

Improved Pharmacokinetic Feasibilities of Mirabegron-1,2-Ethanedisulfonic Acid, Mirabegron-1,5-Naphthalenedisulfonic Acid, and Mirabegron-L-Pyroglutamic Acid as Co-Amorphous Dispersions in Rats and Mice


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals
2.3. LC-MS/MS Analysis of MBR in Biological Samples
2.4. Plasma Protein Binding of MBR Spiked with MBR, MBR-EFA, MBR-NDA, or MBR-PG in Rats and Mice
2.5. Pharmacokinetics of MBR after Administration of MBR, MBR-EFA, MBR-NDA, or MBR-PG in Rats
2.6. Pharmacokinetics of MBR after Administration of MBR, MBR-EFA, MBR-NDA, or MBR-PG in Mice
2.7. Tissue Distribution of MBR after Administration MBR, MBR-EFA, MBR-NDA, or MBR-PG in Mice
2.8. Pharmacokinetic Parameters
2.9. Statistical Analysis
3. Results
3.1. Plasma Protein Binding of MBR in Rat or Mouse Plasma Spiked with MBR, MBR-EFA, MBR-NDA, and MBR-PG
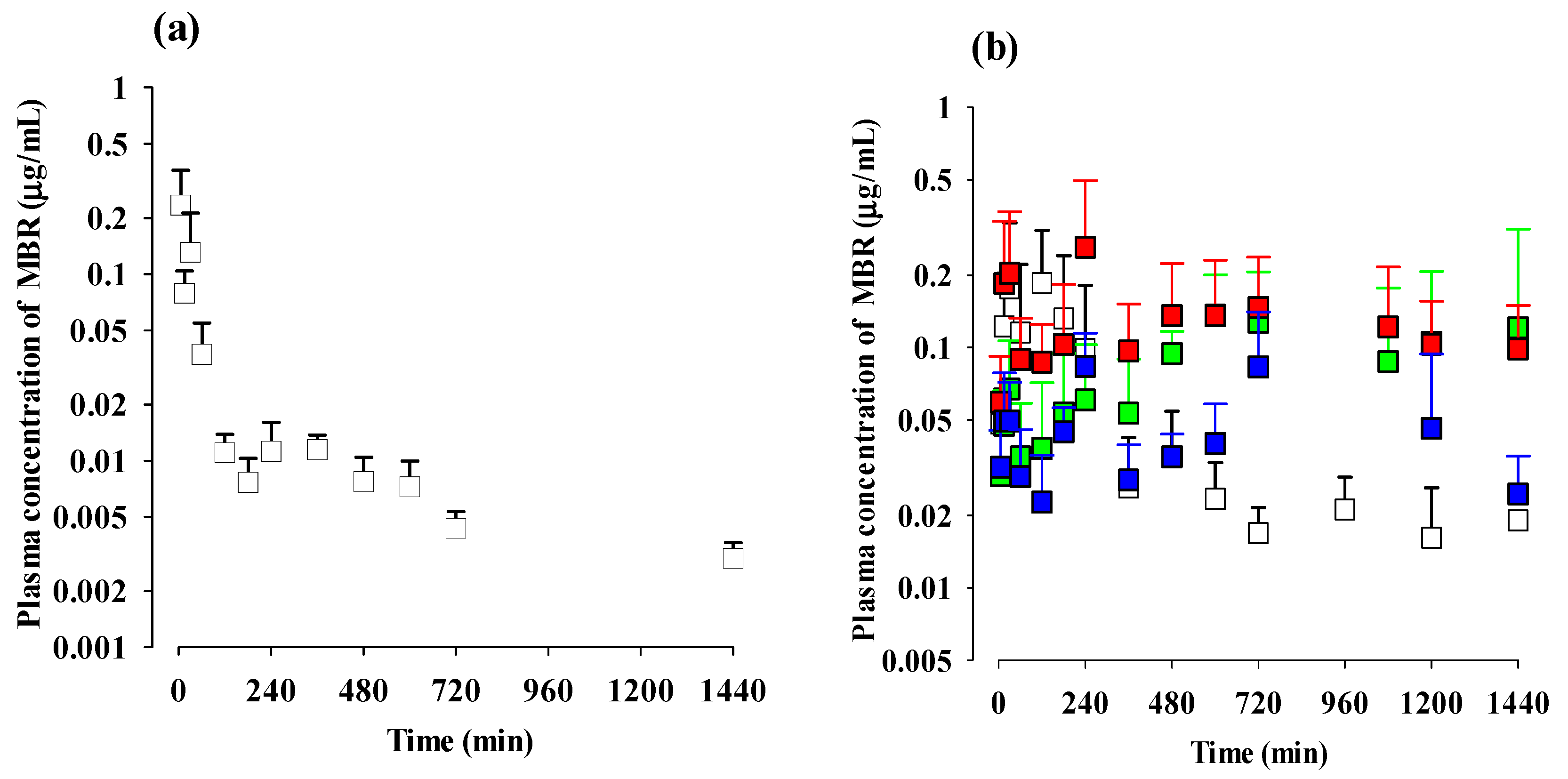
3.2. Pharmacokinetics of MBR after Administration of MBR, MBR-EFA, MBR-NDA, or MBR-PG in Rats
3.3. Pharmacokinetics of MBR after Administration of MBR, MBR-EFA, MBR-NDA, or MBR-PG in Mice
3.4. Tissue Distribution of MBR after Administration of MBR, MBR-EFA, MBR-NDA, or MBR-PG in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, H.L.; Chen, T.C.; Chang, H.M.; Juan, Y.S.; Huang, W.H.; Pan, H.F.; Chang, Y.C.; Wu, C.M.; Wang, Y.L.; Lee, H.Y. Mirabegron is alternative to antimuscarinic agents for overactive bladder without higher risk in hypertension: A systematic review and meta-analysis. World J. Urol. 2018, 36, 1285–1297. [Google Scholar] [CrossRef]
- Kelleher, C.; Hakimi, Z.; Zur, R.; Siddiqui, E.; Maman, K.; Aballéa, S.; Nazir, J.; Chapple, C. Efficacy and tolerability of mirabegron compared with antimuscarinic monotherapy or combination therapies for overactive bladder: A systematic review and network meta-analysis. Eur. Urol. 2018, 74, 324–333. [Google Scholar] [CrossRef]
- Alessio, B.; Pietro, A.; Emanuele, A. Crystalline Forms of Mirabegron Acetate Salt. EP Patent 2-857389, 4 August 2015. [Google Scholar]
- FDA Clinical Pharmacology and Biopharmaceutics Review(s). Application Number: 202611Orig1s000. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202611Orig1s000ClinPharmR.pdf (accessed on 7 March 2012).
- Patel, D.A.; Patel, D.J. Physicochemical characterization and in vitro dissolution enhancement of mirabegron using solid dispersion method. World J. Pharm. 2018, 7, 973–991. [Google Scholar]
- An, J.H.; Lim, C.; Kiyonga, A.N.; Chung, I.H.; Lee, I.K.; Mo, K.; Park, M.; Youn, W.; Choi, W.R.; Suh, Y.G.; et al. Co-amorphous screening for the solubility enhancement of poorly water-soluble mirabegron and investigation of their intermolecular interactions and dissolution behaviors. Pharmaceutics 2018, 10, 149. [Google Scholar] [CrossRef]
- Raut, P.; Gambhire, M.; Panchal, D.; Gambhire, V. Development and optimization of mirabegron solid lipid nanoparticles as an oral drug delivery for overactive bladder. Pharm. Nanotechnol. 2021, 9, 120–129. [Google Scholar] [CrossRef]
- Takusagawa, S.; van Lier, J.J.; Suzuki, K.; Nagata, M.; Meijer, J.; Krauwinkel, W.; Schaddelee, M.; Sekiguchi, M.; Miyashita, A.; Iwatsubo, T.; et al. Absorption, metabolism and excretion of [14C]mirabegron (YM178), a potent and selective β3-adrenoceptor agonist, after oral administration to healthy male volunteers. Drug Metab. Dispos. 2012, 40, 815–824. [Google Scholar] [CrossRef]
- Takusagawa, S.; Ushigome, F.; Nemoto, H.; Takahashi, Y.; Li, Q.; Kerbusch, V.; Miyashita, A.; Iwatsubo, T.; Usui, T. Intestinal absorption mechanism of mirabegron, a potent and selective β3-adrenoceptor agonist: Involvement of human efflux and/or influx transport systems. Mol. Pharm. 2013, 10, 1783–1794. [Google Scholar] [CrossRef]
- Takasu, T.; Sato, S.; Ukai, M.; Maruyama, T.; Shibasaki, M. Remedy for Overactive Bladder Comprising Acetic Acid Anilide Derivative as the Active Ingredient. U.S. Patent 8-835474, 16 September 2014. [Google Scholar]
- Hancock, B.C.; Zografi, G. Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci. 1997, 86, 1–12. [Google Scholar] [CrossRef]
- Hilfiker, R.; Raumer, M.S. Polymorphism in the Pharmaceutical Industry; Wiley-VCH: Weinheim, Germany, 2006; pp. 259–281. [Google Scholar]
- Jaakko, A.; Thomas, R. Towards physico-relevant dissolution testing: The importance of solid-state analysis in dissolution. Dissolut. Technol. 2009, 16, 47–54. [Google Scholar]
- Serajuddin, A.T.M. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 10, 1058–1066. [Google Scholar] [CrossRef]
- Tadini, M.C.; Fernandes, F.S.; Ozelin, S.D.; de Melo, M.R.S.; Mansur, A.L.; de Toledo, T.B.; de Albuquerque, N.C.P.; Tavares, D.C.; Marquele-Oliveira, F.; de Oliveira, A.R.M. Pharmacokinetic study of AmB-NP-GR: A new granule form with amphotericin B to treat leishmaniasis and fungal infections. Eur. J. Pharm. Sci. 2022, 173, 106–173. [Google Scholar] [CrossRef]
- Vanconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef]
- Veith, H.; Wiechert, F.; Luebbert, C.; Sadowski, G. Combining crystalline and polymeric excipients in API solid dispersions–opportunity or risk? Eur. J. Pharm. Biopharm. 2021, 158, 323–335. [Google Scholar] [CrossRef]
- Chiou, W.L.; Riegelman, S. Pharmaceutical applications of solid dispersion systems. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar] [CrossRef]
- Dengale, S.J.; Grohganz, H.; Rades, T.; Löbmann, K. Recent advances in co-amorphous drug formulations. Adv. Drug Deliv. Rev. 2016, 100, 116–125. [Google Scholar] [CrossRef]
- Hancock, B.C.; Shamblin, S.L.; Zografi, G. Molecular mobility of amorphous pharmaceutical solids below their glass transition temperatures. Pharm. Res. 1995, 12, 799–806. [Google Scholar] [CrossRef]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Marsac, P.J.; Shamblin, S.L.; Taylor, L.S. Theoretical and practical approaches for prediction of drug-polymer miscibility and solubility. Pharm. Res. 2006, 23, 2417–2426. [Google Scholar] [CrossRef]
- Liu, J.; Grohganz, H.; Löbmann, K.; Rades, T.; Hempel, N.-J. Co-amorphous drug formulations in numbers: Recent advances in co-amorphous drug formulations with focus on co-formability, molar ratio, preparation methods, physical stability, in vitro and in vivo performance, and new formulation strategies. Pharmaceutics 2021, 13, 389. [Google Scholar] [CrossRef]
- Chung, I.H.; Lee, I.G.; Mo, K.; Han, H.S.; Han, G.R.; Shin, J.W.; Kim, H.J.; Kim, A.R.; Jung, K.; An, J.H. Salts of Amide Derivatives and Method for Preparing the Same. KR Patent 10-2398639, 11 May 2022. [Google Scholar]
- Chen, L.; Zhang, Y. Determination of mirabegron in rat plasma by UPLC–MS/MS after oral and intravenous administration. Rev. Assoc. Med. Bras. 2019, 65, 141–148. [Google Scholar] [CrossRef]
- Han, S.Y.; Chae, H.S.; You, B.H.; Chin, Y.W.; Kim, H.; Choi, H.S.; Choi, Y.H. Lonicera japonica extract increases metformin distribution in the liver without change of systemic exposed metformin in rats. J. Ethnopharmacol. 2019, 238, 111–892. [Google Scholar] [CrossRef] [PubMed]
- You, B.H.; Chin, Y.W.; Kim, H.; Choi, H.S.; Choi, Y.H. Houttuynia cordata extract increased systemic exposure and liver concentrations of metformin through OCTs and MATEs in rats. Phytother. Res. 2018, 32, 1004–1013. [Google Scholar] [CrossRef]
- You, B.H.; Bae, M.; Han, S.Y.; Jung, J.; Jung, K.; Choi, Y.H. Pharmacokinetic feasibility of stability-enhanced solid-state (SESS) tenofovir disoproxil free base crystal. Pharmaceutics 2023, 15, 1392. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; You, B.H.; Yim, S.; Han, S.Y.; Chae, H.S.; Bae, M.; Kim, S.Y.; Yu, J.E.; Jung, J.; Nhoek, P.; et al. Change of metformin concentrations in the liver as a pharmacological target site of metformin after long-term combined treatment with ginseng berry extract. Front. Pharmacol. 2023, 14, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- You, B.H.; BasavanaGowda, M.K.; Lee, J.U.; Chin, Y.W.; Choi, W.J.; Choi, Y.H. Pharmacokinetic properties of moracin C in mice. Planta Med. 2021, 87, 642–651. [Google Scholar] [CrossRef]
- Lee, M.G.; Chiou, W.L. Evaluation of potential causes for the incomplete bioavailability of furosemide: Gastric first-pass metabolism. J. Pharmacokinet. Biopharm. 1983, 11, 623–640. [Google Scholar] [CrossRef]
- Choi, Y.H.; Bae, J.K.; Chae, H.S.; Choi, Y.O.; Nhoek, P.; Choi, J.S.; Chin, Y.W. Isoliquiritigenin ameliorates dextran sulfate sodium-induced colitis through the inhibition of MAPK pathway. Int. Immunopharmacol. 2016, 31, 223–232. [Google Scholar] [CrossRef]
- Cook, D.; Brown, D.; Alexander, R.; March, R.; Morgan, P.; Satterthwaite, G.; Pangalos, M.N. Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework. Nat. Rev. Drug Discov. 2014, 13, 419–431. [Google Scholar] [CrossRef]
- Gabrielsson, J.; Green, A.R.; Van Der Graaf, P.H. Optimising in vivo pharmacology studies–practical PKPD considerations. J. Pharmacol. Toxicol. Methods 2010, 61, 146–156. [Google Scholar] [CrossRef]
- Jia, W.; Du, F.; Liu, X.; Jiang, R.; Xu, F.; Yang, J.; Li, L.; Wang, F.; Olaleye, O.E.; Dong, J.; et al. Renal tubular secretion of tanshinol: Molecular mechanism, impact on its systemic exposure and propensity for dose-related nephrotoxicity and renal herb-drug interactions. Drug Metab. Dispos. 2015, 43, 669–678. [Google Scholar] [CrossRef]
- Lin, J.H. Tissue distribution and pharmacodynamics: A complicated relationship. Cur. Drug Metab. 2006, 7, 39–65. [Google Scholar] [CrossRef] [PubMed]
- Persky, A.M.; Brazeau, G.A.; Hochhaus, G. Pharmacokinetics of the dietary supplement creatine. Clin. Pharmacokinet. 2003, 42, 557–574. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S. Preclinical pharmacokinetics: An approach towards safer and efficacious drug. Curr. Drug Metab. 2006, 7, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Tuntland, T.; Ethell, B.; Kosaka, T.; Blasco, F.; Zang, R.X.; Jain, M.; Gould, T.; Hoffmaster, K. Implementation of pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research. Front. Pharmacol. 2014, 5, 174. [Google Scholar] [CrossRef]
- Kim, D.-S.; Choi, S.; Bae, S.H. Predicting human pharmacokinetics from preclinical data: Absorption. Transl. Clin. Pharmacol. 2020, 28, 126–135. [Google Scholar]
- An, G.; Morris, M.E. A physiologically based pharmacokinetic model of mitoxantrone in mice and scale-up to humans: A semi-mechanistic model incorporating DNA and protein binding. AAPS J. 2012, 14, 352–364. [Google Scholar] [CrossRef]
- Choi, Y.H.; Han, S.Y.; Kim, Y.J.; Kim, Y.M.; Chin, Y.W. Absorption, tissue distribution, tissue metabolism and safety of α-mangostin in mangosteen extract using mouse models. Food Chem. Toxicol. 2014, 66, 140–146. [Google Scholar] [CrossRef]
- Krauwinkel, W.; van Dijk, J.; Schaddelee, M.; Eltink, C.; Meijer, J.; Strabach, G.; van Marle, S.; Kerbusch, V.; van Gelderen, M. Pharmacokinetic properties of mirabegron, a β3-adrenoceptor agonist: Results from two phase I, randomized, multiple-dose studies in healthy young and elderly men and women. Clin. Ther. 2012, 34, 2144–2160. [Google Scholar] [CrossRef]
- Iitsuka, H.; Tokuno, T.; Amada, Y.; Matsushima, H.; Katashima, M.; Sawamoto, T.; Takusagawa, S.; van Gelderen, M.; Tanaka, T.; Miyahara, H. Pharmacokinetics of mirabegron, a β3-adrenoceptor agonist for treatment of overactive bladder, in healthy Japanese male subjects: Results from single- and multiple-dose studies. Clin. Drug Investig. 2014, 34, 27–35. [Google Scholar] [CrossRef]
- Valic, M.S.; Zheng, G. Research tools for extrapolating the disposition and pharmacokinetics of nanomaterials from preclinical animals to humans. Theranostics 2019, 9, 3365–3387. [Google Scholar] [CrossRef]
- FDA Pharmacology Review for Mirabegron. Application Number: 202611Orig1s000. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202611Orig1s000PharmR.pdf (accessed on 25 April 2012).
- Sultana, R.; Sultan, Z. Complexation of andrographolide, mirabegron and suvorexant and bioactivity study of the complexes. Microb. Bioact. 2019, 2, E076–E081. [Google Scholar] [CrossRef]
- Iitsuka, H.; van Gelderen, M.; Katashima, M.; Takusagawa, S.; Sawamoto, T. Pharmacokinetics of mirabegron, a β3-adrenoceptor agonist for treatment of overactive bladder, in healthy east asian subjects. Clin. Ther. 2015, 37, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Boyd, B.J.; Bergström, C.A.S.; Vinarov, Z.; Kuentz, M.; Brouwers, J.; Augustijns, P.; Brandl, M.; Bernkop-Schnürch, A.; Shrestha, N.; Préat, V.; et al. Successful oral delivery of poorly water-soluble drugs both depends on the intraluminal behavior of drugs and of appropriate advanced drug delivery systems. Eur. J. Pharm. Sci. 2019, 137, 1049–1067. [Google Scholar] [CrossRef] [PubMed]
- Sharaf, A.; Hashin, H. Profile of mirabegron in the treatment of overactive bladder: Place in therapy. Drug Des. Devel. Ther. 2017, 20, 463–467. [Google Scholar] [CrossRef]
- Kentaro, K.; Tsuyoshi, M.; Yasuhisa, N.; Kenji, T. Application of a physiologically based pharmacokinetic model for the prediction of mirabegron plasma concentrations in a population with severe renal impairment. Biopharm. Drug Dispos. 2019, 40, 176–187. [Google Scholar]
- EMA Assessment Report. Procedure number: EMEA/H/C/002388. Available online: https://www.ema.europa.eu/en/documents/assessment-report/betmiga-epar-public-assessment-report_en.pdf (accessed on 18 October 2012).
- FDA Highlights of Prescribing Information. MYRBETRIQTM (Mirabegron) Extended-Release Tablets, for Oral Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/202611s011lbl.pdf (accessed on 11 June 2012).
- Sacco, E.; Bientinesi, R. Mirabegron: A review of recent data and its prospects in the management of overactive bladder. Ther. Adb. Urol. 2012, 4, 315–324. [Google Scholar] [CrossRef]
- Roberts, J.; Waller, D.G.; von Renwick, A.G.; O’Shea, N.; Macklin, B.S.; Bulling, M. The effects of co-administration of benzhexol on the peripheral pharmacokinetics of oral levodopa in young volunteers. Br. J. Clin. Pharmacol. 1996, 41, 331–337. [Google Scholar] [CrossRef]
- Davies, N.M.; Takemoto, J.K.; Brocks, D.R.; Yáñez, J.A. Multiple peaking phenomena in pharmacokinetic disposition. Clin. Pharmacokinet. 2010, 49, 351–377. [Google Scholar] [CrossRef]
- Van Duijkeren, E.; Ensink, J.M.; Meijer, L.A. Distribution of orally administered trimethoprim and sulfadiazine into noninfected subcutaneous tissue chambers in adult ponies. J. Vet. Pharmacol. Ther. 2002, 25, 273–277. [Google Scholar] [CrossRef]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Reichel, A.; Lienau, P. Pharmacokinetics in drug discovery: An exposure-centred approach to optimising and predicting drug efficacy and safety. Handb. Exp. Pharmacol. 2016, 232, 235–260. [Google Scholar] [PubMed]
- Nightingale, C.H. Future in vitro and animal studies: Development of pharmacokinetic and pharmacodynamic efficacy predictors for tissue-based antibiotics. Pharmacotherapy 2005, 25, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Dehvari, N.; da Silva Junior, E.D.; Bengtsson, T.; Hutchinson, D.S. Mirabegron: Potential off target effects and uses beyond the bladder. Br. J. Pharmacol. 2018, 175, 4072–4082. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | MBR Rats (n = 14) | MBR-EFA Rats (n = 10) | MBR-NDA Rats (n = 11) | MBR-PG Rats (n = 10) |
|---|---|---|---|---|
| Body weight (g) | 199 ± 37.4 | 217 ± 50.3 | 240 ± 40.2 | 245 ± 44.1 |
| Terminal t1/2 (min) | 279 ± 143 | 240 ± 94.7 | 252 ± 98.1 | 228 ± 75.7 |
| Cmax,1 (μg/mL) | 0.491 ± 0.498 | 0.518 ± 0.224 | 0.577 ± 0.385 | 0.711 ± 0.570 |
| Tmax,1 (min) a | 15 (5–180) | 15 (5–180) | 15 (5–60) | 5 (5–15) |
| Cmax,2 (μg/mL) | 0.491 ± 0.175 | 1.00 ± 0.512 | 0.977 ± 0.255 | 1.09 ± 0.416 |
| Tmax,2 (min) a | 420 (180–720) | 240 (180–600) | 360 (240–480) | 360 (180–600) |
| AUC0–24h (μg min/mL) * | 303 ± 84.7 | 529 ± 225 | 549 ± 122 | 592 ± 203 |
| AUC0-inf (μg min/mL) * | 332 ± 90.3 | 545 ± 221 | 578 ± 123 | 607 ± 210 |
| CL/Finf (mL/min/kg) ** | 96.2 ± 24.4 | 76.3 ± 30.3 | 69.0 ± 18.2 | 55.7 ± 20.9 |
| GI24h (% of dose) * | 20.6 ± 6.88 | 8.71 ± 1.09 | 8.66 ± 2.32 | 8.62 ± 3.22 |
| Ae0–24h (% of dose) * | 16.2 ± 6.61 | 10.6 ± 5.12 | 9.13 ± 4.02 | 8.42 ± 4.95 |
| Relative F24h (%) | 146 | 143 | 195 | |
| Relative Finf (%) | 138 | 137 | 183 |
| Intravenous | Oral | ||||
|---|---|---|---|---|---|
| Parameters | MBR Mice (n = 13) | MBR Mice (n = 16) | MBR-EFA Mice (n = 15) | MBR-NDA Mice (n = 13) | MBR-PG Mice (n = 15) |
| Body weight (g) | 30.3 ± 0.651 | 31.6 ± 2.09 | 31.0 ± 1.13 | 28.7 ± 0.484 | 28.6 ± 0.625 |
| Terminal t1/2 (min) | 729 | ||||
| Cmax,1 (μg/mL) | 0.176 | 0.0672 | 0.203 | 0.0495 | |
| Tmax,1 (min) a | 30 | 30 | 30 | 30 | |
| Cmax,2 (μg/mL) | 0.0350 | 0.136 | 0.261 | 0.0835 | |
| Tmax,2 (min) a | 420 | 600 | 240 | 240 | |
| AUC0–24h (μg min/mL) | 20.5 | 62.2 | 132 | 184 | 71.6 |
| AUC0-inf (μg min/mL) | 23.7 | ||||
| CL (L/min/kg) | 0.211 | ||||
| Vss (L/kg) | 115 | ||||
| GI24h (% of dose) * | 2.32 ± 1.31 | 19.5 ± 13.1 | 1.24 ± 0.510 | 6.54 ± 1.11 | 10.9 ± 7.52 |
| Absolute F24h (%) b | 30.3 | 54.1 | 70.9 | 34.9 | |
| Relative F24h (%) | 178 | 234 | 115 | ||
| Intravenous | Oral | ||||
|---|---|---|---|---|---|
| MBR Mice (n = 15) | MBR Mice (n = 15) | MBR-EFA Mice (n = 15) | MBR-NDA Mice (n = 15) | MBR-PG Mice (n = 15) | |
| Liver * | 318 ± 91.5 | 2062 ± 420 | 4781 ± 2601 | 8025 ± 2412 | 5294 ± 1651 |
| Kidney ** | 2244 ± 585 | 9732 ± 1180 | 9272 ± 3514 | 9326 ± 1831 | |
| Small intestine | 41,729 ± 7923 | 67,826 ± 39,305 | 91,307 ± 64,023 | 84,272 ± 44,757 | |
| Large intestine | 8843 ± 7866 | 53,914 ± 30,115 | 36,058 ± 17,154 | 55,281 ± 27,169 | |
| Stomach | 11,103 ± 1785 | 23,366 ± 13,827 | 15,663 ± 6123 | 13,809 ± 8054 | |
| Lung + | 1567 ± 18.2 | 2764 ± 496 | 6657 ± 3334 | 10,749 ± 2373 | 4442 ± 425 |
| Heart | 313 ± 57.5 | 1562 ± 471 | 3504 ± 2739 | 9371 ± 7647 | 2776 ± 1038 |
| Brain | 102 ± 31.5 | 224 ± 92.9 | 435 ± 51.8 | 325 ± 73.2 | |
| Fat | 102 ± 55.6 | 71.4 ± 61.7 | 142 ± 104 | 117 ± 45.5 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-Y.; You, B.H.; Bae, M.; Han, S.Y.; Jung, K.; Choi, Y.H. Improved Pharmacokinetic Feasibilities of Mirabegron-1,2-Ethanedisulfonic Acid, Mirabegron-1,5-Naphthalenedisulfonic Acid, and Mirabegron-L-Pyroglutamic Acid as Co-Amorphous Dispersions in Rats and Mice. Pharmaceutics 2023, 15, 2277. https://doi.org/10.3390/pharmaceutics15092277
Kim S-Y, You BH, Bae M, Han SY, Jung K, Choi YH. Improved Pharmacokinetic Feasibilities of Mirabegron-1,2-Ethanedisulfonic Acid, Mirabegron-1,5-Naphthalenedisulfonic Acid, and Mirabegron-L-Pyroglutamic Acid as Co-Amorphous Dispersions in Rats and Mice. Pharmaceutics. 2023; 15(9):2277. https://doi.org/10.3390/pharmaceutics15092277
Chicago/Turabian StyleKim, Seo-Yeon, Byung Hoon You, Mingoo Bae, Seung Yon Han, Kiwon Jung, and Young Hee Choi. 2023. "Improved Pharmacokinetic Feasibilities of Mirabegron-1,2-Ethanedisulfonic Acid, Mirabegron-1,5-Naphthalenedisulfonic Acid, and Mirabegron-L-Pyroglutamic Acid as Co-Amorphous Dispersions in Rats and Mice" Pharmaceutics 15, no. 9: 2277. https://doi.org/10.3390/pharmaceutics15092277