone. Commun. Biol. 2022, 5, 798. [Google Scholar] [CrossRef]
- Muntané, G.; Ferrer, I.; Martinez-Vicente, M. α-Synuclein Phosphorylation and Truncation Are Normal Events in the Adult Human Brain. Neuroscience 2012, 200, 106–119. [Google Scholar] [CrossRef]
- Book, A.; Guella, I.; Candido, T.; Brice, A.; Hattori, N.; Jeon, B.; Farrer, M.J. A Meta-Analysis of α-Synuclein Multiplication in Familial Parkinsonism. Front. Neurol. 2018, 9, 1021. [Google Scholar] [CrossRef]
- Edwards, T.L.; Scott, W.K.; Almonte, C.; Burt, A.; Powell, E.H.; Beecham, G.W.; Wang, L.; Züchner, S.; Konidari, I.; Wang, G.; et al. Genome-Wide Association Study Confirms SNPs in SNCA and the MAPT Region as Common Risk Factors for Parkinson Disease. Ann. Hum. Genet. 2010, 74, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wüllner, U. Methylation Regulates Alpha-Synuclein Expression and Is Decreased in Parkinson’s Disease Patients’ Brains. J. Neurosci. 2010, 30, 6355–6359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iakovenko, E.V.; Abramycheva, N.Y.; Fedotova, E.Y.; Illarioshkin, S.N. The SNCA-Rep1 Polymorphic Locus: Association with the Risk of Parkinson’s Disease and SNCA Gene Methylation. Acta Nat. 2020, 12, 105–110. [Google Scholar] [CrossRef]
- Benskey, M.J.; Sellnow, R.C.; Sandoval, I.M.; Sortwell, C.E.; Lipton, J.W.; Manfredsson, F.P. Silencing Alpha Synuclein in Mature Nigral Neurons Results in Rapid Neuroinflammation and Subsequent Toxicity. Front. Mol. Neurosci. 2018, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The Genetic Architecture of Parkinson’s Disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.B.; Hanss, Z.; Krüger, R. The Genetic Architecture of Mitochondrial Dysfunction in Parkinson’s Disease. Cell Tissue Res. 2018, 373, 21–37. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of Novel Risk Loci, Causal Insights, and Heritable Risk for Parkinson’s Disease: A Meta-Genome Wide Association Study. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Wernick, A.I.; Walton, R.L.; Koga, S.; Soto-Beasley, A.I.; Heckman, M.G.; Gan-Or, Z.; Ren, Y.; Rademakers, R.; Uitti, R.J.; Wszolek, Z.K.; et al. GBA Variation and Susceptibility to Multiple System Atrophy. Park. Relat. Disord. 2020, 77, 64–69. [Google Scholar] [CrossRef]
- Granek, Z.; Barczuk, J.; Siwecka, N.; Rozpędek-Kamińska, W.; Kucharska, E.; Majsterek, I. GBA1 Gene Mutations in α-Synucleinopathies—Molecular Mechanisms Underlying Pathology and Their Clinical Significance. Int. J. Mol. Sci. 2023, 24, 2044. [Google Scholar] [CrossRef]
- Braunstein, H.; Maor, G.; Chicco, G.; Filocamo, M.; Zimran, A.; Horowitz, M. UPR Activation and CHOP Mediated Induction of GBA1 Transcription in Gaucher Disease. Blood Cells Mol. Dis. 2018, 68, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, H.J.R.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s IPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojkovska, I.; Wani, W.Y.; Zunke, F.; Belur, N.R.; Pavlenko, E.A.; Mwenda, N.; Sharma, K.; Francelle, L.; Mazzulli, J.R. Rescue of α-Synuclein Aggregation in Parkinson’s Patient Neurons by Synergistic Enhancement of ER Proteostasis and Protein Trafficking. Neuron 2022, 110, 436–451.e11. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Burke, D.; Heales, S.J.R.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H.V. Glucocerebrosidase Deficiency in Substantia Nigra of Parkinson Disease Brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daher, J.P.L. Interaction of LRRK2 and α-Synuclein in Parkinson’s Disease. Adv. Neurobiol. 2017, 14, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Schulte, C.; Gasser, T. Genetic Basis of Parkinson’s Disease: Inheritance, Penetrance, and Expression. Appl. Clin. Genet. 2011, 4, 67–80. [Google Scholar] [CrossRef]
- Schneider, S.A.; Alcalay, R.N. Precision Medicine in Parkinson’s Disease: Emerging Treatments for Genetic Parkinson’s Disease. J. Neurol. 2020, 267, 860–869. [Google Scholar] [CrossRef] [Green Version]
- Ysselstein, D.; Nguyen, M.; Young, T.J.; Severino, A.; Schwake, M.; Merchant, K.; Krainc, D. LRRK2 Kinase Activity Regulates Lysosomal Glucocerebrosidase in Neurons Derived from Parkinson’s Disease Patients. Nat. Commun. 2019, 10, 5570. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Cao, P.; Smith, M.A.; Kramp, K.; Huang, Y.; Hisamoto, N.; Matsumoto, K.; Hatzoglou, M.; Jin, H.; Feng, Z. Dysregulated LRRK2 Signaling in Response to Endoplasmic Reticulum Stress Leads to Dopaminergic Neuron Degeneration in C. Elegans. PLoS ONE 2011, 6, e22354. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Han, J.-H.; Kim, H.; Park, S.M.; Joe, E.-H.; Jou, I. Parkinson’s Disease-Associated LRRK2-G2019S Mutant Acts through Regulation of SERCA Activity to Control ER Stress in Astrocytes. Acta Neuropathol. Commun. 2019, 7, 68. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Han, J.-H.; Joe, E.-H.; Jou, I. Small Heterodimer Partner (SHP) Aggravates ER Stress in Parkinson’s Disease-Linked LRRK2 Mutant Astrocyte by Regulating XBP1 SUMOylation. J. Biomed. Sci. 2021, 28, 51. [Google Scholar] [CrossRef]
- Day, J.O.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12, 1006. [Google Scholar] [CrossRef]
- Schmid, A.W.; Fauvet, B.; Moniatte, M.; Lashuel, H.A. Alpha-Synuclein Post-Translational Modifications as Potential Biomarkers for Parkinson Disease and Other Synucleinopathies. Mol. Cell. Proteom. 2013, 12, 3543–3558. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. Alpha-Synuclein Is Phosphorylated in Synucleinopathy Lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-W.; Chen, W.; Junn, E.; Im, J.-Y.; Grosso, H.; Sonsalla, P.K.; Feng, X.; Ray, N.; Fernandez, J.R.; Chao, Y.; et al. Enhanced Phosphatase Activity Attenuates α-Synucleinopathy in a Mouse Model. J. Neurosci. 2011, 31, 6963–6971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, M.R.; Hanover, J.A. A Little Sugar Goes a Long Way: The Cell Biology of O-GlcNAc. J. Cell Biol. 2015, 208, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Levine, P.M.; De Leon, C.A.; Galesic, A.; Balana, A.; Marotta, N.P.; Lewis, Y.E.; Pratt, M.R. O-GlcNAc Modification Inhibits the Calpain-Mediated Cleavage of α-Synuclein. Bioorg. Med. Chem. 2017, 25, 4977–4982. [Google Scholar] [CrossRef]
- Wani, W.Y.; Ouyang, X.; Benavides, G.A.; Redmann, M.; Cofield, S.S.; Shacka, J.J.; Chatham, J.C.; Darley-Usmar, V.; Zhang, J. O-GlcNAc Regulation of Autophagy and α-Synuclein Homeostasis; Implications for Parkinson’s Disease. Mol. Brain 2017, 10, 32. [Google Scholar] [CrossRef] [Green Version]
- Marotta, N.P.; Lin, Y.H.; Lewis, Y.E.; Ambroso, M.R.; Zaro, B.W.; Roth, M.T.; Arnold, D.B.; Langen, R.; Pratt, M.R. O-GlcNAc Modification Blocks the Aggregation and Toxicity of the Protein α-Synuclein Associated with Parkinson’s Disease. Nat. Chem. 2015, 7, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Mishizen-Eberz, A.J.; Guttmann, R.P.; Giasson, B.I.; Day, G.A.; Hodara, R.; Ischiropoulos, H.; Lee, V.M.-Y.; Trojanowski, J.Q.; Lynch, D.R. Distinct Cleavage Patterns of Normal and Pathologic Forms of Alpha-Synuclein by Calpain I in Vitro. J. Neurochem. 2003, 86, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Dufty, B.M.; Warner, L.R.; Hou, S.T.; Jiang, S.X.; Gomez-Isla, T.; Leenhouts, K.M.; Oxford, J.T.; Feany, M.B.; Masliah, E.; Rohn, T.T. Calpain-Cleavage of α-Synuclein. Am. J. Pathol. 2007, 170, 1725–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishizen-Eberz, A.J.; Norris, E.H.; Giasson, B.I.; Hodara, R.; Ischiropoulos, H.; Lee, V.M.-Y.; Trojanowski, J.Q.; Lynch, D.R. Cleavage of Alpha-Synuclein by Calpain: Potential Role in Degradation of Fibrillized and Nitrated Species of Alpha-Synuclein. Biochemistry 2005, 44, 7818–7829. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Nguyen, L.T.T.; Burlak, C.; Chegini, F.; Guo, F.; Chataway, T.; Ju, S.; Fisher, O.S.; Miller, D.W.; Datta, D.; et al. Caspase-1 Causes Truncation and Aggregation of the Parkinson’s Disease-Associated Protein α-Synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 9587–9592. [Google Scholar] [CrossRef] [PubMed]
- Tatebe, H.; Watanabe, Y.; Kasai, T.; Mizuno, T.; Nakagawa, M.; Tanaka, M.; Tokuda, T. Extracellular Neurosin Degrades α-Synuclein in Cultured Cells. Neurosci. Res. 2010, 67, 341–346. [Google Scholar] [CrossRef]
- Choi, D.-H.; Kim, Y.-J.; Kim, Y.-G.; Joh, T.H.; Beal, M.F.; Kim, Y.-S. Role of Matrix Metalloproteinase 3-Mediated α-Synuclein Cleavage in Dopaminergic Cell Death. J. Biol. Chem. 2011, 286, 14168–14177. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Hebron, M.; Shi, W.; Lonskaya, I.; Moussa, C.E.-H. Ubiquitin Specific Protease-13 Independently Regulates Parkin Ubiquitination and Alpha-Synuclein Clearance in Alpha-Synucleinopathies. Hum. Mol. Genet. 2019, 28, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Razzaq, A.; Ghetti, B.; Lilley, K.S.; Spillantini, M.G. Ubiquitination of Alpha-Synuclein in Lewy Bodies Is a Pathological Event Not Associated with Impairment of Proteasome Function. J. Biol. Chem. 2003, 278, 44405–44411. [Google Scholar] [CrossRef] [Green Version]
- Chavarría, C.; Souza, J.M. Oxidation and Nitration of α-Synuclein and Their Implications in Neurodegenerative Diseases. Arch. Biochem. Biophys. 2013, 533, 25–32. [Google Scholar] [CrossRef]
- Iyer, A.; Sidhu, A.; Subramaniam, V. How Important Is the N-Terminal Acetylation of Alpha-Synuclein for Its Function and Aggregation into Amyloids? Front. Neurosci. 2022, 16, 1003997. [Google Scholar] [CrossRef]
- Rott, R.; Szargel, R.; Shani, V.; Hamza, H.; Savyon, M.; Abd Elghani, F.; Bandopadhyay, R.; Engelender, S. SUMOylation and Ubiquitination Reciprocally Regulate α-Synuclein Degradation and Pathological Aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 13176–13181. [Google Scholar] [CrossRef] [Green Version]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.-H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kügler, S.; Melchior, F.; et al. Sumoylation Inhibits Alpha-Synuclein Aggregation and Toxicity. J. Cell Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Latawiec, D.; Herrera, F.; Bek, A.; Losasso, V.; Candotti, M.; Benetti, F.; Carlino, E.; Kranjc, A.; Lazzarino, M.; Gustincich, S.; et al. Modulation of Alpha-Synuclein Aggregation by Dopamine Analogs. PLoS ONE 2010, 5, e9234. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Rao, S.P.; Kalivendi, S.V. The Deglycase Activity of DJ-1 Mitigates α-Synuclein Glycation and Aggregation in Dopaminergic Cells: Role of Oxidative Stress Mediated Downregulation of DJ-1 in Parkinson’s Disease. Free Radic. Biol. Med. 2019, 135, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Südhof, T.C. α-Synuclein Assembles into Higher-Order Multimers upon Membrane Binding to Promote SNARE Complex Formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein Occurs Physiologically as a Helically Folded Tetramer That Resists Aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.; Südhof, T.C. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Fortin, D.L.; Nemani, V.M.; Voglmaier, S.M.; Anthony, M.D.; Ryan, T.A.; Edwards, R.H. Neural Activity Controls the Synaptic Accumulation of Alpha-Synuclein. J. Neurosci. 2005, 25, 10913–10921. [Google Scholar] [CrossRef] [Green Version]
- Hallacli, E.; Kayatekin, C.; Nazeen, S.; Wang, X.H.; Sheinkopf, Z.; Sathyakumar, S.; Sarkar, S.; Jiang, X.; Dong, X.; Di Maio, R.; et al. The Parkinson’s Disease Protein Alpha-Synuclein Is a Modulator of Processing Bodies and MRNA Stability. Cell 2022, 185, 2035–2056.e33. [Google Scholar] [CrossRef]
- Schaser, A.J.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-Synuclein Is a DNA Binding Protein That Modulates DNA Repair with Implications for Lewy Body Disorders. Sci. Rep. 2019, 9, 10919. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.M.; Yang, D.; Li, X.-Q.; Liu, J.; Back, T.C.; Trivett, A.; Karim, B.; Barbut, D.; Zasloff, M.; Oppenheim, J.J. Alpha Synuclein, the Culprit in Parkinson Disease, Is Required for Normal Immune Function. Cell Rep. 2022, 38, 110090. [Google Scholar] [CrossRef]
- Perez, R.G.; Waymire, J.C.; Lin, E.; Liu, J.J.; Guo, F.; Zigmond, M.J. A Role for α-Synuclein in the Regulation of Dopamine Biosynthesis. J. Neurosci. 2002, 22, 3090–3099. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.; Fuller, N.; Rand, R.P.; St George-Hyslop, P.; Fraser, P.E. Defective Membrane Interactions of Familial Parkinson’s Disease Mutant A30P Alpha-Synuclein. J. Mol. Biol. 2002, 315, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Mor, D.E.; Ugras, S.E.; Daniels, M.J.; Ischiropoulos, H. Dynamic Structural Flexibility of α-Synuclein. Neurobiol. Dis. 2016, 88, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollenhauer, B.; Locascio, J.J.; Schulz-Schaeffer, W.; Sixel-Döring, F.; Trenkwalder, C.; Schlossmacher, M.G. α-Synuclein and Tau Concentrations in Cerebrospinal Fluid of Patients Presenting with Parkinsonism: A Cohort Study. Lancet Neurol. 2011, 10, 230–240. [Google Scholar] [CrossRef]
- Scott, D.A.; Tabarean, I.; Tang, Y.; Cartier, A.; Masliah, E.; Roy, S. A Pathologic Cascade Leading to Synaptic Dysfunction in α-Synuclein-Induced Neurodegeneration. J. Neurosci. 2010, 30, 8083–8095. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.-K.; Choi, M.-G.; Kim, J.-Y.; Yang, Y.; Lai, Y.; Kweon, D.-H.; Lee, N.K.; Shin, Y.-K. Large α-Synuclein Oligomers Inhibit Neuronal SNARE-Mediated Vesicle Docking. Proc. Natl. Acad. Sci. USA 2013, 110, 4087–4092. [Google Scholar] [CrossRef]
- Tulisiak, C.T.; Mercado, G.; Peelaerts, W.; Brundin, L.; Brundin, P. Can Infections Trigger Alpha-Synucleinopathies? Prog. Mol. Biol. Transl. Sci. 2019, 168, 299–322. [Google Scholar] [CrossRef]
- Pan-Montojo, F.; Schwarz, M.; Winkler, C.; Arnhold, M.; O’Sullivan, G.A.; Pal, A.; Said, J.; Marsico, G.; Verbavatz, J.-M.; Rodrigo-Angulo, M.; et al. Environmental Toxins Trigger PD-like Progression via Increased Alpha-Synuclein Release from Enteric Neurons in Mice. Sci. Rep. 2012, 2, 898. [Google Scholar] [CrossRef] [Green Version]
- Moons, R.; Konijnenberg, A.; Mensch, C.; Van Elzen, R.; Johannessen, C.; Maudsley, S.; Lambeir, A.-M.; Sobott, F. Metal Ions Shape α-Synuclein. Sci. Rep. 2020, 10, 16293. [Google Scholar] [CrossRef]
- Mori, A.; Imai, Y.; Hattori, N. Lipids: Key Players That Modulate α-Synuclein Toxicity and Neurodegeneration in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 3301. [Google Scholar] [CrossRef]
- Awasthi, S.; Ying, C.; Li, J.; Mayer, M. Simultaneous Determination of the Size and Shape of Single α-Synuclein Oligomers in Solution. ACS Nano 2023, 17, 12325–12335. [Google Scholar] [CrossRef] [PubMed]
- Cremades, N.; Chen, S.W.; Dobson, C.M. Chapter Three—Structural Characteristics of α-Synuclein Oligomers. In International Review of Cell and Molecular Biology; Sandal, M., Ed.; Early Stage Protein Misfolding and Amyloid Aggregation; Academic Press: Cambridge, MA, USA, 2017; Volume 329, pp. 79–143. [Google Scholar]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The Many Faces of α-Synuclein: From Structure and Toxicity to Therapeutic Target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrasidlo, W.; Tsigelny, I.F.; Price, D.L.; Dutta, G.; Rockenstein, E.; Schwarz, T.C.; Ledolter, K.; Bonhaus, D.; Paulino, A.; Eleuteri, S.; et al. A de Novo Compound Targeting α-Synuclein Improves Deficits in Models of Parkinson’s Disease. Brain 2016, 139, 3217–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural Basis of Membrane Disruption and Cellular Toxicity by α-Synuclein Oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [Green Version]
- Celej, M.S.; Sarroukh, R.; Goormaghtigh, E.; Fidelio, G.D.; Ruysschaert, J.-M.; Raussens, V. Toxic Prefibrillar α-Synuclein Amyloid Oligomers Adopt a Distinctive Antiparallel β-Sheet Structure. Biochem. J. 2012, 443, 719–726. [Google Scholar] [CrossRef]
- Giehm, L.; Svergun, D.I.; Otzen, D.E.; Vestergaard, B. Low-Resolution Structure of a Vesicle Disrupting α-Synuclein Oligomer That Accumulates during Fibrillation. Proc. Natl. Acad. Sci. USA 2011, 108, 3246–3251. [Google Scholar] [CrossRef]
- Bhak, G.; Lee, S.; Kim, T.-H.; Lee, J.-H.; Yang, J.E.; Joo, K.; Lee, J.; Char, K.; Paik, S.R. Morphological Evaluation of Meta-Stable Oligomers of α-Synuclein with Small-Angle Neutron Scattering. Sci. Rep. 2018, 8, 14295. [Google Scholar] [CrossRef] [Green Version]
- Sekiya, H.; Tsuji, A.; Hashimoto, Y.; Takata, M.; Koga, S.; Nishida, K.; Futamura, N.; Kawamoto, M.; Kohara, N.; Dickson, D.W.; et al. Discrepancy between Distribution of Alpha-Synuclein Oligomers and Lewy-Related Pathology in Parkinson’s Disease. Acta Neuropathol. Commun. 2022, 10, 133. [Google Scholar] [CrossRef]
- Koch, J.C.; Bitow, F.; Haack, J.; d’Hedouville, Z.; Zhang, J.-N.; Tönges, L.; Michel, U.; Oliveira, L.M.A.; Jovin, T.M.; Liman, J.; et al. Alpha-Synuclein Affects Neurite Morphology, Autophagy, Vesicle Transport and Axonal Degeneration in CNS Neurons. Cell Death Dis. 2015, 6, e1811. [Google Scholar] [CrossRef] [Green Version]
- Dada, S.T.; Hardenberg, M.C.; Toprakcioglu, Z.; Mrugalla, L.K.; Cali, M.P.; McKeon, M.O.; Klimont, E.; Michaels, T.C.T.; Knowles, T.P.J.; Vendruscolo, M. Spontaneous Nucleation and Fast Aggregate-Dependent Proliferation of α-Synuclein Aggregates within Liquid Condensates at Neutral PH. Proc. Natl. Acad. Sci. USA 2023, 120, e2208792120. [Google Scholar] [CrossRef]
- Ma, L.; Yang, C.; Zhang, X.; Li, Y.; Wang, S.; Zheng, L.; Huang, K. C-Terminal Truncation Exacerbates the Aggregation and Cytotoxicity of α-Synuclein: A Vicious Cycle in Parkinson’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3714–3725. [Google Scholar] [CrossRef] [PubMed]
- Mahul-Mellier, A.-L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The Process of Lewy Body Formation, Rather than Simply α-Synuclein Fibrillization, Is One of the Major Drivers of Neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiechle, M.; Grozdanov, V.; Danzer, K.M. The Role of Lipids in the Initiation of α-Synuclein Misfolding. Front. Cell Dev. Biol. 2020, 8, 562241. [Google Scholar] [CrossRef] [PubMed]
- Ohgita, T.; Namba, N.; Kono, H.; Shimanouchi, T.; Saito, H. Mechanisms of Enhanced Aggregation and Fibril Formation of Parkinson’s Disease-Related Variants of α-Synuclein. Sci. Rep. 2022, 12, 6770. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.; Faidon Brotzakis, Z.; Horne, R.I.; Possenti, A.; Mannini, B.; Cataldi, R.; Nowinska, M.; Staats, R.; Linse, S.; Knowles, T.P.J.; et al. Structure-Based Discovery of Small-Molecule Inhibitors of the Autocatalytic Proliferation of α-Synuclein Aggregates. Mol. Pharm. 2023, 20, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, W.; Antony, T.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Dependence of Alpha-Synuclein Aggregate Morphology on Solution Conditions. J. Mol. Biol. 2002, 322, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Torre-Muruzabal, T.; Van der Perren, A.; Coens, A.; Gelders, G.; Janer, A.B.; Camacho-Garcia, S.; Klingstedt, T.; Nilsson, P.; Stefanova, N.; Melki, R.; et al. Host Oligodendrogliopathy and α-Synuclein Strains Dictate Disease Severity in Multiple System Atrophy. Brain 2023, 146, 237–251. [Google Scholar] [CrossRef]
- Concha-Marambio, L.; Pritzkow, S.; Shahnawaz, M.; Farris, C.M.; Soto, C. Seed Amplification Assay for the Detection of Pathologic Alpha-Synuclein Aggregates in Cerebrospinal Fluid. Nat. Protoc. 2023, 18, 1179–1196. [Google Scholar] [CrossRef]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-Synuclein Strains in Parkinson’s Disease and Multiple System Atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef]
- Majbour, N.; Aasly, J.; Abdi, I.; Ghanem, S.; Erskine, D.; van de Berg, W.; El-Agnaf, O. Disease-Associated α-Synuclein Aggregates as Biomarkers of Parkinson Disease Clinical Stage. Neurology 2022, 99, e2417–e2427. [Google Scholar] [CrossRef]
- Okuzumi, A.; Hatano, T.; Matsumoto, G.; Nojiri, S.; Ueno, S.-I.; Imamichi-Tatano, Y.; Kimura, H.; Kakuta, S.; Kondo, A.; Fukuhara, T.; et al. Propagative α-Synuclein Seeds as Serum Biomarkers for Synucleinopathies. Nat. Med. 2023, 29, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.L.; Schulz-Schaeffer, W.J. Presynaptic Alpha-Synuclein Aggregates, Not Lewy Bodies, Cause Neurodegeneration in Dementia with Lewy Bodies. J. Neurosci. 2007, 27, 1405–1410. [Google Scholar] [CrossRef] [Green Version]
- Luarte, A.; Cornejo, V.H.; Bertin, F.; Gallardo, J.; Couve, A. The Axonal Endoplasmic Reticulum: One Organelle—Many Functions in Development, Maintenance, and Plasticity. Dev. Neurobiol. 2018, 78, 181–208. [Google Scholar] [CrossRef] [PubMed]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein Binds to the ER–Mitochondria Tethering Protein VAPB to Disrupt Ca2+ Homeostasis and Mitochondrial ATP Production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of Toxic α-Synuclein Oligomer within Endoplasmic Reticulum Occurs in α-Synucleinopathy in Vivo. J. Neurosci. 2012, 32, 3301–3305. [Google Scholar] [CrossRef] [Green Version]
- Bellani, S.; Mescola, A.; Ronzitti, G.; Tsushima, H.; Tilve, S.; Canale, C.; Valtorta, F.; Chieregatti, E. GRP78 Clustering at the Cell Surface of Neurons Transduces the Action of Exogenous Alpha-Synuclein. Cell Death Differ. 2014, 21, 1971–1983. [Google Scholar] [CrossRef]
- Gorbatyuk, M.S.; Shabashvili, A.; Chen, W.; Meyers, C.; Sullivan, L.F.; Salganik, M.; Lin, J.H.; Lewin, A.S.; Muzyczka, N.; Gorbatyuk, O.S. Glucose Regulated Protein 78 Diminishes α-Synuclein Neurotoxicity in a Rat Model of Parkinson Disease. Mol. Ther. 2012, 20, 1327–1337. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Pytel, D.; Riese, M.J.; Vaites, L.P.; Singh, N.; Koretzky, G.A.; Witze, E.S.; Diehl, J.A. PERK Utilizes Intrinsic Lipid Kinase Activity To Generate Phosphatidic Acid, Mediate Akt Activation, and Promote Adipocyte Differentiation. Mol. Cell Biol. 2012, 32, 2268–2278. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Liu, J.; Gao, J.; Sun, Y.; Zhang, L.; Song, H.; Xue, L.; Zhan, L.; Gao, G.; Ke, Z.; et al. IRE1 Promotes Neurodegeneration through Autophagy-Dependent Neuron Death in the Drosophila Model of Parkinson’s Disease. Cell Death Dis. 2019, 10, 800. [Google Scholar] [CrossRef] [Green Version]
- Credle, J.J.; Forcelli, P.A.; Delannoy, M.; Oaks, A.W.; Permaul, E.; Berry, D.L.; Duka, V.; Wills, J.; Sidhu, A. α-Synuclein-Mediated Inhibition of ATF6 Processing into COPII Vesicles Disrupts UPR Signaling in Parkinson’s Disease. Neurobiol. Dis. 2015, 76, 112–125. [Google Scholar] [CrossRef]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, X.; Li, Q.; Wen, X.; Xie, J.; Wang, Y.; Song, N. Extracellular α-Synuclein Modulates Iron Metabolism Related Proteins via Endoplasmic Reticulum Stress in MES23.5 Dopaminergic Cells. Neurochem. Res. 2021, 46, 1502–1513. [Google Scholar] [CrossRef] [PubMed]
- Mercado, G.; Castillo, V.; Soto, P.; López, N.; Axten, J.M.; Sardi, S.P.; Hoozemans, J.J.M.; Hetz, C. Targeting PERK Signaling with the Small Molecule GSK2606414 Prevents Neurodegeneration in a Model of Parkinson’s Disease. Neurobiol. Dis. 2018, 112, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.; Mancini, M.; Ibraheem, P.L.; Aryal, S.; Mesini, C.; Patel, J.C.; Penhos, E.; Rahman, N.; Mamcarz, M.; Santini, E.; et al. Cell-Type-Specific Disruption of PERK-EIF2α Signaling in Dopaminergic Neurons Alters Motor and Cognitive Function. Mol. Psychiatry 2021, 26, 6427–6450. [Google Scholar] [CrossRef]
- Elvira, R.; Cha, S.J.; Noh, G.-M.; Kim, K.; Han, J. PERK-Mediated EIF2α Phosphorylation Contributes to The Protection of Dopaminergic Neurons from Chronic Heat Stress in Drosophila. Int. J. Mol. Sci. 2020, 21, 845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugallo, R.; Marlin, E.; Baltanás, A.; Toledo, E.; Ferrero, R.; Vinueza-Gavilanes, R.; Larrea, L.; Arrasate, M.; Aragón, T. Fine Tuning of the Unfolded Protein Response by ISRIB Improves Neuronal Survival in a Model of Amyotrophic Lateral Sclerosis. Cell Death Dis. 2020, 11, 397. [Google Scholar] [CrossRef]
- Xu, B.; Wang, F.; Wu, S.-W.; Deng, Y.; Liu, W.; Feng, S.; Yang, T.-Y.; Xu, Z.-F. α-Synuclein Is Involved in Manganese-Induced ER Stress via PERK Signal Pathway in Organotypic Brain Slice Cultures. Mol. Neurobiol. 2014, 49, 399–412. [Google Scholar] [CrossRef]
- Raj, A.; Banerjee, R.; Santhoshkumar, R.; Sagar, C.; Datta, I. Presence of Extracellular Alpha-Synuclein Aggregates Trigger Astrocytic Degeneration Through Enhanced Membrane Rigidity and Deregulation of Store-Operated Calcium Entry (SOCE) into the Endoplasmic Reticulum. Mol. Neurobiol. 2023. [Google Scholar] [CrossRef]
- Castillo-Carranza, D.L.; Zhang, Y.; Guerrero-Muñoz, M.J.; Kayed, R.; Rincon-Limas, D.E.; Fernandez-Funez, P. Differential Activation of the ER Stress Factor XBP1 by Oligomeric Assemblies. Neurochem. Res. 2012, 37, 1707–1717. [Google Scholar] [CrossRef] [Green Version]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J. Alpha-Synuclein Induces the Unfolded Protein Response in Parkinson’s Disease SNCA Triplication IPSC-Derived Neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef] [Green Version]
- Valdés, P.; Mercado, G.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Galleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of Dopaminergic Neuron Survival by the Unfolded Protein Response Transcription Factor XBP1. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Imai, Y. Pael Receptor, Endoplasmic Reticulum Stress, and Parkinson’s Disease. J. Neurol. 2003, 250 (Suppl. 3), III25–III29. [Google Scholar] [CrossRef] [PubMed]
- Ugolino, J.; Fang, S.; Kubisch, C.; Monteiro, M.J. Mutant Atp13a2 Proteins Involved in Parkinsonism Are Degraded by ER-Associated Degradation and Sensitize Cells to ER-Stress Induced Cell Death. Hum. Mol. Genet. 2011, 20, 3565–3577. [Google Scholar] [CrossRef] [Green Version]
- Toyofuku, T.; Okamoto, Y.; Ishikawa, T.; Sasawatari, S.; Kumanogoh, A. LRRK2 Regulates Endoplasmic Reticulum-Mitochondrial Tethering through the PERK-Mediated Ubiquitination Pathway. EMBO J. 2020, 39, e100875. [Google Scholar] [CrossRef]
- Amodio, G.; Moltedo, O.; Fasano, D.; Zerillo, L.; Oliveti, M.; Di Pietro, P.; Faraonio, R.; Barone, P.; Pellecchia, M.T.; De Rosa, A.; et al. PERK-Mediated Unfolded Protein Response Activation and Oxidative Stress in PARK20 Fibroblasts. Front. Neurosci. 2019, 13, 673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Kim, K.S.; Iyirhiaro, G.O.; Marcogliese, P.C.; Callaghan, S.M.; Qu, D.; Kim, W.J.; Slack, R.S.; Park, D.S. DJ-1 Modulates the Unfolded Protein Response and Cell Death via Upregulation of ATF4 Following ER Stress. Cell Death Dis. 2019, 10, 135. [Google Scholar] [CrossRef] [Green Version]
- Bouman, L.; Schlierf, A.; Lutz, A.K.; Shan, J.; Deinlein, A.; Kast, J.; Galehdar, Z.; Palmisano, V.; Patenge, N.; Berg, D.; et al. Parkin Is Transcriptionally Regulated by ATF4: Evidence for an Interconnection between Mitochondrial Stress and ER Stress. Cell Death Differ. 2011, 18, 769–782. [Google Scholar] [CrossRef] [Green Version]
- Duplan, E.; Giaime, E.; Viotti, J.; Sévalle, J.; Corti, O.; Brice, A.; Ariga, H.; Qi, L.; Checler, F.; Alves da Costa, C. ER-Stress-Associated Functional Link between Parkin and DJ-1 via a Transcriptional Cascade Involving the Tumor Suppressor P53 and the Spliced X-Box Binding Protein XBP-1. J. Cell Sci. 2013, 126, 2124–2133. [Google Scholar] [CrossRef] [Green Version]
- Martínez, G.; Vidal, R.L.; Mardones, P.; Serrano, F.G.; Ardiles, A.O.; Wirth, C.; Valdés, P.; Thielen, P.; Schneider, B.L.; Kerr, B.; et al. Regulation of Memory Formation by the Transcription Factor XBP1. Cell Rep. 2016, 14, 1382–1394. [Google Scholar] [CrossRef] [Green Version]
- Parkash, V.; Lindholm, P.; Peränen, J.; Kalkkinen, N.; Oksanen, E.; Saarma, M.; Leppänen, V.-M.; Goldman, A. The Structure of the Conserved Neurotrophic Factors MANF and CDNF Explains Why They Are Bifunctional. Protein Eng. Des. Sel. 2009, 22, 233–241. [Google Scholar] [CrossRef]
- Mavroeidi, P.; Xilouri, M. Neurons and Glia Interplay in α-Synucleinopathies. Int. J. Mol. Sci. 2021, 22, 4994. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, I.M.; Marmion, D.J.; Meyers, K.T.; Manfredsson, F.P. Gene Therapy to Modulate Alpha-Synuclein in Synucleinopathies. J. Park. Dis. 2021, 11, S189–S197. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Corona, D.; Vázquez-Hernández, N.; Escobedo, L.; Orozco-Barrios, C.E.; Ayala-Davila, J.; Moreno, M.G.; Amaro-Lara, M.E.; Flores-Martinez, Y.M.; Espadas-Alvarez, A.J.; Fernandez-Parrilla, M.A.; et al. Neurturin Overexpression in Dopaminergic Neurons Induces Presynaptic and Postsynaptic Structural Changes in Rats with Chronic 6-Hydroxydopamine Lesion. PLoS ONE 2017, 12, e0188239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Gao, Y.; Chang, N. Nurr1 Overexpression Exerts Neuroprotective and Anti-Inflammatory Roles via down-Regulating CCL2 Expression in Both in Vivo and in Vitro Parkinson’s Disease Models. Biochem. Biophys. Res. Commun. 2017, 482, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Mastaglia, F.L.; Yau, W.Y.; Chen, S.; Wilton, S.D.; Akkari, P.A. Targeted Molecular Therapeutics for Parkinson’s Disease: A Role for Antisense Oligonucleotides? Mov. Disord. 2022, 37, 2184–2190. [Google Scholar] [CrossRef] [PubMed]
- Ntetsika, T.; Papathoma, P.-E.; Markaki, I. Novel Targeted Therapies for Parkinson’s Disease. Mol. Med. 2021, 27, 17. [Google Scholar] [CrossRef]
- Lassot, I.; Mora, S.; Lesage, S.; Zieba, B.A.; Coque, E.; Condroyer, C.; Bossowski, J.P.; Mojsa, B.; Marelli, C.; Soulet, C.; et al. The E3 Ubiquitin Ligases TRIM17 and TRIM41 Modulate α-Synuclein Expression by Regulating ZSCAN21. Cell Rep. 2018, 25, 2484–2496.e9. [Google Scholar] [CrossRef] [Green Version]
- Ndemazie, N.B.; Inkoom, A.; Morfaw, E.F.; Smith, T.; Aghimien, M.; Ebesoh, D.; Agyare, E. Multi-Disciplinary Approach for Drug and Gene Delivery Systems to the Brain. AAPS PharmSciTech 2021, 23, 11. [Google Scholar] [CrossRef]
- Fu, C.; Xiang, Y.; Li, X.; Fu, A. Targeted Transport of Nanocarriers into Brain for Theranosis with Rabies Virus Glycoprotein-Derived Peptide. Mater. Sci. Eng. C 2018, 87, 155–166. [Google Scholar] [CrossRef]
- Spencer, B.; Trinh, I.; Rockenstein, E.; Mante, M.; Florio, J.; Adame, A.; El-Agnaf, O.M.A.; Kim, C.; Masliah, E.; Rissman, R.A. Systemic Peptide Mediated Delivery of an SiRNA Targeting α-Syn in the CNS Ameliorates the Neurodegenerative Process in a Transgenic Model of Lewy Body Disease. Neurobiol. Dis. 2019, 127, 163–177. [Google Scholar] [CrossRef]
- Alarcón-Arís, D.; Recasens, A.; Galofré, M.; Carballo-Carbajal, I.; Zacchi, N.; Ruiz-Bronchal, E.; Pavia-Collado, R.; Chica, R.; Ferrés-Coy, A.; Santos, M.; et al. Selective α-Synuclein Knockdown in Monoamine Neurons by Intranasal Oligonucleotide Delivery: Potential Therapy for Parkinson’s Disease. Mol. Ther. 2018, 26, 550–567. [Google Scholar] [CrossRef] [Green Version]
- Camilleri, M.; Subramanian, T.; Pagan, F.; Isaacson, S.; Gil, R.; Hauser, R.A.; Feldman, M.; Goldstein, M.; Kumar, R.; Truong, D.; et al. Oral ENT-01 Targets Enteric Neurons to Treat Constipation in Parkinson Disease: A Randomized Controlled Trial. Ann. Intern. Med. 2022, 175, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Barbour, R.; Kling, K.; Anderson, J.P.; Banducci, K.; Cole, T.; Diep, L.; Fox, M.; Goldstein, J.M.; Soriano, F.; Seubert, P.; et al. Red Blood Cells Are the Major Source of Alpha-Synuclein in Blood. Neurodegener. Dis. 2008, 5, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, O.S.; Li, S.; Nash, K.; Gorbatyuk, M.; Lewin, A.S.; Sullivan, L.F.; Mandel, R.J.; Chen, W.; Meyers, C.; Manfredsson, F.P.; et al. In Vivo RNAi-Mediated Alpha-Synuclein Silencing Induces Nigrostriatal Degeneration. Mol. Ther. 2010, 18, 1450–1457. [Google Scholar] [CrossRef]
- Zharikov, A.; Bai, Q.; De Miranda, B.R.; Van Laar, A.; Greenamyre, J.T.; Burton, E.A. Long-Term RNAi Knockdown of α-Synuclein in the Adult Rat Substantia Nigra without Neurodegeneration. Neurobiol. Dis. 2019, 125, 146–153. [Google Scholar] [CrossRef]
- Zhang, P.; Park, H.-J.; Zhang, J.; Junn, E.; Andrews, R.J.; Velagapudi, S.P.; Abegg, D.; Vishnu, K.; Costales, M.G.; Childs-Disney, J.L.; et al. Translation of the Intrinsically Disordered Protein α-Synuclein Is Inhibited by a Small Molecule Targeting Its Structured MRNA. Proc. Natl. Acad. Sci. USA 2020, 117, 1457–1467. [Google Scholar] [CrossRef] [Green Version]
- Mikkilineni, S.; Cantuti-Castelvetri, I.; Cahill, C.M.; Balliedier, A.; Greig, N.H.; Rogers, J.T. The Anticholinesterase Phenserine and Its Enantiomer Posiphen as 5′Untranslated-Region-Directed Translation Blockers of the Parkinson’s Alpha Synuclein Expression. Park. Dis. 2012, 2012, e142372. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.; Bjørnevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. Β2-Adrenoreceptor Is a Regulator of the α-Synuclein Gene Driving Risk of Parkinson’s Disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgianni, F.; Ernst, P.; Dell’Aniello, S.; Suissa, S.; Renoux, C. Β2-Agonists and the Incidence of Parkinson Disease. Am. J. Epidemiol. 2020, 189, 801–810. [Google Scholar] [CrossRef]
- Li, J.; Zhu, M.; Manning-Bog, A.B.; Di Monte, D.A.; Fink, A.L. Dopamine and L-Dopa Disaggregate Amyloid Fibrils: Implications for Parkinson’s and Alzheimer’s Disease. FASEB J. 2004, 18, 962–964. [Google Scholar] [CrossRef]
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine Induces Soluble α-Synuclein Oligomers and Nigrostriatal Degeneration. Nat. Neurosci. 2017, 20, 1560–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.-T.; Lin, D.-H.; Luo, X.-Y.; Zhang, F.; Ji, L.-N.; Du, H.-N.; Song, G.-Q.; Hu, J.; Zhou, J.-W.; Hu, H.-Y. Inhibition of α-Synuclein Fibrillization by Dopamine Analogs via Reaction with the Amino Groups of α-Synuclein. FEBS J. 2005, 272, 3661–3672. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Wang, X.; Islam, M.R.; Akash, S.; Supti, F.A.; Mitu, M.I.; Harun-Or-Rashid, M.; Aktar, M.N.; Khatun Kali, M.S.; Jahan, F.I.; et al. Multifunctional Role of Natural Products for the Treatment of Parkinson’s Disease: At a Glance. Front. Pharmacol. 2022, 13, 976385. [Google Scholar] [CrossRef] [PubMed]
- Rathore, A.S.; Singh, S.S.; Birla, H.; Zahra, W.; Keshri, P.K.; Dilnashin, H.; Singh, R.; Singh, S.; Singh, S.P. Curcumin Modulates P62-Keap1-Nrf2-Mediated Autophagy in Rotenone-Induced Parkinson’s Disease Mouse Models. ACS Chem. Neurosci. 2023, 14, 1412–1423. [Google Scholar] [CrossRef]
- Gaballah, H.H.; Zakaria, S.S.; Elbatsh, M.M.; Tahoon, N.M. Modulatory Effects of Resveratrol on Endoplasmic Reticulum Stress-Associated Apoptosis and Oxido-Inflammatory Markers in a Rat Model of Rotenone-Induced Parkinson’s Disease. Chem. Biol. Interact. 2016, 251, 10–16. [Google Scholar] [CrossRef]
- Liu, Q.; Zhu, D.; Jiang, P.; Tang, X.; Lang, Q.; Yu, Q.; Zhang, S.; Che, Y.; Feng, X. Resveratrol Synergizes with Low Doses of L-DOPA to Improve MPTP-Induced Parkinson Disease in Mice. Behav. Brain Res. 2019, 367, 10–18. [Google Scholar] [CrossRef]
- Liu, Y.; Carver, J.A.; Calabrese, A.N.; Pukala, T.L. Gallic Acid Interacts with α-Synuclein to Prevent the Structural Collapse Necessary for Its Aggregation. Biochim. Biophys. Acta 2014, 1844, 1481–1485. [Google Scholar] [CrossRef]
- Zhu, M.; Rajamani, S.; Kaylor, J.; Han, S.; Zhou, F.; Fink, A.L. The Flavonoid Baicalein Inhibits Fibrillation of α-Synuclein and Disaggregates Existing Fibrils*. J. Biol. Chem. 2004, 279, 26846–26857. [Google Scholar] [CrossRef] [Green Version]
- Grønnemose, A.L.; Østerlund, E.C.; Otzen, D.E.; Jørgensen, T.J.D. EGCG Has Dual and Opposing Effects on the N-Terminal Region of Self-Associating α-Synuclein Oligomers. J. Mol. Biol. 2022, 434, 167855. [Google Scholar] [CrossRef]
- Daniels, M.J.; Nourse, J.B.; Kim, H.; Sainati, V.; Schiavina, M.; Murrali, M.G.; Pan, B.; Ferrie, J.J.; Haney, C.M.; Moons, R.; et al. Cyclized NDGA Modifies Dynamic α-Synuclein Monomers Preventing Aggregation and Toxicity. Sci. Rep. 2019, 9, 2937. [Google Scholar] [CrossRef] [Green Version]
- Pandey, N.; Strider, J.; Nolan, W.C.; Yan, S.X.; Galvin, J.E. Curcumin Inhibits Aggregation of Alpha-Synuclein. Acta Neuropathol. 2008, 115, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumar, R.; Kumari, M.; Kumari, R.; Saha, S.; Bhavesh, N.S.; Maiti, T.K. Ellagic Acid Inhibits α-Synuclein Aggregation at Multiple Stages and Reduces Its Cytotoxicity. ACS Chem. Neurosci. 2021, 12, 1919–1930. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Uversky, V.N.; Huang, M.; Kang, H.; Xu, F.; Liu, X.; Lian, L.; Liang, Q.; Jiang, H.; Liu, A.; et al. Baicalein Inhibits α-Synuclein Oligomer Formation and Prevents Progression of α-Synuclein Accumulation in a Rotenone Mouse Model of Parkinson’s Disease. Biochim. Biophys. Acta 2016, 1862, 1883–1890. [Google Scholar] [CrossRef]
- Ardah, M.T.; Paleologou, K.E.; Lv, G.; Menon, S.A.; Abul Khair, S.B.; Lu, J.-H.; Safieh-Garabedian, B.; Al-Hayani, A.A.; Eliezer, D.; Li, M.; et al. Ginsenoside Rb1 Inhibits Fibrillation and Toxicity of Alpha-Synuclein and Disaggregates Preformed Fibrils. Neurobiol. Dis. 2015, 74, 89–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Müller, M.B.D.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.A.; Cascella, R.; et al. A Natural Product Inhibits the Initiation of α-Synuclein Aggregation and Suppresses Its Toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017. [Google Scholar] [CrossRef]
- Limbocker, R.; Mannini, B.; Ruggeri, F.S.; Cascella, R.; Xu, C.K.; Perni, M.; Chia, S.; Chen, S.W.; Habchi, J.; Bigi, A.; et al. Trodusquemine Displaces Protein Misfolded Oligomers from Cell Membranes and Abrogates Their Cytotoxicity through a Generic Mechanism. Commun. Biol. 2020, 3, 435. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG Redirects Amyloidogenic Polypeptides into Unstructured, off-Pathway Oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Ono, K.; Yamada, M. Antioxidant Compounds Have Potent Anti-Fibrillogenic and Fibril-Destabilizing Effects for Alpha-Synuclein Fibrils in Vitro. J. Neurochem. 2006, 97, 105–115. [Google Scholar] [CrossRef]
- Morshedi, D.; Aliakbari, F.; Tayaranian-Marvian, A.; Fassihi, A.; Pan-Montojo, F.; Pérez-Sánchez, H. Cuminaldehyde as the Major Component of Cuminum Cyminum, a Natural Aldehyde with Inhibitory Effect on Alpha-Synuclein Fibrillation and Cytotoxicity. J. Food Sci. 2015, 80, H2336–H2345. [Google Scholar] [CrossRef]
- Yamamoto, H.; Matsumura, R.; Nakashima, M.; Adachi, M.; Ogawa, K.; Hongo, K.; Mizobata, T.; Kawata, Y. Effects of the Polyphenols Delphinidin and Rosmarinic Acid on the Inducible Intra-Cellular Aggregation of Alpha-Synuclein in Model Neuron Cells. Appl. Biochem. Biotechnol. 2023, 195, 4134–4147. [Google Scholar] [CrossRef]
- Kavya, R.; Aouti, S.; Jos, S.; Prasad, T.K.; Kumuda, K.N.; Unni, S.; Padmanabhan, B.; Kamariah, N.; Padavattan, S.; Mythri, R.B. High-Affinity Binding of Celastrol to Monomeric α-Synuclein Mitigates in Vitro Aggregation. J. Biomol. Struct. Dyn. 2023, 1–11. [Google Scholar] [CrossRef]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-Activated AMPK/SIRT1/Autophagy in Cellular Models of Parkinson’s Disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Rosado-Ramos, R.; Poças, G.M.; Marques, D.; Foito, A.; Sevillano, D.M.; Lopes-da-Silva, M.; Gonçalves, L.G.; Menezes, R.; Ottens, M.; Stewart, D.; et al. Genipin Prevents Alpha-Synuclein Aggregation and Toxicity by Affecting Endocytosis, Metabolism and Lipid Storage. Nat. Commun. 2023, 14, 1918. [Google Scholar] [CrossRef]
- Lu, J.-H.; Tan, J.-Q.; Durairajan, S.S.K.; Liu, L.-F.; Zhang, Z.-H.; Ma, L.; Shen, H.-M.; Chan, H.Y.E.; Li, M. Isorhynchophylline, a Natural Alkaloid, Promotes the Degradation of Alpha-Synuclein in Neuronal Cells via Inducing Autophagy. Autophagy 2012, 8, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doytchinova, I.; Atanasova, M.; Salamanova, E.; Ivanov, S.; Dimitrov, I. Curcumin Inhibits the Primary Nucleation of Amyloid-Beta Peptide: A Molecular Dynamics Study. Biomolecules 2020, 10, 1323. [Google Scholar] [CrossRef]
- Andrich, K.; Bieschke, J. The Effect of (−)-Epigallo-Catechin-(3)-Gallate on Amyloidogenic Proteins Suggests a Common Mechanism. Adv. Exp. Med. Biol. 2015, 863, 139–161. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tong, Q.; Ma, S.-R.; Zhao, Z.-X.; Pan, L.-B.; Cong, L.; Han, P.; Peng, R.; Yu, H.; Lin, Y.; et al. Oral Berberine Improves Brain Dopa/Dopamine Levels to Ameliorate Parkinson’s Disease by Regulating Gut Microbiota. Signal Transduct. Target. Ther. 2021, 6, 1–20. [Google Scholar] [CrossRef]
- Zarmouh, N.O.; Messeha, S.S.; Elshami, F.M.; Soliman, K.F.A. Evaluation of the Isoflavone Genistein as Reversible Human Monoamine Oxidase-A and -B Inhibitor. Evid. Based Complement. Altern. Med. 2016, 2016, 1423052. [Google Scholar] [CrossRef] [Green Version]
- Limbocker, R.; Errico, S.; Barbut, D.; Knowles, T.P.J.; Vendruscolo, M.; Chiti, F.; Zasloff, M. Squalamine and Trodusquemine: Two Natural Products for Neurodegenerative Diseases, from Physical Chemistry to the Clinic. Nat. Prod. Rep. 2022, 39, 742–753. [Google Scholar] [CrossRef]
- Ardah, M.T.; Paleologou, K.E.; Lv, G.; Abul Khair, S.B.; Kazim, A.S.; Minhas, S.T.; Al-Tel, T.H.; Al-Hayani, A.A.; Haque, M.E.; Eliezer, D.; et al. Structure Activity Relationship of Phenolic Acid Inhibitors of α-Synuclein Fibril Formation and Toxicity. Front. Aging Neurosci. 2014, 6, 197. [Google Scholar] [CrossRef] [Green Version]
- Ahsan, N.; Mishra, S.; Jain, M.K.; Surolia, A.; Gupta, S. Curcumin Pyrazole and Its Derivative (N-(3-Nitrophenylpyrazole) Curcumin Inhibit Aggregation, Disrupt Fibrils and Modulate Toxicity of Wild Type and Mutant α-Synuclein. Sci. Rep. 2015, 5, 9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taebnia, N.; Morshedi, D.; Yaghmaei, S.; Aliakbari, F.; Rahimi, F.; Arpanaei, A. Curcumin-Loaded Amine-Functionalized Mesoporous Silica Nanoparticles Inhibit α-Synuclein Fibrillation and Reduce Its Cytotoxicity-Associated Effects. Langmuir 2016, 32, 13394–13402. [Google Scholar] [CrossRef] [PubMed]
- Bollimpelli, V.S.; Kumar, P.; Kumari, S.; Kondapi, A.K. Neuroprotective Effect of Curcumin-Loaded Lactoferrin Nano Particles against Rotenone Induced Neurotoxicity. Neurochem. Int. 2016, 95, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, M.; Rajamani, S.; Uversky, V.N.; Fink, A.L. Rifampicin Inhibits Alpha-Synuclein Fibrillation and Disaggregates Fibrils. Chem. Biol. 2004, 11, 1513–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedel, M.; Goldbaum, O.; Schwarz, L.; Schmitt, S.; Richter-Landsberg, C. 17-AAG Induces Cytoplasmic Alpha-Synuclein Aggregate Clearance by Induction of Autophagy. PLoS ONE 2010, 5, e8753. [Google Scholar] [CrossRef] [Green Version]
- Masuda, M.; Suzuki, N.; Taniguchi, S.; Oikawa, T.; Nonaka, T.; Iwatsubo, T.; Hisanaga, S.; Goedert, M.; Hasegawa, M. Small Molecule Inhibitors of Alpha-Synuclein Filament Assembly. Biochemistry 2006, 45, 6085–6094. [Google Scholar] [CrossRef]
- Trusch, F.; Kowski, K.; Bravo-Rodriguez, K.; Beuck, C.; Sowislok, A.; Wettig, B.; Matena, A.; Sanchez-Garcia, E.; Meyer, H.; Schrader, T.; et al. Molecular Tweezers Target a Protein–Protein Interface and Thereby Modulate Complex Formation. Chem. Commun. 2016, 52, 14141–14144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, S.; Lopes, D.H.J.; Du, Z.; Pang, E.S.; Shanmugam, A.; Lomakin, A.; Talbiersky, P.; Tennstaedt, A.; McDaniel, K.; Bakshi, R.; et al. Lysine-Specific Molecular Tweezers Are Broad-Spectrum Inhibitors of Assembly and Toxicity of Amyloid Proteins. J. Am. Chem. Soc. 2011, 133, 16958–16969. [Google Scholar] [CrossRef] [Green Version]
- Attar, A.; Bitan, G. Disrupting Self-Assembly and Toxicity of Amyloidogenic Protein Oligomers by “Molecular Tweezers”—From the Test Tube to Animal Models. Curr. Pharm. Des. 2014, 20, 2469–2483. [Google Scholar] [CrossRef] [Green Version]
- Bengoa-Vergniory, N.; Faggiani, E.; Ramos-Gonzalez, P.; Kirkiz, E.; Connor-Robson, N.; Brown, L.V.; Siddique, I.; Li, Z.; Vingill, S.; Cioroch, M.; et al. CLR01 Protects Dopaminergic Neurons in Vitro and in Mouse Models of Parkinson’s Disease. Nat. Commun. 2020, 11, 4885. [Google Scholar] [CrossRef]
- Prabhudesai, S.; Sinha, S.; Attar, A.; Kotagiri, A.; Fitzmaurice, A.G.; Lakshmanan, R.; Ivanova, M.I.; Loo, J.A.; Klärner, F.-G.; Schrader, T.; et al. A Novel “Molecular Tweezer” Inhibitor of α-Synuclein Neurotoxicity in Vitro and in Vivo. Neurotherapeutics 2012, 9, 464–476. [Google Scholar] [CrossRef] [Green Version]
- Richter, F.; Subramaniam, S.R.; Magen, I.; Lee, P.; Hayes, J.; Attar, A.; Zhu, C.; Franich, N.R.; Bove, N.; De La Rosa, K.; et al. A Molecular Tweezer Ameliorates Motor Deficits in Mice Overexpressing α-Synuclein. Neurotherapeutics 2017, 14, 1107–1119. [Google Scholar] [CrossRef] [Green Version]
- Tatenhorst, L.; Eckermann, K.; Dambeck, V.; Fonseca-Ornelas, L.; Walle, H.; Lopes da Fonseca, T.; Koch, J.C.; Becker, S.; Tönges, L.; Bähr, M.; et al. Fasudil Attenuates Aggregation of α-Synuclein in Models of Parkinson’s Disease. Acta Neuropathol. Commun. 2016, 4, 39. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Qu, J.; Xue, F.; Zheng, Y.; Yang, B.; Chang, Y.; Yang, H.; Zhang, J. Novel DNA Aptamers for Parkinson’s Disease Treatment Inhibit α-Synuclein Aggregation and Facilitate Its Degradation. Mol. Ther. Nucleic Acids 2018, 11, 228–242. [Google Scholar] [CrossRef] [Green Version]
- Ogen-Shtern, N.; Ben David, T.; Lederkremer, G.Z. Protein Aggregation and ER Stress. Brain Res. 2016, 1648, 658–666. [Google Scholar] [CrossRef]
- Cox, D.; Selig, E.; Griffin, M.D.W.; Carver, J.A.; Ecroyd, H. Small Heat-Shock Proteins Prevent α-Synuclein Aggregation via Transient Interactions and Their Efficacy Is Affected by the Rate of Aggregation. J. Biol. Chem. 2016, 291, 22618–22629. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The Co-Chaperone Carboxyl Terminus of Hsp70-Interacting Protein (CHIP) Mediates Alpha-Synuclein Degradation Decisions between Proteasomal and Lysosomal Pathways. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef] [Green Version]
- Durrenberger, P.F.; Filiou, M.D.; Moran, L.B.; Michael, G.J.; Novoselov, S.; Cheetham, M.E.; Clark, P.; Pearce, R.K.B.; Graeber, M.B. DnaJB6 Is Present in the Core of Lewy Bodies and Is Highly Up-Regulated in Parkinsonian Astrocytes. J. Neurosci. Res. 2009, 87, 238–245. [Google Scholar] [CrossRef]
- Bohush, A.; Niewiadomska, G.; Weis, S.; Filipek, A. HSP90 and Its Novel Co-Chaperones, SGT1 and CHP-1, in Brain of Patients with Parkinson’s Disease and Dementia with Lewy Bodies. J. Park. Dis. 2019, 9, 97–107. [Google Scholar] [CrossRef]
- Ping, L.; Duong, D.M.; Yin, L.; Gearing, M.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Global Quantitative Analysis of the Human Brain Proteome in Alzheimer’s and Parkinson’s Disease. Sci. Data 2018, 5, 180036. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.C.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M.-Y. Interactions between HSP70 and the hydrophobic core of alpha–synuclein inhibit fibril assembly. Biochemistry 2008, 47, 12614–12625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.; Berthet, A.; Citron, Y.R.; Tsiolaki, P.L.; Stanley, R.; Gestwicki, J.E.; Agard, D.A.; McConlogue, L. Hsp70 Chaperone Blocks α-Synuclein Oligomer Formation via a Novel Engagement Mechanism. J. Biol. Chem. 2021, 296, 100613. [Google Scholar] [CrossRef] [PubMed]
- Putcha, P.; Danzer, K.M.; Kranich, L.R.; Scott, A.; Silinski, M.; Mabbett, S.; Hicks, C.D.; Veal, J.M.; Steed, P.M.; Hyman, B.T.; et al. Brain-Permeable Small-Molecule Inhibitors of Hsp90 Prevent α-Synuclein Oligomer Formation and Rescue α-Synuclein-Induced Toxicity. J. Pharmacol. Exp. Ther. 2010, 332, 849–857. [Google Scholar] [CrossRef] [Green Version]
- McFarland, N.R.; Dimant, H.; Kibuuka, L.; Ebrahimi-Fakhari, D.; Desjardins, C.A.; Danzer, K.M.; Danzer, M.; Fan, Z.; Schwarzschild, M.A.; Hirst, W.; et al. Chronic Treatment with Novel Small Molecule Hsp90 Inhibitors Rescues Striatal Dopamine Levels but Not α-Synuclein-Induced Neuronal Cell Loss. PLoS ONE 2014, 9, e86048. [Google Scholar] [CrossRef]
- Bendikov-Bar, I.; Maor, G.; Filocamo, M.; Horowitz, M. Ambroxol as a Pharmacological Chaperone for Mutant Glucocerebrosidase. Blood Cells Mol. Dis. 2013, 50, 141–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migdalska-Richards, A.; Ko, W.K.D.; Li, Q.; Bezard, E.; Schapira, A.H.V. Oral Ambroxol Increases Brain Glucocerebrosidase Activity in a Nonhuman Primate. Synapse 2017, 71, e21967. [Google Scholar] [CrossRef] [Green Version]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hällqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the Treatment of Patients with Parkinson Disease With and Without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. 2020, 77, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Aflaki, E.; Borger, D.K.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.J.; Westbroek, W.; Zheng, W.; Sullivan, P.; et al. A New Glucocerebrosidase Chaperone Reduces α-Synuclein and Glycolipid Levels in IPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef]
- Aflaki, E.; Moaven, N.; Borger, D.K.; Lopez, G.; Westbroek, W.; Chae, J.J.; Marugan, J.; Patnaik, S.; Maniwang, E.; Gonzalez, A.N.; et al. Lysosomal Storage and Impaired Autophagy Lead to Inflammasome Activation in Gaucher Macrophages. Aging Cell 2016, 15, 77–88. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Jeon, S.; Zheng, J.; Song, P.; Silverman, R.B.; Krainc, D. A Modulator of Wild-Type Glucocerebrosidase Improves Pathogenic Phenotypes in Dopaminergic Neuronal Models of Parkinson’s Disease. Sci. Transl. Med. 2019, 11, eaau6870. [Google Scholar] [CrossRef]
- den Heijer, J.M.; Kruithof, A.C.; van Amerongen, G.; de Kam, M.L.; Thijssen, E.; Grievink, H.W.; Moerland, M.; Walker, M.; Been, K.; Skerlj, R.; et al. A Randomized Single and Multiple Ascending Dose Study in Healthy Volunteers of LTI-291, a Centrally Penetrant Glucocerebrosidase Activator. Br. J. Clin. Pharmacol. 2021, 87, 3561–3573. [Google Scholar] [CrossRef]
- Kornhaber, G.J.; Tropak, M.B.; Maegawa, G.H.; Tuske, S.J.; Coales, S.J.; Mahuran, D.J.; Hamuro, Y. Isofagomine Induced Stabilization of Glucocerebrosidase. Chembiochem 2008, 9, 2643–2649. [Google Scholar] [CrossRef]
- Sun, Y.; Ran, H.; Liou, B.; Quinn, B.; Zamzow, M.; Zhang, W.; Bielawski, J.; Kitatani, K.; Setchell, K.D.R.; Hannun, Y.A.; et al. Isofagomine In Vivo Effects in a Neuronopathic Gaucher Disease Mouse. PLoS ONE 2011, 6, e19037. [Google Scholar] [CrossRef] [PubMed]
- Schidlitzki, A.; Stanojlovic, M.; Fournier, C.; Käufer, C.; Feja, M.; Gericke, B.; Garzotti, M.; Welford, R.W.D.; Steiner, M.A.; Angot, E.; et al. Double-Edged Effects of Venglustat on Behavior and Pathology in Mice Overexpressing α-Synuclein. Mov. Disord. 2023, 38, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E.; et al. Glucosylceramide Synthase Inhibition Alleviates Aberrations in Synucleinopathy Models. Proc. Natl. Acad. Sci. USA 2017, 114, 2699–2704. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, C.F.; Hijaz, B.A.; Singh, V.; Gcwensa, N.Z.; Kelly, K.; Boyden, E.S.; West, A.B.; Sarkar, D.; Volpicelli-Daley, L.A. Inhibition of LRRK2 Kinase Activity Promotes Anterograde Axonal Transport and Presynaptic Targeting of α-Synuclein. Acta Neuropathol. Commun. 2021, 9, 180. [Google Scholar] [CrossRef]
- Chei, W.S.; Lee, J.-W.; Kim, J.B.; Suh, J. Cell-Penetration by Co(III)Cyclen-Based Peptide-Cleaving Catalysts Selective for Pathogenic Proteins of Amyloidoses. Bioorg. Med. Chem. 2010, 18, 5248–5253. [Google Scholar] [CrossRef]
- Price, D.L.; Koike, M.A.; Khan, A.; Wrasidlo, W.; Rockenstein, E.; Masliah, E.; Bonhaus, D. The Small Molecule Alpha-Synuclein Misfolding Inhibitor, NPT200-11, Produces Multiple Benefits in an Animal Model of Parkinson’s Disease. Sci. Rep. 2018, 8, 16165. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Johnson, R.; Wittmer, C.; Maile, M.; Tatsukawa, K.; Wong, J.L.; Gill, M.B.; Stocking, E.M.; Natala, S.R.; Paulino, A.D.; et al. NPT520-34 Improves Neuropathology and Motor Deficits in a Transgenic Mouse Model of Parkinson’s Disease. Brain 2021, 144, 3692–3709. [Google Scholar] [CrossRef]
- Krishnan, R.; Tsubery, H.; Proschitsky, M.Y.; Asp, E.; Lulu, M.; Gilead, S.; Gartner, M.; Waltho, J.P.; Davis, P.J.; Hounslow, A.M.; et al. A Bacteriophage Capsid Protein Provides a General Amyloid Interaction Motif (GAIM) That Binds and Remodels Misfolded Protein Assemblies. J. Mol. Biol. 2014, 426, 2500–2519. [Google Scholar] [CrossRef]
- Fisher, R.A.; Gannon, K.S.; Krishnan, R.; Levenson, J.M.; Tsubery, H.; Asp, E.; Carroll, J.C.; Cullen, V.; Gartner, M.; Gilead, S.; et al. O1-05-01: Discovery, Preclinical Development, and Clinical Trial Approach for Npt088, a General Amyloid Interaction Motif (GAIM)-Immunoglobulin Fusion. Alzheimer’s Dement. 2015, 11, P135. [Google Scholar] [CrossRef]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A Novel Oligomer Modulator for Disease-Modifying Therapy of Neurodegenerative Diseases Such as Prion and Parkinson’s Disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef] [Green Version]
- Antonschmidt, L.; Matthes, D.; Dervişoğlu, R.; Frieg, B.; Dienemann, C.; Leonov, A.; Nimerovsky, E.; Sant, V.; Ryazanov, S.; Giese, A.; et al. The Clinical Drug Candidate Anle138b Binds in a Cavity of Lipidic α-Synuclein Fibrils. Nat. Commun. 2022, 13, 5385. [Google Scholar] [CrossRef] [PubMed]
- Deeg, A.A.; Reiner, A.M.; Schmidt, F.; Schueder, F.; Ryazanov, S.; Ruf, V.C.; Giller, K.; Becker, S.; Leonov, A.; Griesinger, C.; et al. Anle138b and Related Compounds Are Aggregation Specific Fluorescence Markers and Reveal High Affinity Binding to α-Synuclein Aggregates. Biochim. Biophys. Acta 2015, 1850, 1884–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, J.; Schmidt, F.; Boehm, C.; Prix, C.; Bötzel, K.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A. The Oligomer Modulator Anle138b Inhibits Disease Progression in a Parkinson Mouse Model Even with Treatment Started after Disease Onset. Acta Neuropathol. 2014, 127, 779–780. [Google Scholar] [CrossRef] [Green Version]
- Brendel, M.; Deussing, M.; Blume, T.; Kaiser, L.; Probst, F.; Overhoff, F.; Peters, F.; von Ungern-Sternberg, B.; Ryazanov, S.; Leonov, A.; et al. Late-Stage Anle138b Treatment Ameliorates Tau Pathology and Metabolic Decline in a Mouse Model of Human Alzheimer’s Disease Tau. Alzheimers Res. Ther. 2019, 11, 67. [Google Scholar] [CrossRef] [Green Version]
- Heras-Garvin, A.; Weckbecker, D.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A.; Wenning, G.K.; Stefanova, N. Anle138b Modulates A-synuclein Oligomerization and Prevents Motor Decline and Neurodegeneration in a Mouse Model of Multiple System Atrophy. Mov. Disord. 2019, 34, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellner, L.; Kuzdas-Wood, D.; Levin, J.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A.; Wenning, G.K.; Stefanova, N. Anle138b Partly Ameliorates Motor Deficits Despite Failure of Neuroprotection in a Model of Advanced Multiple System Atrophy. Front. Neurosci. 2016, 10, 710–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallabh, S.M.; Zou, D.; Pitstick, R.; O’Moore, J.; Peters, J.; Silvius, D.; Kriz, J.; Jackson, W.S.; Carlson, G.A.; Minikel, E.V.; et al. Therapeutic Trial of Anle138b in Mouse Models of Genetic Prion Disease. J. Virol. 2023, 97, e0167222. [Google Scholar] [CrossRef]
- Levin, J.; Sing, N.; Melbourne, S.; Morgan, A.; Mariner, C.; Spillantini, M.G.; Wegrzynowicz, M.; Dalley, J.W.; Langer, S.; Ryazanov, S.; et al. Safety, Tolerability and Pharmacokinetics of the Oligomer Modulator Anle138b with Exposure Levels Sufficient for Therapeutic Efficacy in a Murine Parkinson Model: A Randomised, Double-Blind, Placebo-Controlled Phase 1a Trial. eBioMedicine 2022, 80, 104021. [Google Scholar] [CrossRef]
- Peña-Díaz, S.; Pujols, J.; Vasili, E.; Pinheiro, F.; Santos, J.; Manglano-Artuñedo, Z.; Outeiro, T.F.; Ventura, S. The Small Aromatic Compound SynuClean-D Inhibits the Aggregation and Seeded Polymerization of Multiple α-Synuclein Strains. J. Biol. Chem. 2022, 298, 101902. [Google Scholar] [CrossRef] [PubMed]
- Pujols, J.; Peña-Díaz, S.; Lázaro, D.F.; Peccati, F.; Pinheiro, F.; González, D.; Carija, A.; Navarro, S.; Conde-Giménez, M.; García, J.; et al. Small Molecule Inhibits α-Synuclein Aggregation, Disrupts Amyloid Fibrils, and Prevents Degeneration of Dopaminergic Neurons. Proc. Natl. Acad. Sci. USA 2018, 115, 10481–10486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña-Díaz, S.; Pujols, J.; Pinheiro, F.; Santos, J.; Pallarés, I.; Navarro, S.; Conde-Gimenez, M.; García, J.; Salvatella, X.; Dalfó, E.; et al. Inhibition of α-Synuclein Aggregation and Mature Fibril Disassembling with a Minimalistic Compound, ZPDm. Front. Bioeng. Biotechnol. 2020, 8, 588947. [Google Scholar] [CrossRef] [PubMed]
- Peña-Díaz, S.; Pujols, J.; Conde-Giménez, M.; Čarija, A.; Dalfo, E.; García, J.; Navarro, S.; Pinheiro, F.; Santos, J.; Salvatella, X.; et al. ZPD-2, a Small Compound That Inhibits α-Synuclein Amyloid Aggregation and Its Seeded Polymerization. Front. Mol. Neurosci. 2019, 12, 306. [Google Scholar] [CrossRef]
- Gitto, R.; Vittorio, S.; Bucolo, F.; Peña-Díaz, S.; Siracusa, R.; Cuzzocrea, S.; Ventura, S.; Di Paola, R.; De Luca, L. Discovery of Neuroprotective Agents Based on a 5-(4-Pyridinyl)-1,2,4-Triazole Scaffold. ACS Chem. Neurosci. 2022, 13, 581–586. [Google Scholar] [CrossRef]
- Wang, Q.; Yao, S.; Yang, Z.; Zhou, C.; Zhang, Y.; Zhang, Y.; Zhang, L.; Li, J.; Xu, Z.; Zhu, W.; et al. Pharmacological Characterization of the Small Molecule 03A10 as an Inhibitor of α-Synuclein Aggregation for Parkinson’s Disease Treatment. Acta Pharmacol. Sin. 2023, 44, 1122–1134. [Google Scholar] [CrossRef]
- Zhang, J.; Jasutkar, H.G.; Mouradian, M.M. Targeting Transglutaminase 2 as a Potential Disease Modifying Therapeutic Strategy for Synucleinopathies. Neural Regen. Res. 2021, 16, 1560–1561. [Google Scholar] [CrossRef]
- Paillusson, S.; Tasselli, M.; Lebouvier, T.; Mahé, M.M.; Chevalier, J.; Biraud, M.; Cario-Toumaniantz, C.; Neunlist, M.; Derkinderen, P. α-Synuclein Expression Is Induced by Depolarization and Cyclic AMP in Enteric Neurons. J. Neurochem. 2010, 115, 694–706. [Google Scholar] [CrossRef]
- Rostami, J.; Jäntti, M.; Cui, H.; Rinne, M.K.; Kukkonen, J.P.; Falk, A.; Erlandsson, A.; Myöhänen, T. Prolyl Oligopeptidase Inhibition by KYP-2407 Increases Alpha-Synuclein Fibril Degradation in Neuron-like Cells. Biomed. Pharmacother. 2020, 131, 110788. [Google Scholar] [CrossRef]
- Braun, A.R.; Liao, E.E.; Horvath, M.; Kalra, P.; Acosta, K.; Young, M.C.; Kochen, N.N.; Lo, C.H.; Brown, R.; Evans, M.D.; et al. Potent Inhibitors of Toxic Alpha-Synuclein Identified via Cellular Time-Resolved FRET Biosensors. NPJ Park. Dis. 2021, 7, 1–17. [Google Scholar] [CrossRef]
- Cankara, F.N.; Kuş, M.S.; Günaydın, C.; Şafak, S.; Bilge, S.S.; Ozmen, O.; Tural, E.; Kortholt, A. The Beneficial Effect of Salubrinal on Neuroinflammation and Neuronal Loss in Intranigral LPS-Induced Hemi-Parkinson Disease Model in Rats. Immunopharmacol. Immunotoxicol. 2022, 44, 168–177. [Google Scholar] [CrossRef]
- Harrison, C. A PERK for the Therapy of Prion Disease. Nat. Rev. Drug Discov. 2013, 12, 906. [Google Scholar] [CrossRef]
- Sen, T.; Gupta, R.; Kaiser, H.; Sen, N. Activation of PERK Elicits Memory Impairment through Inactivation of CREB and Downregulation of PSD95 After Traumatic Brain Injury. J. Neurosci. 2017, 37, 5900–5911. [Google Scholar] [CrossRef]
- Lusa, W.; Rozpędek-Kamińska, W.; Siwecka, N.; Galita, G.; Majsterek, I.; Kucharska, E. Small-molecule PKR-like Endoplasmic Reticulum Kinase Inhibitors as a Novel Targeted Therapy for Parkinson’s Disease. Mol. Med. Rep. 2023, 27, 102. [Google Scholar] [CrossRef]
- Halliday, M.; Radford, H.; Sekine, Y.; Moreno, J.; Verity, N.; le Quesne, J.; Ortori, C.A.; Barrett, D.A.; Fromont, C.; Fischer, P.M.; et al. Partial Restoration of Protein Synthesis Rates by the Small Molecule ISRIB Prevents Neurodegeneration without Pancreatic Toxicity. Cell Death Dis. 2015, 6, e1672. [Google Scholar] [CrossRef] [Green Version]
- Halliday, M.; Radford, H.; Zents, K.A.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M.; et al. Repurposed Drugs Targeting EIF2α-P-Mediated Translational Repression Prevent Neurodegeneration in Mice. Brain 2017, 140, 1768–1783. [Google Scholar] [CrossRef] [Green Version]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing Proteostasis Diseases by Selective Inhibition of a Phosphatase Regulatory Subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Popko, B.; Tixier, E.; Roos, R.P. Guanabenz, Which Enhances the Unfolded Protein Response, Ameliorates Mutant SOD1-Induced Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2014, 71, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Zhang, M.; Zhang, Y.-Q.; Xu, Z.-H. JNK Pathway: Diseases and Therapeutic Potential. Acta Pharmacol. Sin. 2007, 28, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Esteves, A.R.; Arduíno, D.M.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Oxidative Stress Involvement in Alpha-Synuclein Oligomerization in Parkinson’s Disease Cybrids. Antioxid. Redox Signal 2009, 11, 439–448. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N.; et al. Targeting Chelatable Iron as a Therapeutic Modality in Parkinson’s Disease. Antioxid. Redox Signal 2014, 21, 195–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain Iron Chelation by Deferiprone in a Phase 2 Randomised Double-Blinded Placebo Controlled Clinical Trial in Parkinson’s Disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devos, D.; Labreuche, J.; Rascol, O.; Corvol, J.-C.; Duhamel, A.; Guyon Delannoy, P.; Poewe, W.; Compta, Y.; Pavese, N.; Růžička, E.; et al. Trial of Deferiprone in Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 2045–2055. [Google Scholar] [CrossRef]
- Febbraro, F.; Andersen, K.J.; Sanchez-Guajardo, V.; Tentillier, N.; Romero-Ramos, M. Chronic Intranasal Deferoxamine Ameliorates Motor Defects and Pathology in the α-Synuclein RAAV Parkinson’s Model. Exp. Neurol. 2013, 247, 45–58. [Google Scholar] [CrossRef]
- Das, B.; Rajagopalan, S.; Joshi, G.S.; Xu, L.; Luo, D.; Andersen, J.K.; Todi, S.V.; Dutta, A.K. A Novel Iron (II) Preferring Dopamine Agonist Chelator D-607 Significantly Suppresses α-Syn- and MPTP-Induced Toxicities in Vivo. Neuropharmacology 2017, 123, 88–99. [Google Scholar] [CrossRef]
- Heras-Garvin, A.; Refolo, V.; Schmidt, C.; Malfertheiner, K.; Wenning, G.K.; Bradbury, M.; Stamler, D.; Stefanova, N. ATH434 Reduces α-Synuclein-Related Neurodegeneration in a Murine Model of Multiple System Atrophy. Mov. Disord. 2021, 36, 2605–2614. [Google Scholar] [CrossRef]
- Finkelstein, D.I.; Shukla, J.J.; Cherny, R.A.; Billings, J.L.; Saleh, E.; Stefanova, N.; Barnham, K.J.; Adlard, P.A. The Compound ATH434 Prevents Alpha-Synuclein Toxicity in a Murine Model of Multiple System Atrophy. J. Parkinsons Dis. 2022, 12, 105–115. [Google Scholar] [CrossRef]
- Finkelstein, D.I.; Billings, J.L.; Adlard, P.A.; Ayton, S.; Sedjahtera, A.; Masters, C.L.; Wilkins, S.; Shackleford, D.M.; Charman, S.A.; Bal, W.; et al. The Novel Compound PBT434 Prevents Iron Mediated Neurodegeneration and Alpha-Synuclein Toxicity in Multiple Models of Parkinson’s Disease. Acta Neuropathol. Commun. 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamler, D.; Bradbury, M.; Wong, C.; Offman, E. A First in Human Study of PBT434, a Novel Small Molecule Inhibitor of α-Synuclein Aggregation (S4.001). Neurology 2019, 92 (Suppl. 15), S4.001. [Google Scholar]
- Shi, L.; Huang, C.; Luo, Q.; Xia, Y.; Liu, W.; Zeng, W.; Cheng, A.; Shi, R.; Zhengli, C. Clioquinol Improves Motor and Non-Motor Deficits in MPTP-Induced Monkey Model of Parkinson’s Disease through AKT/MTOR Pathway. Aging 2020, 12, 9515–9533. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Fridkin, M.; Zheng, H. Bifunctional Drug Derivatives of MAO-B Inhibitor Rasagiline and Iron Chelator VK-28 as a More Effective Approach to Treatment of Brain Ageing and Ageing Neurodegenerative Diseases. Mech. Ageing Dev. 2005, 126, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Gal, S.; Weiner, L.M.; Bar-Am, O.; Warshawsky, A.; Fridkin, M.; Youdim, M.B.H. Novel Multifunctional Neuroprotective Iron Chelator-Monoamine Oxidase Inhibitor Drugs for Neurodegenerative Diseases: In Vitro Studies on Antioxidant Activity, Prevention of Lipid Peroxide Formation and Monoamine Oxidase Inhibition. J. Neurochem. 2005, 95, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Dunkel, P.; Chai, C.L.; Sperlágh, B.; Huleatt, P.B.; Mátyus, P. Clinical Utility of Neuroprotective Agents in Neurodegenerative Diseases: Current Status of Drug Development for Alzheimer’s, Parkinson’s and Huntington’s Diseases, and Amyotrophic Lateral Sclerosis. Expert. Opin. Investig. Drugs 2012, 21, 1267–1308. [Google Scholar] [CrossRef]
- Mohamad Najib, N.H.; Yahaya, M.F.; Das, S.; Teoh, S.L. The Effects of Metallothionein in Paraquat-Induced Parkinson Disease Model of Zebrafish. Int. J. Neurosci. 2021, 133, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Asanuma, M. Multifunctional Metallothioneins as a Target for Neuroprotection in Parkinson’s Disease. Antioxidants 2023, 12, 894. [Google Scholar] [CrossRef]
- Choi, J.S.; Park, C.; Jeong, J.W. AMP-Activated Protein Kinase Is Activated in Parkinson’s Disease Models Mediated by 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine. Biochem. Biophys. Res. Commun. 2010, 391, 147–151. [Google Scholar] [CrossRef]
- Cullen, V.; Lindfors, M.; Ng, J.; Paetau, A.; Swinton, E.; Kolodziej, P.; Boston, H.; Saftig, P.; Woulfe, J.; Feany, M.B.; et al. Cathepsin D Expression Level Affects Alpha-Synuclein Processing, Aggregation, and Toxicity in Vivo. Mol. Brain 2009, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, X.-X.; Wang, S.-F.; Tan, Y.; Song, J.-X.; Zhu, Z.; Wang, Z.-Y.; Wu, M.-Y.; Cai, C.-Z.; Huang, Z.-J.; Tan, J.-Q.; et al. Pharmacological Enhancement of TFEB-Mediated Autophagy Alleviated Neuronal Death in Oxidative Stress-Induced Parkinson’s Disease Models. Cell Death Dis. 2020, 11, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Issa, A.-R.; Sun, J.; Petitgas, C.; Mesquita, A.; Dulac, A.; Robin, M.; Mollereau, B.; Jenny, A.; Chérif-Zahar, B.; Birman, S. The Lysosomal Membrane Protein LAMP2A Promotes Autophagic Flux and Prevents SNCA-Induced Parkinson Disease-like Symptoms in the Drosophila Brain. Autophagy 2018, 14, 1898–1910. [Google Scholar] [CrossRef] [Green Version]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 Gene Transfer Activates Autophagy and Ameliorates the Neurodegenerative Pathology in Alpha-Synuclein Models of Parkinson’s and Lewy Body Diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [Green Version]
- Lee, F.K.M.; Wong, A.K.Y.; Lee, Y.W.; Wan, O.W.; Chan, H.Y.E.; Chung, K.K.K. The Role of Ubiquitin Linkages on Alpha-Synuclein Induced-Toxicity in a Drosophila Model of Parkinson’s Disease. J. Neurochem. 2009, 110, 208–219. [Google Scholar] [CrossRef]
- Li, T.; Feng, Y.; Yang, R.; Wu, L.; Li, R.; Huang, L.; Yang, Q.; Chen, J. Salidroside Promotes the Pathological α-Synuclein Clearance Through Ubiquitin-Proteasome System in SH-SY5Y Cells. Front. Pharmacol. 2018, 9, 377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boselli, M.; Lee, B.-H.; Robert, J.; Prado, M.A.; Min, S.-W.; Cheng, C.; Silva, M.C.; Seong, C.; Elsasser, S.; Hatle, K.M.; et al. An Inhibitor of the Proteasomal Deubiquitinating Enzyme USP14 Induces Tau Elimination in Cultured Neurons. J. Biol. Chem. 2017, 292, 19209–19225. [Google Scholar] [CrossRef] [Green Version]
- Kiprowska, M.J.; Stepanova, A.; Todaro, D.R.; Galkin, A.; Haas, A.; Wilson, S.M.; Figueiredo-Pereira, M.E. Neurotoxic Mechanisms by Which the USP14 Inhibitor IU1 Depletes Ubiquitinated Proteins and Tau in Rat Cerebral Cortical Neurons: Relevance to Alzheimer’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1157–1170. [Google Scholar] [CrossRef]
- Zhou, H.; Shao, M.; Guo, B.; Li, C.; Lu, Y.; Yang, X.; Li, S.; Li, H.; Zhu, Q.; Zhong, H.; et al. Tetramethylpyrazine Analogue T-006 Promotes the Clearance of Alpha-Synuclein by Enhancing Proteasome Activity in Parkinson’s Disease Models. Neurotherapeutics 2019, 16, 1225–1236. [Google Scholar] [CrossRef]
- Chatterjee, D.; Bhatt, M.; Butler, D.; De Genst, E.; Dobson, C.M.; Messer, A.; Kordower, J.H. Proteasome-Targeted Nanobodies Alleviate Pathology and Functional Decline in an α-Synuclein-Based Parkinson’s Disease Model. NPJ Park. Dis. 2018, 4, 25. [Google Scholar] [CrossRef] [Green Version]
- Bové, J.; Martínez-Vicente, M.; Vila, M. Fighting Neurodegeneration with Rapamycin: Mechanistic Insights. Nat. Rev. Neurosci. 2011, 12, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Rockenstein, E.; Ostroff, G.; Dikengil, F.; Rus, F.; Mante, M.; Florio, J.; Adame, A.; Trinh, I.; Kim, C.; Overk, C.; et al. Combined Active Humoral and Cellular Immunization Approaches for the Treatment of Synucleinopathies. J. Neurosci. 2018, 38, 1000–1014. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Tyson, T.; George, S.; Hildebrandt, E.N.; Steiner, J.A.; Madaj, Z.; Schulz, E.; Machiela, E.; McDonald, W.G.; Escobar Galvis, M.L.; et al. Mitochondrial Pyruvate Carrier Regulates Autophagy, Inflammation, and Neurodegeneration in Experimental Models of Parkinson’s Disease. Sci. Transl. Med. 2016, 8, 368ra174. [Google Scholar] [CrossRef]
- Siracusa, R.; Paterniti, I.; Cordaro, M.; Crupi, R.; Bruschetta, G.; Campolo, M.; Cuzzocrea, S.; Esposito, E. Neuroprotective Effects of Temsirolimus in Animal Models of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 2403–2419. [Google Scholar] [CrossRef]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a Novel MTOR-Independent Autophagy Enhancer, Accelerates the Clearance of Mutant Huntingtin and Alpha-Synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [Green Version]
- Redmann, M.; Wani, W.Y.; Volpicelli-Daley, L.; Darley-Usmar, V.; Zhang, J. Trehalose Does Not Improve Neuronal Survival on Exposure to Alpha-Synuclein Pre-Formed Fibrils. Redox Biol. 2017, 11, 429–437. [Google Scholar] [CrossRef]
- Song, W.; Wang, F.; Lotfi, P.; Sardiello, M.; Segatori, L. 2-Hydroxypropyl-β-Cyclodextrin Promotes Transcription Factor EB-Mediated Activation of Autophagy. J. Biol. Chem. 2014, 289, 10211–10222. [Google Scholar] [CrossRef] [Green Version]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical Modulation of Chaperone-Mediated Autophagy by Retinoic Acid Derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Pupyshev, A.B.; Tikhonova, M.A.; Akopyan, A.A.; Tenditnik, M.V.; Dubrovina, N.I.; Korolenko, T.A. Therapeutic Activation of Autophagy by Combined Treatment with Rapamycin and Trehalose in a Mouse MPTP-Induced Model of Parkinson’s Disease. Pharmacol. Biochem. Behav. 2019, 177, 1–11. [Google Scholar] [CrossRef]
- Ko, H.S.; Lee, Y.; Shin, J.-H.; Karuppagounder, S.S.; Gadad, B.S.; Koleske, A.J.; Pletnikova, O.; Troncoso, J.C.; Dawson, V.L.; Dawson, T.M. Phosphorylation by the C-Abl Protein Tyrosine Kinase Inhibits Parkin’s Ubiquitination and Protective Function. Proc. Natl. Acad. Sci. USA 2010, 107, 16691–16696. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.-L.; Fauvet, B.; Gysbers, A.; Dikiy, I.; Oueslati, A.; Georgeon, S.; Lamontanara, A.J.; Bisquertt, A.; Eliezer, D.; Masliah, E.; et al. C-Abl Phosphorylates α-Synuclein and Regulates Its Degradation: Implication for α-Synuclein Clearance and Contribution to the Pathogenesis of Parkinson’s Disease. Hum. Mol. Genet. 2014, 23, 2858–2879. [Google Scholar] [CrossRef] [Green Version]
- Pagan, F.L.; Hebron, M.L.; Wilmarth, B.; Torres-Yaghi, Y.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, N.J.; Anjum, M.; Arellano, J.; et al. Nilotinib Effects on Safety, Tolerability, and Potential Biomarkers in Parkinson Disease: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2020, 77, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Espay, A.J.; Hauser, R.A.; Armstrong, M.J. The Narrowing Path for Nilotinib and Other Potential Disease-Modifying Therapies for Parkinson Disease. JAMA Neurol. 2020, 77, 295–297. [Google Scholar] [CrossRef]
- Danzer, K.M.; Ruf, W.P.; Putcha, P.; Joyner, D.; Hashimoto, T.; Glabe, C.; Hyman, B.T.; McLean, P.J. Heat-Shock Protein 70 Modulates Toxic Extracellular α-Synuclein Oligomers and Rescues Trans-Synaptic Toxicity. FASEB J. 2011, 25, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Kasai, T.; Tokuda, T.; Yamaguchi, N.; Watanabe, Y.; Kametani, F.; Nakagawa, M.; Mizuno, T. Cleavage of Normal and Pathological Forms of Alpha-Synuclein by Neurosin in Vitro. Neurosci. Lett. 2008, 436, 52–56. [Google Scholar] [CrossRef]
- Pampalakis, G.; Sykioti, V.-S.; Ximerakis, M.; Stefanakou-Kalakou, I.; Melki, R.; Vekrellis, K.; Sotiropoulou, G. KLK6 Proteolysis Is Implicated in the Turnover and Uptake of Extracellular Alpha-Synuclein Species. Oncotarget 2016, 8, 14502–14515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.-J.; Woo, M.-S.; Moon, P.-G.; Baek, M.-C.; Choi, I.-Y.; Kim, W.-K.; Junn, E.; Kim, H.-S. Alpha-Synuclein Activates Microglia by Inducing the Expressions of Matrix Metalloproteinases and the Subsequent Activation of Protease-Activated Receptor-1. J. Immunol. 2010, 185, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, J.Y.; Park, S.M.; Lee, C.-H.; Um, J.W.; Lee, H.J.; Kim, J.; Oh, Y.J.; Lee, S.-T.; Paik, S.R.; Chung, K.C. Proteolytic Cleavage of Extracellular Secreted {alpha}-Synuclein via Matrix Metalloproteinases. J. Biol. Chem. 2005, 280, 25216–25224. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.S.; Choi, Y.R.; Park, J.-Y.; Lee, J.-H.; Kim, D.K.; Lee, S.-J.; Paik, S.R.; Jou, I.; Park, S.M. Proteolytic Cleavage of Extracellular α-Synuclein by Plasmin. J. Biol. Chem. 2012, 287, 24862–24872. [Google Scholar] [CrossRef] [Green Version]
- Tucker, H.M.; Kihiko, M.; Caldwell, J.N.; Wright, S.; Kawarabayashi, T.; Price, D.; Walker, D.; Scheff, S.; McGillis, J.P.; Rydel, R.E.; et al. The Plasmin System Is Induced by and Degrades Amyloid-β Aggregates. J. Neurosci. 2000, 20, 3937–3946. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, K.; Yamada, T.; Tsujioka, Y.; Taguchi, J.; Takahashi, M.; Tsuboi, Y.; Fujino, Y.; Nakajima, M.; Yamamoto, T.; Akatsu, H.; et al. Localization of a Novel Type Trypsin-like Serine Protease, Neurosin, in Brain Tissues of Alzheimer’s Disease and Parkinson’s Disease. Psychiatry Clin. Neurosci. 2000, 54, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Joo, S.H.; Kwon, K.J.; Kim, J.W.; Kim, J.W.; Hasan, M.R.; Lee, H.-J.; Han, S.-H.; Shin, C.Y. Regulation of Matrix Metalloproteinase-9 and Tissue Plasminogen Activator Activity by Alpha-Synuclein in Rat Primary Glial Cells. Neurosci. Lett. 2010, 469, 352–356. [Google Scholar] [CrossRef]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan Sulfate Proteoglycans Mediate Internalization and Propagation of Specific Proteopathic Seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef]
- Freeman, D.; Cedillos, R.; Choyke, S.; Lukic, Z.; McGuire, K.; Marvin, S.; Burrage, A.M.; Sudholt, S.; Rana, A.; O’Connor, C.; et al. Alpha-Synuclein Induces Lysosomal Rupture and Cathepsin Dependent Reactive Oxygen Species Following Endocytosis. PLoS ONE 2013, 8, e62143. [Google Scholar] [CrossRef]
- Jiang, P.; Gan, M.; Yen, S.-H.; McLean, P.J.; Dickson, D.W. Impaired Endo-Lysosomal Membrane Integrity Accelerates the Seeding Progression of α-Synuclein Aggregates. Sci. Rep. 2017, 7, 7690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabey, J.M.; Nissipeanu, P.; Korczyn, A.D. Efficacy of Memantine, an NMDA Receptor Antagonist, in the Treatment of Parkinson’s Disease. J. Neural Transm. Park. Dis. Dement. Sect. 1992, 4, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Sheng, M. NMDA Receptors in Nervous System Diseases. Neuropharmacology 2013, 74, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.-I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.-H.; et al. Pathological α-Synuclein Transmission Initiated by Binding Lymphocyte-Activation Gene 3. Science 2016, 353, aah3374. [Google Scholar] [CrossRef] [Green Version]
- Emmenegger, M.; De Cecco, E.; Hruska-Plochan, M.; Eninger, T.; Schneider, M.M.; Barth, M.; Tantardini, E.; de Rossi, P.; Bacioglu, M.; Langston, R.G.; et al. LAG3 Is Not Expressed in Human and Murine Neurons and Does Not Modulate A-synucleinopathies. EMBO Mol. Med. 2021, 13, e14745. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.G.; Béraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of MyD88-Dependent TLR1/2 Signaling by Misfolded α-Synuclein, a Protein Linked to Neurodegenerative Disorders. Sci. Signal 2015, 8, ra45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Spencer, B.; Rockenstein, E.; Yamakado, H.; Mante, M.; Adame, A.; Fields, J.A.; Masliah, D.; Iba, M.; Lee, H.-J.; et al. Immunotherapy Targeting Toll-like Receptor 2 Alleviates Neurodegeneration in Models of Synucleinopathy by Modulating α-Synuclein Transmission and Neuroinflammation. Mol. Neurodegener. 2018, 13, 43. [Google Scholar] [CrossRef]
- Ren, L.; Yi, J.; Yang, J.; Li, P.; Cheng, X.; Mao, P. Nonsteroidal Anti-Inflammatory Drugs Use and Risk of Parkinson Disease: A Dose-Response Meta-Analysis. Medicine 2018, 97, e12172. [Google Scholar] [CrossRef]
- Brakedal, B.; Tzoulis, C.; Tysnes, O.-B.; Haugarvoll, K. NSAID Use Is Not Associated with Parkinson’s Disease Incidence: A Norwegian Prescription Database Study. PLoS ONE 2021, 16, e0256602. [Google Scholar] [CrossRef]
- Oueslati, A.; Schneider, B.L.; Aebischer, P.; Lashuel, H.A. Polo-like Kinase 2 Regulates Selective Autophagic α-Synuclein Clearance and Suppresses Its Toxicity in Vivo. Proc. Natl. Acad. Sci. USA 2013, 110, E3945–E3954. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, J.; Park, H.-J.; Park, E.S.; Oh, S.; Zheng, H.; Junn, E.; Voronkov, M.; Stock, J.B.; Mouradian, M.M. Synergistic Neuroprotection by Coffee Components Eicosanoyl-5-Hydroxytryptamide and Caffeine in Models of Parkinson’s Disease and DLB. Proc. Natl. Acad. Sci. USA 2018, 115, E12053–E12062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Revuelta, B.I.; Hettich, M.M.; Ciociaro, A.; Rotermund, C.; Kahle, P.J.; Krauss, S.; Di Monte, D.A. Metformin Lowers Ser-129 Phosphorylated α-Synuclein Levels via MTOR-Dependent Protein Phosphatase 2A Activation. Cell Death Dis. 2014, 5, e1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katila, N.; Bhurtel, S.; Shadfar, S.; Srivastav, S.; Neupane, S.; Ojha, U.; Jeong, G.-S.; Choi, D.-Y. Metformin Lowers α-Synuclein Phosphorylation and Upregulates Neurotrophic Factor in the MPTP Mouse Model of Parkinson’s Disease. Neuropharmacology 2017, 125, 396–407. [Google Scholar] [CrossRef]
- Gao, Y.; Kabotyanski, E.B.; Shepherd, J.H.; Villegas, E.; Acosta, D.; Hamor, C.; Sun, T.; Montmeyor-Garcia, C.; He, X.; Dobrolecki, L.E.; et al. Tumor Suppressor PLK2 May Serve as a Biomarker in Triple-Negative Breast Cancer for Improved Response to PLK1 Therapeutics. Cancer Res. Commun. 2021, 1, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Huang, P.; Liang, J.; Fu, W.; Guo, Z.; Xu, L. Microcystin-LR (MCLR) Induces a Compensation of PP2A Activity Mediated by A4 Protein in HEK293 Cells. Int. J. Biol. Sci. 2011, 7, 740–752. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.-M.; Kotzbauer, P.T.; Uryu, K.; Leight, S.; Trojanowski, J.Q.; Lee, V.M.-Y. Neuroinflammation and Oxidation/Nitration of Alpha-Synuclein Linked to Dopaminergic Neurodegeneration. J. Neurosci. 2008, 28, 7687–7698. [Google Scholar] [CrossRef] [Green Version]
- Boza-Serrano, A.; Reyes, J.F.; Rey, N.L.; Leffler, H.; Bousset, L.; Nilsson, U.; Brundin, P.; Venero, J.L.; Burguillos, M.A.; Deierborg, T. The Role of Galectin-3 in α-Synuclein-Induced Microglial Activation. Acta Neuropathol. Commun. 2014, 2, 156. [Google Scholar] [CrossRef]
- Parlak, H.; Ozkan, A.; Dilmac, S.; Tanriover, G.; Ozsoy, O.; Agar, A. Neuronal Nitric Oxide Synthase Phosphorylation Induced by Docosahexaenoic Acid Protects Dopaminergic Neurons in an Experimental Model of Parkinson’s Disease. Folia Histochem. Cytobiol. 2018, 56, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Gao, W.; Shan, X.; Wang, C.; Wang, H.; Shao, Z.; Dou, S.; Jiang, Y.; Wang, C.; Cheng, B. Apelin-36 Mediates Neuroprotective Effects by Regulating Oxidative Stress, Autophagy and Apoptosis in MPTP-Induced Parkinson’s Disease Model Mice. Brain Res. 2020, 1726, 146493. [Google Scholar] [CrossRef]
- Lee, J.A.; Kim, D.J.; Hwang, O. KMS99220 Exerts Anti-Inflammatory Effects, Activates the Nrf2 Signaling and Interferes with IKK, JNK and P38 MAPK via HO-1. Mol. Cells 2019, 42, 702–710. [Google Scholar] [CrossRef]
- Hung, L.W.; Villemagne, V.L.; Cheng, L.; Sherratt, N.A.; Ayton, S.; White, A.R.; Crouch, P.J.; Lim, S.; Leong, S.L.; Wilkins, S.; et al. The Hypoxia Imaging Agent CuII(Atsm) Is Neuroprotective and Improves Motor and Cognitive Functions in Multiple Animal Models of Parkinson’s Disease. J. Exp. Med. 2012, 209, 837–854. [Google Scholar] [CrossRef] [PubMed]
- Abdeen, A.H.; Trist, B.G.; Double, K.L. Empirical Evidence for Biometal Dysregulation in Parkinson’s Disease from a Systematic Review and Bradford Hill Analysis. NPJ Park. Dis. 2022, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Selnick, H.G.; Hess, J.F.; Tang, C.; Liu, K.; Schachter, J.B.; Ballard, J.E.; Marcus, J.; Klein, D.J.; Wang, X.; Pearson, M.; et al. Discovery of MK-8719, a Potent O-GlcNAcase Inhibitor as a Potential Treatment for Tauopathies. J. Med. Chem. 2019, 62, 10062–10097. [Google Scholar] [CrossRef] [Green Version]
- Hastings, N.B.; Wang, X.; Song, L.; Butts, B.D.; Grotz, D.; Hargreaves, R.; Fred Hess, J.; Hong, K.-L.K.; Huang, C.R.-R.; Hyde, L.; et al. Inhibition of O-GlcNAcase Leads to Elevation of O-GlcNAc Tau and Reduction of Tauopathy and Cerebrospinal Fluid Tau in RTg4510 Mice. Mol. Neurodegener. 2017, 12, 39. [Google Scholar] [CrossRef] [Green Version]
- Permanne, B.; Sand, A.; Ousson, S.; Nény, M.; Hantson, J.; Schubert, R.; Wiessner, C.; Quattropani, A.; Beher, D. O-GlcNAcase Inhibitor ASN90 Is a Multimodal Drug Candidate for Tau and α-Synuclein Proteinopathies. ACS Chem. Neurosci. 2022, 13, 1296–1314. [Google Scholar] [CrossRef]
- Vicente Miranda, H.; Szego, É.M.; Oliveira, L.M.A.; Breda, C.; Darendelioglu, E.; de Oliveira, R.M.; Ferreira, D.G.; Gomes, M.A.; Rott, R.; Oliveira, M.; et al. Glycation Potentiates α-Synuclein-Associated Neurodegeneration in Synucleinopathies. Brain 2017, 140, 1399–1419. [Google Scholar] [CrossRef] [Green Version]
- Diepenbroek, M.; Casadei, N.; Esmer, H.; Saido, T.C.; Takano, J.; Kahle, P.J.; Nixon, R.A.; Rao, M.V.; Melki, R.; Pieri, L.; et al. Overexpression of the Calpain-Specific Inhibitor Calpastatin Reduces Human Alpha-Synuclein Processing, Aggregation and Synaptic Impairment in [A30P]ASyn Transgenic Mice. Hum. Mol. Genet. 2014, 23, 3975–3989. [Google Scholar] [CrossRef] [PubMed]
- Hassen, G.W.; Kesner, L.; Stracher, A.; Shulman, A.; Rockenstein, E.; Mante, M.; Adame, A.; Overk, C.; Rissman, R.A.; Masliah, E. Effects of Novel Calpain Inhibitors in Transgenic Animal Model of Parkinson’s Disease/Dementia with Lewy Bodies. Sci. Rep. 2018, 8, 18083. [Google Scholar] [CrossRef] [Green Version]
- Nuber, S.; Nam, A.Y.; Rajsombath, M.M.; Cirka, H.; Hronowski, X.; Wang, J.; Hodgetts, K.; Kalinichenko, L.S.; Müller, C.P.; Lambrecht, V.; et al. A Stearoyl–Coenzyme A Desaturase Inhibitor Prevents Multiple Parkinson Disease Phenotypes in A-Synuclein Mice. Ann. Neurol. 2021, 89, 74–90. [Google Scholar] [CrossRef]
- Tardiff, D.F.; Lucas, M.; Wrona, I.; Chang, B.; Chung, C.Y.; Le Bourdonnec, B.; Rhodes, K.J.; Scannevin, R.H. Non-Clinical Pharmacology of YTX-7739: A Clinical Stage Stearoyl-CoA Desaturase Inhibitor Being Developed for Parkinson’s Disease. Mol. Neurobiol. 2022, 59, 2171–2189. [Google Scholar] [CrossRef]
- Daffu, G.; del Pozo, C.H.; O’Shea, K.M.; Ananthakrishnan, R.; Ramasamy, R.; Schmidt, A.M. Radical Roles for RAGE in the Pathogenesis of Oxidative Stress in Cardiovascular Diseases and Beyond. Int. J. Mol. Sci. 2013, 14, 19891–19910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Shi, C.; Xu, Y. Rab GTPases: The Key Players in the Molecular Pathway of Parkinson’s Disease. Front. Cell Neurosci. 2017, 11, 81. [Google Scholar] [CrossRef] [Green Version]
- Donmez, G.; Outeiro, T.F. SIRT1 and SIRT2: Emerging Targets in Neurodegeneration. EMBO Mol. Med. 2013, 5, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Kam, T.-I.; Mao, X.; Park, H.; Chou, S.-C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly (ADP-Ribose) Drives Pathologic α-Synuclein Neurodegeneration in Parkinson’s Disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.; Wang, J.; He, M.; Xiong, M.; Tian, Y.; Liu, C.; Zhang, Z. Lovastatin Alleviates α-Synuclein Aggregation and Phosphorylation in Cellular Models of Synucleinopathy. Front. Mol. Neurosci. 2021, 14, 682320. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, M.; Shen, L.; Zhu, Y.; Ma, H.; Liu, B.; Ouyang, L.; Guo, W.; Xu, Q.; Sun, Y. SHP2-Mediated Mitophagy Boosted by Lovastatin in Neuronal Cells Alleviates Parkinsonism in Mice. Signal Transduct. Target. Ther. 2021, 6, 34. [Google Scholar] [CrossRef]
- Albert, K.; Raymundo, D.P.; Panhelainen, A.; Eesmaa, A.; Shvachiy, L.; Araújo, G.R.; Chmielarz, P.; Yan, X.; Singh, A.; Cordeiro, Y.; et al. Cerebral Dopamine Neurotrophic Factor Reduces α-Synuclein Aggregation and Propagation and Alleviates Behavioral Alterations in Vivo. Mol. Ther. 2021, 29, 2821–2840. [Google Scholar] [CrossRef]
- Zhang, J.-X.; Zhou, K.-G.; Yin, Y.-X.; Jin, L.-J.; Tong, W.-F.; Guo, J.; Yu, L.-H.; Ye, X.-C.; Jiang, M. Mesencephalic Astrocyte-Derived Neurotrophic Factor (MANF) Prevents the Neuroinflammation Induced Dopaminergic Neurodegeneration. Exp. Gerontol. 2023, 171, 112037. [Google Scholar] [CrossRef]
- Cheng, L.; Zhao, H.; Zhang, W.; Liu, B.; Liu, Y.; Guo, Y.; Nie, L. Overexpression of Conserved Dopamine Neurotrophic Factor (CDNF) in Astrocytes Alleviates Endoplasmic Reticulum Stress-Induced Cell Damage and Inflammatory Cytokine Secretion. Biochem. Biophys. Res. Commun. 2013, 435, 34–39. [Google Scholar] [CrossRef]
- Schneeberger, A.; Mandler, M.; Mattner, F.; Schmidt, W. Vaccination for Parkinson’s Disease. Park. Relat. Disord. 2012, 18 (Suppl. 1), S11–S13. [Google Scholar] [CrossRef]
- Ugen, K.E.; Lin, X.; Bai, G.; Liang, Z.; Cai, J.; Li, K.; Song, S.; Cao, C.; Sanchez-Ramos, J. Evaluation of an α Synuclein Sensitized Dendritic Cell Based Vaccine in a Transgenic Mouse Model of Parkinson Disease. Hum. Vaccin. Immunother. 2015, 11, 922–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallab, M.; Herrera-Vaquero, M.; Johannesson, M.; Eriksson, F.; Sigvardson, J.; Poewe, W.; Wenning, G.K.; Nordström, E.; Stefanova, N. Region-Specific Effects of Immunotherapy with Antibodies Targeting α-Synuclein in a Transgenic Model of Synucleinopathy. Front. Neurosci. 2018, 12, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masliah, E.; Rockenstein, E.; Mante, M.; Crews, L.; Spencer, B.; Adame, A.; Patrick, C.; Trejo, M.; Ubhi, K.; Rohn, T.T.; et al. Passive Immunization Reduces Behavioral and Neuropathological Deficits in an Alpha-Synuclein Transgenic Model of Lewy Body Disease. PLoS ONE 2011, 6, e19338. [Google Scholar] [CrossRef] [Green Version]
- Messer, A.; Butler, D.C. Optimizing Intracellular Antibodies (Intrabodies/Nanobodies) to Treat Neurodegenerative Disorders. Neurobiol. Dis. 2020, 134, 104619. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Weinreb, P.H.; Lansbury, P.T. The Core Alzheimer’s Peptide NAC Forms Amyloid Fibrils Which Seed and Are Seeded by β-Amyloid: Is NAC a Common Trigger or Target in Neurodegenerative Disease? Chem. Biol. 1995, 2, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Hervé, C.; Laupèze, B.; Del Giudice, G.; Didierlaurent, A.M.; Tavares Da Silva, F. The How’s and What’s of Vaccine Reactogenicity. NPJ Vaccines 2019, 4, 39. [Google Scholar] [CrossRef] [Green Version]
- Spencer, B.; Valera, E.; Rockenstein, E.; Overk, C.; Mante, M.; Adame, A.; Zago, W.; Seubert, P.; Barbour, R.; Schenk, D.; et al. Anti-α-Synuclein Immunotherapy Reduces α-Synuclein Propagation in the Axon and Degeneration in a Combined Viral Vector and Transgenic Model of Synucleinopathy. Acta Neuropathol. Commun. 2017, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Hovakimyan, A.; Zagorski, K.; Antonyan, T.; Petrushina, I.; Davtyan, H.; Chailyan, G.; Hasselmann, J.; Iba, M.; Adame, A.; et al. Efficacy and Immunogenicity of MultiTEP-Based DNA Vaccines Targeting Human α-Synuclein: Prelude for IND Enabling Studies. NPJ Vaccines 2022, 7, 1–16. [Google Scholar] [CrossRef]
- Volc, D.; Poewe, W.; Kutzelnigg, A.; Lührs, P.; Thun-Hohenstein, C.; Schneeberger, A.; Galabova, G.; Majbour, N.; Vaikath, N.; El-Agnaf, O.; et al. Safety and Immunogenicity of the α-Synuclein Active Immunotherapeutic PD01A in Patients with Parkinson’s Disease: A Randomised, Single-Blinded, Phase 1 Trial. Lancet Neurol. 2020, 19, 591–600. [Google Scholar] [CrossRef]
- Meissner, W.G.; Traon, A.P.-L.; Foubert-Samier, A.; Galabova, G.; Galitzky, M.; Kutzelnigg, A.; Laurens, B.; Lührs, P.; Medori, R.; Péran, P.; et al. A Phase 1 Randomized Trial of Specific Active α-Synuclein Immunotherapies PD01A and PD03A in Multiple System Atrophy. Mov. Disord. 2020, 35, 1957–1965. [Google Scholar] [CrossRef]
- Poewe, W.; Volc, D.; Seppi, K.; Medori, R.; Lührs, P.; Kutzelnigg, A.; Djamshidian, A.; Thun-Hohenstein, C.; Meissner, W.G.; Rascol, O.; et al. Safety and Tolerability of Active Immunotherapy Targeting α-Synuclein with PD03A in Patients with Early Parkinson’s Disease: A Randomized, Placebo-Controlled, Phase 1 Study. J. Park. Dis. 2021, 11, 1079–1089. [Google Scholar] [CrossRef]
- Yu, H.J.; Thijssen, E.; van Brummelen, E.; van der Plas, J.L.; Radanovic, I.; Moerland, M.; Hsieh, E.; Groeneveld, G.J.; Dodart, J.-C. A Randomized First-in-Human Study With UB-312, a UBITh® α-Synuclein Peptide Vaccine. Mov. Disord. 2022, 37, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Ghochikyan, A.; Petrushina, I.; Davtyan, H.; Hovakimyan, A.; Saing, T.; Davtyan, A.; Cribbs, D.H.; Agadjanyan, M.G. Immunogenicity of Epitope Vaccines Targeting Different B Cell Antigenic Determinants of Human α-Synuclein: Feasibility Study. Neurosci. Lett. 2014, 560, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.-J.; Lee, H.-J.; Rockenstein, E.; Ho, D.-H.; Park, E.-B.; Yang, N.-Y.; Desplats, P.; Masliah, E.; Lee, S.-J. Antibody-Aided Clearance of Extracellular α-Synuclein Prevents Cell-to-Cell Aggregate Transmission. J. Neurosci. 2012, 32, 13454–13469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Games, D.; Valera, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Patrick, C.; Ubhi, K.; Nuber, S.; Sacayon, P.; et al. Reducing C-Terminal-Truncated Alpha-Synuclein by Immunotherapy Attenuates Neurodegeneration and Propagation in Parkinson’s Disease-Like Models. J. Neurosci. 2014, 34, 9441–9454. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Taylor, K.I.; Anzures-Cabrera, J.; Marchesi, M.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Azulay, J.-P.; et al. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 421–432. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.J.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of Oligomeric Forms of Alpha-Synuclein Protein in Human Plasma as a Potential Biomarker for Parkinson’s Disease. FASEB J. 2006, 20, 419–425. [Google Scholar] [CrossRef]
- Tran, H.T.; Chung, C.H.-Y.; Iba, M.; Zhang, B.; Trojanowski, J.Q.; Luk, K.C.; Lee, V.M.Y. A-Synuclein Immunotherapy Blocks Uptake and Templated Propagation of Misfolded α-Synuclein and Neurodegeneration. Cell Rep. 2014, 7, 2054–2065. [Google Scholar] [CrossRef] [Green Version]
- Shahaduzzaman, M.; Nash, K.; Hudson, C.; Sharif, M.; Grimmig, B.; Lin, X.; Bai, G.; Liu, H.; Ugen, K.E.; Cao, C.; et al. Anti-Human α-Synuclein N-Terminal Peptide Antibody Protects against Dopaminergic Cell Death and Ameliorates Behavioral Deficits in an AAV-α-Synuclein Rat Model of Parkinson’s Disease. PLoS ONE 2015, 10, e0116841. [Google Scholar] [CrossRef]
- Spencer, B.; Williams, S.; Rockenstein, E.; Valera, E.; Xin, W.; Mante, M.; Florio, J.; Adame, A.; Masliah, E.; Sierks, M.R. α-Synuclein Conformational Antibodies Fused to Penetratin Are Effective in Models of Lewy Body Disease. Ann. Clin. Transl. Neurol. 2016, 3, 588–606. [Google Scholar] [CrossRef]
- Zha, J.; Liu, X.-M.; Zhu, J.; Liu, S.-Y.; Lu, S.; Xu, P.-X.; Yu, X.-L.; Liu, R.-T. A ScFv Antibody Targeting Common Oligomeric Epitope Has Potential for Treating Several Amyloidoses. Sci. Rep. 2016, 6, 36631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal Analysis Reveals High Prevalence of Epstein-Barr Virus Associated with Multiple Sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Siderowf, A.; Concha-Marambio, L.; Lafontant, D.-E.; Farris, C.M.; Ma, Y.; Urenia, P.A.; Nguyen, H.; Alcalay, R.N.; Chahine, L.M.; Foroud, T.; et al. Assessment of Heterogeneity among Participants in the Parkinson’s Progression Markers Initiative Cohort Using α-Synuclein Seed Amplification: A Cross-Sectional Study. Lancet Neurol. 2023, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Pineda-Pardo, J.A.; Gasca-Salas, C.; Fernández-Rodríguez, B.; Rodríguez-Rojas, R.; Del Álamo, M.; Obeso, I.; Hernández-Fernández, F.; Trompeta, C.; Martínez-Fernández, R.; Matarazzo, M.; et al. Striatal Blood-Brain Barrier Opening in Parkinson’s Disease Dementia: A Pilot Exploratory Study. Mov. Disord. 2022, 37, 2057–2065. [Google Scholar] [CrossRef]
- Morizane, A. Cell Therapy for Parkinson’s Disease with Induced Pluripotent Stem Cells. Inflamm. Regen. 2023, 43, 16. [Google Scholar] [CrossRef]
- Hiller, B.M.; Marmion, D.J.; Thompson, C.A.; Elliott, N.A.; Federoff, H.; Brundin, P.; Mattis, V.B.; McMahon, C.W.; Kordower, J.H. Optimizing Maturity and Dose of IPSC-Derived Dopamine Progenitor Cell Therapy for Parkinson’s Disease. NPJ Regen. Med. 2022, 7, 24. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Active Immunization | ||||
| Target | Compound Name | Type | Current Status | Results |
| C-terminus (aa 110–130); aggregates | PD01A | α-syn-mimicking peptide with adjuvant | CT phase I; Completed | safe, well-tolerated, substantial immune response in PD [391] and early MSA [392] |
| C-terminus (aa 110–130); aggregates | PD03A | α-syn-mimicking peptide with adjuvant | CT phase I; Completed | safe, well-tolerated, substantial immune response in early PD and early MSA [393] |
| C-terminus (10 aa fragment); aggregates | UB-312 | α-syn-mimicking peptide with adjuvant | CT phase I; completed (PD) CT phase II; recruiting (PD, MSA) | safe, well-tolerated, substantial immune response in healthy volunteers [394] |
| C-terminus (aa 126–140); aggregates | α-Syn126-140-P30 | α-syn-mimicking peptide with adjuvant | Preclinial | substantial immune response in mice [395] |
| C-terminus (aa 85–99, 109–126, 126–140); aggregates | PV-1950D | α-syn DNA | Preclinical | ↓ total and PK-resistant α-syn level in tg mice ↓ neuroinflammation in tg mice [390] |
| Passive Immunization | ||||
| Target | Compound Name | Type | Current Status | Results |
| C-terminus (aa 120–140); monomers and aggregates | Ab274 | Murine IgG2a | Preclinical | ↓ α-syn level in cells ↓ neuroinflammation in cells ↑ behavioral and motor function in tg mice [396] |
| C-terminus (aa 118–126); monomers | 9E4, 5C1, 5D12 | Murine IgG1 | Preclinical | ↓ α-syn level (also truncated forms) in cells ↓ neuroinflammation in cells ↑ behavioral and motor function in tg mice [397] |
| C-terminus (aa 91–99); monomers | 1H7 | |||
| C-terminus (aa 118–126); aggregates | PRX002/prasinezumab | Human IgG1 (humanized version of 9E4) | CT phase II; active, not recruiting | no efficacy over placebo and safety concerns [398] |
| C-terminus (aa 127–140); aggregates | Syn-O1 Syn-O2 Syn-O4 Syn-F1 Syn-F2 | IgG2b IgG1 IgG1 IgG1 IgG2a | Preclinical | ↓ α-syn level ↓ neuroinflammation ↑ behavioral function in tg mice [399] |
| C-terminus (aa 121–127); soluble aggregates | ABBV-0805/ BAN0805/mAb47 | Human IgG1 (humanized version of mAb47) | CT phase I; withdrawn | study withdrawn due to strategic considerations [NCT04127695] |
| N-terminus (aa 1–10); aggregates | BIIB054/cinpanemab | Human IgG1 | CT phase II; discontinued | study did not meet its primary and secondary outcome measures [NCT03318523] |
| C-terminus (aa 102–130); monomers and aggregates | MEDI1341 | Human IgG1 | CT phase I; completed (PD) CT phase II; recruiting (MSA) | data unpublished [NCT04449484] |
| C-terminus (aa 121–125); insoluble aggregates | Syn211 | Murine IgG1 | Preclinical | ↓ α-syn propagation in cells ↓ α-syn oligomer toxicity in cells ↑ motor function in PFF-inoculated mice (Syn303 only) [400] |
| N-terminus (aa 1–5); insoluble aggregates | Syn303 | |||
| N-terminus (aa 16–35); aggregates | AB1 | Goat IgG | Preclinical | ↓ α-syn levels in tg rats ↓ neuroinflammation in tg rats ↑ behavioral function in tg rats [401] |
| Unknown; oligomers | syn-D5 syn-10H | scFv | Preclinical | ↓ α-syn aggregation in cells ↓ α-syn oligomer toxicity in cells ↑ behavioral function in tg mice [402] |
| NAC region (aa 71–77); oligomers | W20 | scFv | Preclinical | ↓ neuroinflammation in tg mice ↑ cognitive and motor function in tg mice [403] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siwecka, N.; Saramowicz, K.; Galita, G.; Rozpędek-Kamińska, W.; Majsterek, I. Inhibition of Protein Aggregation and Endoplasmic Reticulum Stress as a Targeted Therapy for α-Synucleinopathy. Pharmaceutics 2023, 15, 2051. https://doi.org/10.3390/pharmaceutics15082051
Siwecka N, Saramowicz K, Galita G, Rozpędek-Kamińska W, Majsterek I. Inhibition of Protein Aggregation and Endoplasmic Reticulum Stress as a Targeted Therapy for α-Synucleinopathy. Pharmaceutics. 2023; 15(8):2051. https://doi.org/10.3390/pharmaceutics15082051
Chicago/Turabian StyleSiwecka, Natalia, Kamil Saramowicz, Grzegorz Galita, Wioletta Rozpędek-Kamińska, and Ireneusz Majsterek. 2023. "Inhibition of Protein Aggregation and Endoplasmic Reticulum Stress as a Targeted Therapy for α-Synucleinopathy" Pharmaceutics 15, no. 8: 2051. https://doi.org/10.3390/pharmaceutics15082051