
Advances in Pancreatic Cancer Treatment by Nano-Based Drug Delivery Systems

 , and
, and
Abstract
:1. Introduction
2. Methodology
3. Anatomophisiology of the Pancreas
- Proteolytic enzymes, whose function is the digestion of proteins. They are divided into exopeptidases (such as carboxypeptidase), which act on the chemical bonds between amino acids, from a terminal end of the protein, and into endopeptidases (such as chymotrypsin and trypsin), which degrade proteins by cleaving chemical bonds between the amino acids of the protein molecule [25,29];
- Lipolytic enzymes, or lipases, whose function is to digest lipids or fats. The lipases hydrolyze the fats, transforming them into glycerol-free fatty acids, easily assimilated by cells. In addition to this function, they also break down neutral fats or triglycerides into fatty acids and glycerin [25,26,27,28,29,30,31].
- Nucleases enzyme, that promote the digestion of nucleic acids. Ribonuclease cleaves RNA molecules in the sugar ribose and the nitrogenous bases adenine, cytosine, guanine, and uracil, while the deoxyribonuclease digests the DNA molecules in the sugar deoxyribose and the nitrogen bases cytosine, adenine, guanine, and thymine. There are enzymes of two types (α and β) that catalyze the hydrolysis of the phosphodiester bonds [25,32].
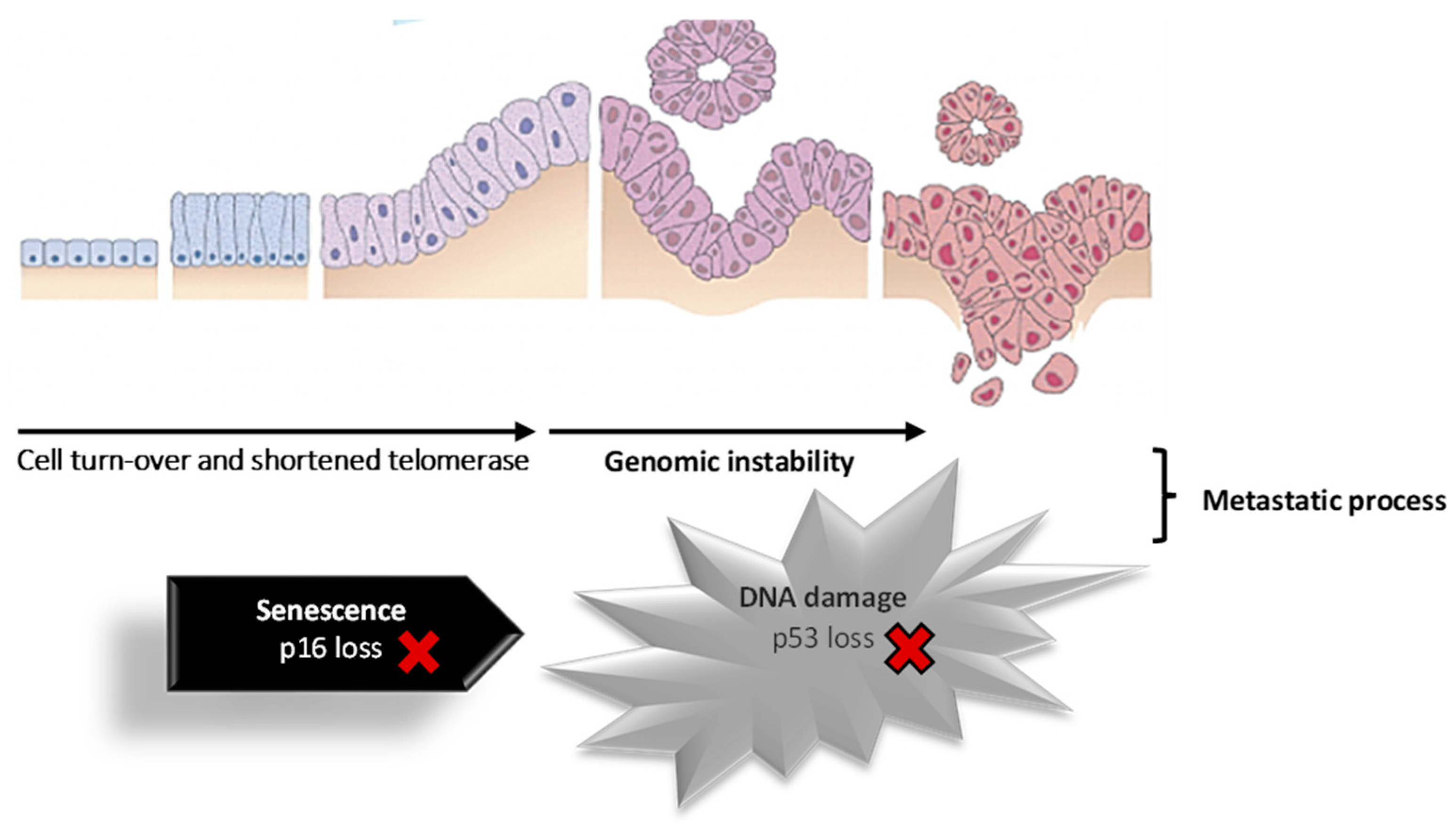
4. Pancreatic Cancer
- Functional NETs: Approximately 50% of neuroendocrine tumors produce hormones that are released into the bloodstream, leading to the onset of symptoms (for example, gastrinomas, insulinomas, glucagonomas, somatostatinomas, VIPomas—vasoactive intestinal peptides, and the PPomas—pancreatic polypeptides).
- Non-functional NETs: This type of tumor typically does not produce hormones in levels high enough to cause noticeable symptoms, which makes them more likely to develop into cancer as they remain asymptomatic for a longer period.
- Carcinoid tumors: This type of tumor does not often originate in the pancreas, as they are more commonly found in other parts of the digestive system. These tumors typically produce serotonin (5-HT) or its precursor, 5-hydroxytryptophan (5-HTP).
5. Conventional Treatment of Pancreatic Cancer
6. Advantages and Disadvantages of Nano-Based Drug Delivery Systems
7. Nano-Based Drug Delivery Systems for Pancreatic Cancer Treatment
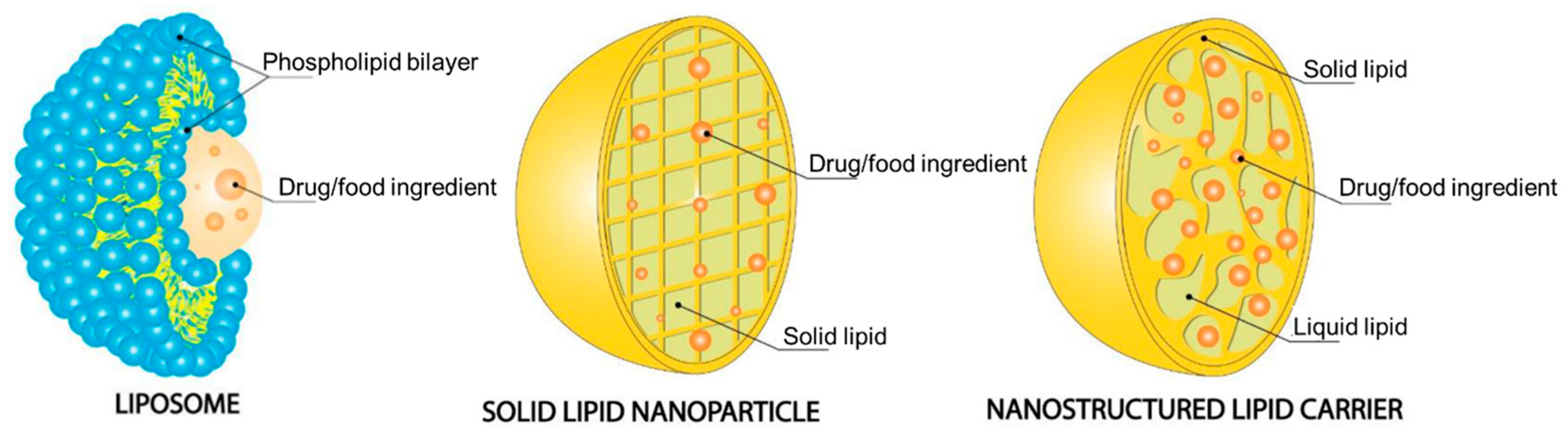
7.1. Lipid-Based Nanoparticles
7.1.1. Liposomes
7.1.2. Solid Lipid Nanoparticles
7.1.3. Nanostructured Lipid Carriers
7.2. Hybrid Nanoparticles
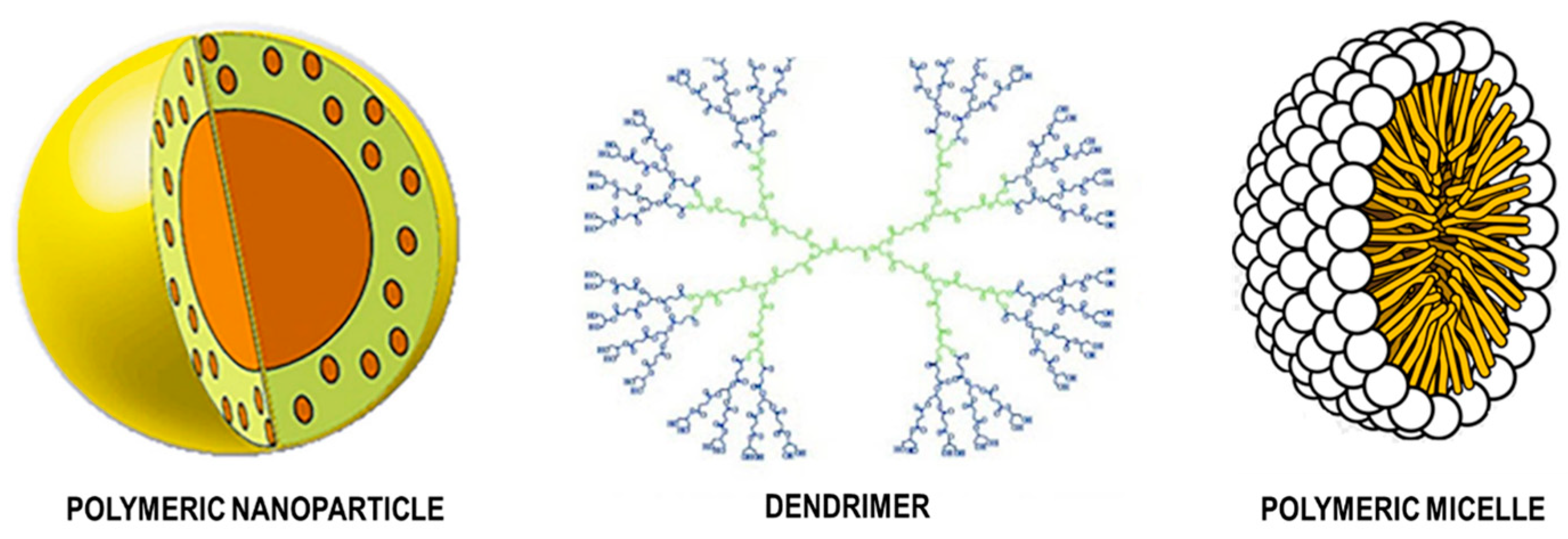
7.3. Polymer Nanoparticles
7.3.1. Natural Polymeric Nanoparticles
7.3.2. Synthetic Polymeric Nanoparticles
7.4. Inorganic Nanoparticles
8. Toxicity Concerns in Nanoparticle Delivery Systems
9. Walkthrough on Pipeline Products
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations and Acronyms
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Löhr, J.M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.L.; Heinemann, V. Addressing the Challenges of Pancreatic Cancer: Future Directions for Improving Outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Michalski, C.; Friess, H.; Buchler, M.W. Pancreatic Cancer. Pancreas 2006, 33, 111–118. [Google Scholar] [CrossRef]
- Qiu, D.; Katanoda, K.; Marugame, T.; Sobue, T. A Joinpoint Regression Analysis of Long-Term Trends in Cancer Mortality in Japan (1958–2004). Int. J. Cancer 2009, 124, 443–448. [Google Scholar] [CrossRef]
- Wang, L.; Yang, G.; Li, H.; Lu, X. The Changing Pancreatic Cancer Mortality in China (1991–2000). Zhonghua Nei Ke Za Zhi 2005, 44, 509–513. [Google Scholar]
- Malvezzi, M.; Carioli, G.; Bertuccio, P.; Rosso, T.; Bofetta, P.; Levi, F.; La Vecchia, C.; Negri, E. European Cancer Mortality Predictions for the Year 2016 with Focus on Leukemias. Ann. Oncol. Adv. Access 2016, 33, 725–731. [Google Scholar] [CrossRef]
- Mathers, C.D.; Fat, D.M.; Inoue, M.; Rao, C.; Lopez, A.D. Counting the Dead and What They Died from: An Assessment of the Global Status of Cause of Death Data. Bull. World Health Organ. 2005, 83, 171–177. [Google Scholar]
- World Health Organization. Pancreas Fact Sheet. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/13-Pancreas-fact-sheet.pdf (accessed on 2 August 2022).
- Yu, X.; Zhang, Y.; Chen, C.; Yao, Q.; Li, M. Targeted Drug Delivery in Pancreatic Cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2010, 1805, 97–104. [Google Scholar] [CrossRef]
- McCarroll, J.; Teo, J.; Boyer, C.; Goldstein, D.; Kavallaris, M.; Phillips, P.A. Potential Applications of Nanotechnology for the Diagnosis and Treatment of Pancreatic Cancer. Front. Physiol. 2014, 5, 2. [Google Scholar] [CrossRef]
- Yu, X.; Jin, C. Application of Albumin-Based Nanoparticles in the Management of Cancer. J. Mater. Sci. Mater. Med. 2016, 27, 4. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhu, W.; Di, Y.; Gu, J.; Guo, Z.; Li, H.; Fu, D.; Jin, C. Triple-Functional Albumin-Based Nanoparticles for Combined Chemotherapy and Photodynamic Therapy of Pancreatic Cancer with Lymphatic Metastases. Int. J. Nanomed. 2017, 12, 6771–6785. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Treating Pancreatic Cancer. Available online: https://www.cancer.org/cancer/types/pancreatic-cancer/treating.html (accessed on 26 July 2023).
- Liu, L.; Kshirsagar, P.G.; Gautam, S.K.; Gulati, M.; Wafa, E.I.; Christiansen, J.C.; White, B.M.; Mallapragada, S.K.; Wannemuehler, M.J.; Kumar, S.; et al. Nanocarriers for Pancreatic Cancer Imaging, Treatments, and Immunotherapies. Theranostics 2022, 12, 1030. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Yu, H. Gemcitabine Conjugated Chitosan and Double Antibodies (Abc-GC-Gemcitabine Nanoparticles) Enhanced Cytoplasmic Uptake of Gemcitabine and Inhibit Proliferation and Metastasis in Human SW1990 Pancreatic Cancer Cells. Med. Sci. Monit. 2017, 23, 1613–1620. [Google Scholar] [CrossRef]
- Gmeiner, W.H.; Ghosh, S. Nanotechnology for Cancer Treatment. Nanotechnol. Rev. 2015, 3, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Buzea, C.; Pacheco, I.I.; Robbie, K. Nanomaterials and Nanoparticles: Sources and Toxicity. Biointerphases 2007, 2, 17–71. [Google Scholar] [CrossRef]
- Gianella, A.; Jarzyna, P.A.; Mani, V.; Ramachandran, S.; Tang, J.; Kann, B.; Dijk, W.J.R.; Thijssen, V.L.; Arjan, W.; Storm, G.; et al. A Multifunctional Nanoemulsion Platform for Imaging Guided Therapy Evaluated in Experimental Cancer. Am. Chem. Soc. Nanotechnol. 2011, 5, 4422–4433. [Google Scholar] [CrossRef]
- Tanaka, T.; Decuzzi, P. Nanotechnology for Breast Cancer Therapy. Springer Sci. 2009, 11, 49–63. [Google Scholar] [CrossRef]
- Qi Yao, W.; Yan-Ming, X.; Andy, T.Y.L. Recent Progress of Nanocarrier-Based Therapy for Solid Malignancies. Cancers 2020, 12, 2783. [Google Scholar] [CrossRef]
- Santos-Rebelo, A.; Garcia, C.; Eleutério, C.; Bastos, A.; Coelho, S.C.; Coelho, M.A.N.; Molpeceres, J.; Viana, A.S.; Ascensão, L.; Pinto, J.F.; et al. Development of Parvifloron D-Loaded Smart Nanoparticles to Target Pancreatic Cancer. Pharmaceutics 2018, 10, 216. [Google Scholar] [CrossRef]
- Gali-Muhtasib, H.; Hmadi, R.; Kareh, M.; Tohme, R.; Darwiche, N. Cell Death Mechanisms of Plant-Derived Anticancer Drugs: Beyond Apoptosis. Apoptosis 2015, 20, 1531–1562. [Google Scholar] [CrossRef]
- Seeley, R.; Stephens, T.; Tate, P. Anatomia & Fisiologia, 8th ed.; Lusociência, Ed.; The McGraw-Hill: New York, NY, USA, 2011. [Google Scholar]
- Fonseca, L. Application of Nanotechnological Delivery Systems in Pancreatic Cancer Therapy; Universidade Lusófona de Humanidades e Tecnologias: Lisbon, Portugal, 2018. [Google Scholar]
- Röder, P.V.; Wu, B.; Liu, Y.; Han, W. Pancreatic Regulation of Glucose Homeostasis. Exp. Mol. Med. 2016, 48, 219. [Google Scholar] [CrossRef] [PubMed]
- Kostov, D.; Kobakov, G.; Yankov, D. Involvement of Regional Lymph Nodes in Patients with Pancreatic Head Adenocarcinoma. Surg. Chron. 2015, 20, 265–269. [Google Scholar]
- Pallagi, P.; Hegyi, P.; Rakonczay, Z. The Physiology and Pathophysiology of Pancreatic Ductal Secretion. Pancreas 2015, 44, 1211–1233. [Google Scholar] [CrossRef]
- Fieker, A.; Philpott, J.; Armand, M. Enzyme Replacement Therapy for Pancreatic Insufficiency: Present and Future. Clin. Exp. Gastroenterol. 2011, 4, 55–73. [Google Scholar] [CrossRef]
- Konkit, M.; Kim, W. Activities of Amylase, Proteinase, and Lipase Enzymes from Lactococcus Chungangensis and Its Application in Dairy Products. J. Dairy. Sci. 2016, 99, 4999–5007. [Google Scholar] [CrossRef]
- Hameed, A.M.; Lam, V.W.T.; Pleass, H.C. Significant Elevations of Serum Lipase Not Caused by Pancreatitis: A Systematic Review. Int. Hepato-Pancreato-Biliary Assoc. 2015, 17, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, S.; Libonati, M. Structure-Function Relationships in Human Ribonucleases: Main Distinctive Features of the Major RNase Types. Fed. Eur. Biochem. Soc. Lett. 1997, 404, 1–5. [Google Scholar] [CrossRef]
- Freychet, L.; Rizkalla, S.W.; Desplanque, N.; Basdevant, A.; Zirinis, P.; Tchobroutsky, G.; Slama, G. Effect of Intranasal Glucagon on Blood Glucose Levels in Healthy Subjects and Hypoglycaemic Patients with Insulin-Dependent Diabetes. Lancet 1988, 1, 1364–1366. [Google Scholar] [CrossRef]
- Göke, B. Islet Cell Function: α and β Cells—Partners towards Normoglycaemia. Int. J. Clin. Pract. 2008, 62, 2–7. [Google Scholar] [CrossRef]
- Biolo, G.; Fleming, R.Y.D.; Wolfe, R.R. Physiologic Hyperinsulinemia Stimulates Protein Synthesis and Enhances Transport of Selected Amino Acids in Human Skeletal Muscle. J. Clin. Investig. 1995, 95, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Zisman, A.; Peroni, O.D.; Abel, E.D.; Michael, M.D.; Mauvais-Jarvis, F.; Lowell, B.B.; Wojtaszewski, J.F.P.; Hirshman, M.F.; Virkamaki, A.; Goodyear, L.J.; et al. Targeted Disruption of the Glucose Transporter 4 Selectively in Muscle Causes Insulin Resistance and Glucose Intolerance. Nat. Med. 2000, 6, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Takei, M.; Ishii, H.; Sato, Y. Glucose-Stimulated Insulin Secretion: A Newer Perspective. J. Diabetes Investig. 2013, 4, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R.; Liddle, R.A. Neural and Hormonal Regulation of Pancreatic Secretion. Curr. Opin. Gastroenterol. 2009, 25, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Coleman, F.H.; Travagli, R.A. Cholecystokinin-8s Excites Identified Rat Pancreatic-Projecting Vagal Motoneurons. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G484–G492. [Google Scholar] [CrossRef]
- RA, L. Integrated Actions of Cholecystokinin on the Gastrointestinal Tract: Use of the Cholecystokinin Bioassay. Gastroenterol. Clin. N. Am. 1989, 18, 735–756. [Google Scholar]
- Reynolds, R.B.; Folloder, J. Clinical Management of Pancreatic Cancer. J. Adv. Pract. Oncol. 2014, 5, 356–364. [Google Scholar] [CrossRef]
- Thomasset, S.C.; Lobo, D.N. Pancreatic Cancer. Surgery 2010, 28, 198–204. [Google Scholar] [CrossRef]
- Moran, A.; O’Hara, C.; Khan, S.; Shack, L.; Woodward, E.; Maher, E.R.; Lalloo, F.; Evans, D.G.R. Risk of Cancer Other than Breast or Ovarian in Individuals with BRCA1 and BRCA2 Mutations. Fam. Cancer 2012, 11, 235–242. [Google Scholar] [CrossRef]
- Lochan, R.; Reeves, H.L.; Daly, A.K.; Charnley, R.M. The Role of Tobacco-Derived Carcinogens in Pancreas Cancer. ISRN Oncol. 2011, 2011, 249235. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer Incidence and Mortality Worldwide: Sources, Methods and Major Patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Hruban, R.H. Pathology and Molecular Genetics of Pancreatic Neoplasms. Cancer J. 2012, 18, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Rishi, A.; Goggins, M.; Wood, L.D.; Hruban, H.; Sol, T.; Pancreatic, G.; Sol, T.; Pancreatic, G.; Hopkins, J. Pathological and Molecular Evaluation of Pancreatic Neoplasms. Semin. Oncol. 2016, 42, 28–39. [Google Scholar] [CrossRef]
- Jiao, Y.; Yonescu, R.; Offerhaus, G.; Al, E. Whole Exome Sequencing of Pancreatic Neoplasms with Acinar Differentiation. J. Pathol. 2014, 232, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Goral, V. Pancreatic Cancer: Pathogenesis and Diagnosis. Asian Pac. J. Cancer Prev. 2015, 16, 5619–5624. [Google Scholar] [CrossRef] [PubMed]
- Gnoni, A.; Licchetta, A.; Scarpa, A.; Azzariti, A.; Brunetti, A.E.; Simone, G.; Nardulli, P.; Santini, D.; Aieta, M.; Delcuratolo, S.; et al. Carcinogenesis of Pancreatic Adenocarcinoma: Precursor Lesions. Int. J. Mol. Sci. 2013, 14, 19731–19762. [Google Scholar] [CrossRef] [PubMed]
- Iacobuzio-Donahue, C.A.; Velculescu, V.E.; Wolfgang, C.L.; Hruban, R.H. Genetic Basis of Pancreas Cancer Development and Progression: Insights from Whole-Exome and Whole-Genome Sequencing. Clin. Cancer Res. 2012, 18, 4257–4265. [Google Scholar] [CrossRef]
- Chakraborty, S.; Baine, M.J.; Sasson, A.R.; Surinder, K. Current Status of Molecular Markers for Early Detection of Sporadic Pancreatic Cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2011, 1815, 44–64. [Google Scholar] [CrossRef]
- Chu, L.C.; Singhi, A.D.; Hruban, R.H.; Fishman, E.K. Characterization of Pancreatic Serous Cystadenoma on Dual- Phase Multidetector Computed Tomography Linda. J. Comput. Assist. Tomogr. 2014, 38, 258–263. [Google Scholar] [CrossRef]
- Farrell, J.J. Prevalence, Diagnosis and Management of Pancreatic Cystic Neoplasms: Current Status and Future Directions. Gut Liver 2015, 9, 571–589. [Google Scholar] [CrossRef]
- Yamada, N.; Okuse, C.; Nomoto, M.; Orita, M.; Katakura, Y.; Ishii, T.; Shinmyo, T.; Osada, H.; Maeda, I.; Yotsuyanagi, H.; et al. Obstructive Jaundice Caused by Secondary Pancreatic Tumor from Malignant Solitary Fibrous Tumor of Pleura: A Case Report. World J. Gastroenterol. 2006, 12, 4922–4926. [Google Scholar] [CrossRef] [PubMed]
- Karoumpalis, I.; Christodoulou, D.K. Cystic Lesions of the Pancreas. Ann. Gastroenterol. 2016, 2, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, C.; Herman, J.; Laheru, D.; Klein, A.; Erdek, M.A.; Fishman, E.K.; Hruban, R.H. Recent Progress in Pancreatic Cancer. CA Cancer J. Clin. 2014, 63, 318–348. [Google Scholar] [CrossRef] [PubMed]
- Ro, C.; Chai, W.; Yu, V.E.; Yu, R. Pancreatic Neuroendocrine Tumors: Biology, Diagnosis, and Treatment. Chin. J. Cancer 2013, 32, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Vinik, A.I.; Raymond, E. Pancreatic Neuroendocrine Tumors: Approach to Treatment with Focus on Sunitinib. Ther. Adv. Gastroenterol. 2013, 6, 396–411. [Google Scholar] [CrossRef]
- Huang, C.; Xie, K. Crosstalk of Sp1 and Stat3 Signaling in Pancreatic Cancer Pathogenesis. Cytokine Growth Factor. Rev. 2012, 23, 25–35. [Google Scholar] [CrossRef]
- Iovanna, J.; Mallmann, M.C.; Gonçalves, A.; Turrini, O.; Dagorn, J.-C. Current Knowledge on Pancreatic Cancer. Front. Oncol. 2012, 2, 6. [Google Scholar] [CrossRef]
- Sarkar, F.; Banerjee, S.; Li, Y. Pancreatic Cancer: Pathogenesis, Prevention and Treatment. Toxicol. Appl. Pharmacol. 2007, 224, 326–336. [Google Scholar] [CrossRef]
- Jamieson, N.B.; Morran, D.C.; Morton, J.P.; Ali, A.; Dickson, E.J.; Carter, C.R.; Sansom, O.J.; Evans, T.R.J.; McKay, C.J.; Oien, K.A. MicroRNA Molecular Profiles Associated with Diagnosis, Clinicopathologic Criteria, and Overall Survival in Patients with Resectable Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2012, 18, 534–545. [Google Scholar] [CrossRef]
- Hong, S.-M.; Park, J.Y.; Hruban, R.H.; Goggins, M. Molecular Signatures of Pancreatic Cancer. Arch. Pathol. Lab. Med. 2011, 135, 716–727. [Google Scholar] [CrossRef]
- Lee, E.S.; Lee, J.M. Imaging Diagnosis of Pancreatic Cancer: A State-of-the-Art Review. World J. Gastroenterol. 2014, 20, 7864–7877. [Google Scholar] [CrossRef] [PubMed]
- Mikata, R.; Ishihara, T.; Tada, M.; Tawada, K.; Saito, M.; Kurosawa, J.; Yokosuka, O. Clinical Usefulness of Repeated Pancreatic Juice Cytology via Endoscopic Naso-Pancreatic Drainage Tube in Patients with Pancreatic Cancer. J. Gastroenterol. 2013, 48, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Grapa, C.M.; Mocan, L.; Crisan, D.; Florea, M.; Mocan, T. Biomarkers in Pancreatic Cancer as Analytic Targets for Nanomediated Imaging and Therapy. Materials 2021, 14, 3083. [Google Scholar] [CrossRef]
- Cristina, J.-L.; Carolina, T.; Raul, O.; Carmelo, D.; Joaquina, M.-G.; Consolacion, M.; Jose, C.P.; Octavio, C. Proteomic Biomarkers in Body Fluids Associated with Pancreatic Cancer. Oncotarget 2018, 9, 16573–16587. [Google Scholar] [CrossRef]
- Qiao, X.W.; Qiu, L.; Chen, Y.J.; Meng, C.T.; Sun, Z.; Bai, C.M.; Zhao, D.C.; Zhang, T.P.; Zhao, Y.P.; Song, Y.L.; et al. Chromogranin A Is a Reliable Serum Diagnostic Biomarker for Pancreatic Neuroendocrine Tumors but Not for Insulinomas. BMC Endocr. Disord. 2014, 14, 64. [Google Scholar] [CrossRef]
- Li, J.; Wientjes, M.G.; Au, J.L.-S. Pancreatic Cancer: Pathobiology, Treatment Options, and Drug Delivery. AAPS J. 2010, 12, 223–232. [Google Scholar] [CrossRef]
- Burris, H., III; Moore, M.; Andersen, J.; Green, J.; Rothenberg, M.; Modiano, M. Improvements in Survival and Clinical Benefit with Gemcitabine as First-Line Therapy for Patients with Advanced Pancreas Cancer: A Randomized Trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Au, M.; Emeto, T.I.; Power, J.; Vangaveti, V.N. Emerging Therapeutic Potential of Nanoparticles in Pancreatic Cancer: A Systematic Review of Clinical Trials. Biomedicines 2016, 4, 20. [Google Scholar] [CrossRef]
- Louvet, C.; Labianca, R.; Hammel, P.; Lledo, G.; Zampino, M.; Andre, T.; Zaniboni, A.; Ducreux, M.; Aitini, E.; Taïeb, J.; et al. Gemcitabine in Combination with Oxaliplatin Compared with Gemcitabine Alone in Locally Advanced or Metastatic Pancreatic Cancer: Results of a GERCOR and GISCAD Phase III Trial. J. Clin. Oncol. Oncol. 2005, 23, 3509–3516. [Google Scholar] [CrossRef]
- Moore, M.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.; Gallinger, S.A.E. Erlotinib plus Gemcitabine Compared with Gemcitabine Alone in Patients with Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Eltawil, K.M.; Renfrew, P.D.; Walsh, M.J.; Molinari, M. Advances in Diagnosis, Treatment and Palliation of Pancreatic Carcinoma: 1990–2010. World J. Gastroenterol. 2011, 17, 867–897. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, E.G.; Coveler, A.L. Pancreatic Cancer: Optimizing Treatment Options, New, and Emerging Targeted Therapies. Drug Des. Dev. Ther. 2015, 9, 3529–3545. [Google Scholar] [CrossRef] [PubMed]
- Yue, Q.; Gao, G.; Zou, G.; Yu, H.; Zheng, X. Natural Products as Adjunctive Treatment for Pancreatic Cancer: Recent Trends and Advancements. Biomed Res. Int. 2017, 2017, 8412508. [Google Scholar] [CrossRef]
- Weckbecker, G.; Raulf, F.; Stolz, B.; Bruns, C. Somatostatin Analogs for Diagnosis and Treatment of Cancer. Pharmacol. Ther. 1994, 60, 245–264. [Google Scholar] [CrossRef]
- Szende, B.; Zalatnai, A.; Schally, A.V. Programmed Cell Death (Apoptosis) in Pancreatic Cancers of Hamsters after Treatment with Analogs of Both Luteinizing Hormone-Releasing Hormone and Somatostatin. Proc. Natl. Acad. Sci. USA 1989, 86, 1643–1647. [Google Scholar] [CrossRef]
- Keskin, O.; Yalcin, S. A Review of the Use of Somatostatin Analogs in Oncology. Onco Targets Ther. 2013, 6, 471–483. [Google Scholar] [CrossRef]
- Ebert, M.; Friess, H.; Beger, H.G.; Buchler, M.W. Role of Octreotide in the Treatment of Pancreatic Cancer. Digestion 1994, 55 (Suppl. S1), 48–51. [Google Scholar] [CrossRef]
- Rosenberg, L.; Barkun, A.N.; Denis, M.H.; Pollak, M. Low Dose Octreotide and Tamoxifen in the Treatment of Adenocarcinoma of the Pancreas. Cancer 1995, 75, 23–28. [Google Scholar] [CrossRef]
- Benz, C.; Hollander, C.; Miller, B. Endocrine-Responsive Pancreatic Carcinoma: Steroid Binding and Cytotoxicity Studies in Human Tumor Cell Lines. Cancer Res. 1986, 46, 2276–2281. [Google Scholar]
- Zaniboni, A.; Meriggi, F.; Arcangeli, G.; Alghisi, A.; Huscher, C.; Marini, G. Leuprolide and Tamoxifen in the Treatment of Pancreatic Cancer: Phase II Study. Eur. J. Cancer 1994, 30A, 128. [Google Scholar] [CrossRef]
- Hays, J.B.; Brent, E. Inhibition of Growth of Pancreatic Carcinomas in Animal Models by Analogs of Hypothalamic Hormones. Proc. Natl. Acad. Sci. USA 1984, 81, 423–425. [Google Scholar]
- Szepeshazi, K.; Schally, R.V.; Cai, R.Z.; Radulovic, S.; Milovanovic, S.; Szoke, B. Inhibitory Effect of Bombesin/Gastrin-Releasing Peptide Antagonist RC-3095 and High Dose of Somatostatin Analogue RC-160 on Nitrosamine-Induced Pancreatic Cancers in Hamsters. Cancer Res. 1991, 51, 5980–5986. [Google Scholar] [PubMed]
- Kotteas, E.; Saif, M.W.; Syrigos, K. Immunotherapy for Pancreatic Cancer. J. Cancer Res. Clin. Oncol. 2016, 142, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting Focal Adhesion Kinase Renders Pancreatic Cancers Responsive to Checkpoint Immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Zhang, Y.; Velez-Delgado, A.; Mathew, E.; Li, D.; Mendez, F.M.; Flannagan, K.; Rhim, A.D.; Simeone, D.M.; Beatty, G.L.; Di Magliano, M.P. Myeloid Cells Are Required for PD-1/PD-L1 Checkpoint Activation and the Establishment of an Immunosuppressive Environment in Pancreatic Cancer. Gut 2017, 66, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Manzur, A.; Oluwasanmi, A.; Moss, D.; Curtis, A.; Hoskins, C. Nanotechnologies in Pancreatic Cancer Therapy. Pharmaceutics 2017, 9, 39. [Google Scholar] [CrossRef]
- Jain, K.K. Advances in the Field of Nanooncology. BMC Med. 2010, 8, 83. [Google Scholar] [CrossRef]
- Wolinsky, J.B.; Colson, Y.L.; Grinstaff, M.W. Local Drug Delivery Strategies for Cancer Treatment: Gels, Nanoparticles, Polymeric Films, Rods, and Wafers. J. Control. Release 2012, 159, 14–26. [Google Scholar] [CrossRef]
- Grapa, C.M.; Mocan, T.; Gonciar, D.; Zdrehus, C.; Mosteanu, O.; Pop, T.; Mocan, L. Epidermal Growth Factor Receptor and Its Role in Pancreatic Cancer Treatment Mediated by Nanoparticles. Int. J. Nanomed. 2019, 14, 9693. [Google Scholar] [CrossRef]
- Li, Y.J.; Wu, J.Y.; Wang, J.M.; Xiang, D.X. Emerging Nanomedicine-Based Strategies for Preventing Metastasis of Pancreatic Cancer. J. Control. Release 2020, 320, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Xiangsheng, L.; Jinhong, J.; Huan, M. Transcytosis—An Effective Targeting Strategy That Is Complementary to “EPR Effect” for Pancreatic Cancer Nano Drug Delivery. Theranostics 2019, 9, 8018–8025. [Google Scholar] [CrossRef]
- Hoskins, C.; Malekigorji, M.; Curtis, A.D.M.; Hoskins, C. The Use of Iron Oxide Nanoparticles for Pancreatic Cancer Therapy. J. Nanomed. Res. 2014, 1, 4. [Google Scholar] [CrossRef]
- Heinz, H.; Pramanik, C.; Heinz, O.; Ding, Y.; Mishra, R.K.; Marchon, D.; Flatt, R.J.; Estrela-lopis, I.; Llop, J.; Moya, S.; et al. Surface Science Reports Nanoparticle Decoration with Surfactants: Molecular Interactions, Assembly, and Applications. Surf. Sci. Rep. 2017, 72, 1–58. [Google Scholar] [CrossRef]
- Laurent, S.; Mahmoudi, M. Superparamagnetic Iron Oxide Nanoparticles: Promises for Diagnosis and Treatment of Cancer. Int. J. Mol. Epidemiol. Genet. 2011, 2, 367–390. [Google Scholar] [PubMed]
- Ma, P.; Mumper, R.J. Paclitaxel Nano-Delivery Systems: A Comprehensive Review. J. Nanomed. Nanotechnol. 2013, 4, 1000164. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, N.; Kennedy, A.M.; Shea, J.E.; Scaife, C.L.; Nam, K.H. Ultrasonic Nanotherapy of Pancreatic Cancer: Lessons from Ultrasound Imaging. Mol. Pharm. 2010, 7, 22. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Frese, K.K.; Neesse, A.; Cook, N.; Bapiro, T.E.; Lolkema, M.P.; Jodrell, D.I.; Tuveson, D.A. Nab-Paclitaxel Potentiates Gemcitabine Activity by Reducing Cytidine Deaminase Levels in a Mouse Model of Pancreatic Cancer. Cancer Discov. 2012, 2, 260–269. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ramanathan, R.K.; Borad, M.J.; Laheru, D.A.; Smith, L.S.; Wood, T.E.; Korn, R.L.; Desai, N.; Trieu, V.; Iglesias, J.L.; et al. Gemcitabine plus Nab-Paclitaxel Is an Active Regimen in Patients with Advanced Pancreatic Cancer: A Phase I/II Trial. J. Clin. Oncol. 2011, 29, 4548–4554. [Google Scholar] [CrossRef]
- Galanis, E.; Carlson, S.K.; Foster, N.R.; Lowe, V.; Quevedo, F.; McWilliams, R.R.; Grothey, A.; Jatoi, A.; Alberts, S.R.; Rubin, J. Phase I Trial of a Pathotropic Retroviral Vector Expressing a Cytocidal Cyclin G1 Construct (Rexin-G) in Patients with Advanced Pancreatic Cancer. Mol. Ther. 2008, 16, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Rana, T.M. Illuminating the Silence: Understanding the Structure and Function of Small RNAs. Nat. Rev. Mol. Cell Biol. 2007, 8, 23–36. [Google Scholar] [CrossRef]
- Eriksson, M.; Taskinen, M.; Leppä, S. Mitogen Activated Protein Kinase-Dependent Activation of c-Jun and c-Fos Is Required for Neuronal Differentiation but Not for Growth and Stress Response in PC12 Cells. J. Cell Physiol. 2006, 207, 12–22. [Google Scholar] [CrossRef]
- Baigude, H.; Rana, T.M. Delivery of Therapeutic RNAi by Nanovehicles. ChemBioChem 2009, 10, 2449–2454. [Google Scholar] [CrossRef]
- Zimmermann, T.S.; Lee, A.C.H.; Akinc, A.; Bramlage, B.; Bumcrot, D.; Fedoruk, M.N.; Harborth, J.; Heyes, J.A.; Jeffs, L.B.; John, M.; et al. RNAi-Mediated Gene Silencing in Non-Human Primates. Nature 2006, 441, 111–114. [Google Scholar] [CrossRef]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.J.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D. Evidence of RNAi in Humans from Systemically Administered SiRNA via Targeted Nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.; Levins, C.G.; Cortez, C.; Langer, R.; Anderson, D.G. Lipid-Based Nanotherapeutics for SiRNA Delivery. J. Int. Med. 2010, 267, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Jindal, A.; Sarkar, S.; Alam, A. Nanomaterials-Mediated Immunomodulation for Cancer Therapeutics. Front. Chem. 2021, 9, 629635. [Google Scholar] [CrossRef]
- Pirollo, K.F.; Rait, A.; Zhou, Q.; Sung, H.H.; Dagata, J.A.; Zon, G.; Hogrefe, R.I.; Palchik, G.; Chang, E.H. Materializing the Potential of Small Interfering RNA via a Tumor-Targeting Nanodelivery System. Cancer Res. 2007, 67, 2938–2943. [Google Scholar] [CrossRef]
- Pavan, P.A.; Rachael, M.C.; Sara, S.H.; Scott, E.M. Nanomedicine Strategies to Overcome the Pathophysiological Barriers of Pancreatic Cancer. Nat. Rev. Clin. Oncol. 2016, 13, 750–765. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Li, C.-P.; Bodoky, G.; Dean, A.; Shan, Y.-S.; Jameson, G.; Macarulla, T.; Lee, K.-H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal Irinotecan with Fluorouracil and Folinic Acid in Metastatic Pancreatic Cancer after Previous Gemcitabine-Based Therapy (NAPOLI-1): A Global, Randomised, Open-Label, Phase 3 Trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Ashish, V.K.; Jaeyeon, K.; Stephan, G.K.; Nancy, P.; Jason, C.; Daryl, C.D.; Ulrik, B.N.; Jonathan, B.F. Preclinical Activity of Nanoliposomal Irinotecan Is Governed by Tumor Deposition and Intratumor Prodrug Conversion. Cancer Res. 2014, 74, 7003–7013. [Google Scholar] [CrossRef]
- Matsumoto, T.; Komori, T.; Yoshino, Y.; Ioroi, T.; Kitahashi, T.; Kitahara, H.; Ono, K.; Higuchi, T.; Sakabe, M.; Kori, H.; et al. A Liposomal Gemcitabine, FF-10832, Improves Plasma Stability, Tumor Targeting, and Antitumor Efficacy of Gemcitabine in Pancreatic Cancer Xenograft Models. Pharm. Res. 2021, 38, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-J.; Liang, T.-L.; Chang, Y.-Y.; Liu, D.-Z.; Fan, J.-Y.; Roffler, S.R.; Lin, S.-Y. Development of Irinotecan Liposome Armed with Dual-Target Anti-Epidermal Growth Factor Receptor and Anti-Fibroblast Activation Protein-Specific Antibody for Pancreatic Cancer Treatment. Pharmaceutics 2022, 14, 1202. [Google Scholar] [CrossRef] [PubMed]
- Affram, K.O.; Smith, T.; Ofori, E.; Krishnan, S.; Underwood, P.; Trevino, J.G.; Agyare, E. Cytotoxic Effects of Gemcitabine-Loaded Solid Lipid Nanoparticles in Pancreatic Cancer Cells. J. Drug Deliv. Sci. Technol. 2020, 55, 101374. [Google Scholar] [CrossRef] [PubMed]
- Sutaria, D.; Grandhi, B.K.; Thakkar, A.; Wang, J.; Prabhu, S. Chemoprevention of Pancreatic Cancer Using Solid-Lipid Nanoparticulate Delivery of a Novel Aspirin, Curcumin and Sulforaphane Drug Combination Regimen. Int. J. Oncol. 2012, 41, 2260–2268. [Google Scholar] [CrossRef]
- Pramanik, D.; Campbell, N.R.; Karikari, C.; Chivukula, R.; Kent, O.A.; Mendell, J.T.; Maitra, A. Restitution of Tumor Suppressor MicroRNAs Using a Systemic Nanovector Inhibits Pancreatic Cancer Growth in Mice. Mol. Cancer Ther. 2011, 10, 1470–1480. [Google Scholar] [CrossRef]
- Lu, Z.; Su, J.; Li, Z.; Zhan, Y.; Ye, D. Hyaluronic Acid-Coated, Prodrug-Based Nanostructured Lipid Carriers for Enhanced Pancreatic Cancer Therapy. Drug Dev. Ind. Pharm. 2016, 43, 160–170. [Google Scholar] [CrossRef]
- Zhao, X.; Li, F.; Li, Y.; Wang, H.; Ren, H.; Chen, J.; Nie, G.; Hao, J. Co-Delivery of HIF1α SiRNA and Gemcitabine via Biocompatible Lipid-Polymer Hybrid Nanoparticles for Effective Treatment of Pancreatic Cancer. Biomaterials 2015, 46, 13–25. [Google Scholar] [CrossRef]
- Yu, Q.; Qiu, Y.; Li, J.; Tang, X.; Wang, X.; Cun, X.; Xu, S.; Liu, Y.; Li, M.; Zhang, Z.; et al. Targeting Cancer-Associated Fibroblasts by Dual-Responsive Lipid-Albumin Nanoparticles to Enhance Drug Perfusion for Pancreatic Tumor Therapy. J. Control. Release 2020, 321, 564–575. [Google Scholar] [CrossRef]
- Singh, A.; Xu, J.; Mattheolabakis, G.; Amiji, M. EGFR-Targeted Gelatin Nanoparticles for Systemic Administration of Gemcitabine in an Orthotopic Pancreatic Cancer Model. Nanomedicine 2016, 12, 589–600. [Google Scholar] [CrossRef]
- David, K.I.; Jaidev, L.R.; Sethuraman, S.; Krishnan, U.M. Dual Drug Loaded Chitosan Nanoparticles-Sugar-Coated Arsenal against Pancreatic Cancer. Colloids Surf. B Biointerfaces 2015, 135, 689–698. [Google Scholar] [CrossRef]
- Fan, F.; Jin, L.; Yang, L. PH-Sensitive Nanoparticles Composed Solely of Membrane-Disruptive Macromolecules for Treating Pancreatic Cancer. ACS Appl. Mater. Interfaces 2021, 13, 12824–12835. [Google Scholar] [CrossRef]
- Xin, X.; Lin, F.; Wang, Q.; Yin, L.; Mahato, R.I. ROS-Responsive Polymeric Micelles for Triggered Simultaneous Delivery of PLK1 Inhibitor/MiR-34a and Effective Synergistic Therapy in Pancreatic Cancer. ACS Appl. Mater. Interfaces 2019, 11, 14647–14659. [Google Scholar] [CrossRef]
- Wang, G.; Zhou, Z.; Zhao, Z.; Li, Q.; Wu, Y.; Yan, S.; Shen, Y.; Huang, P. Enzyme-Triggered Transcytosis of Dendrimer-Drug Conjugate for Deep Penetration into Pancreatic Tumors. ACS Nano 2020, 14, 4890–4904. [Google Scholar] [CrossRef]
- Tong, Q.-S.; Miao, W.-M.; Huang, H.; Luo, J.-Q.; Liu, R.; Huang, Y.-C.; Zhao, D.-K.; Shen, S.; Du, J.-Z.; Wang, J. A Tumor-Penetrating Nanomedicine Improves the Chemoimmunotherapy of Pancreatic Cancer. Small 2021, 17, 2101208. [Google Scholar] [CrossRef]
- Mousa, D.S.; El-Far, A.H.; Saddiq, A.A.; Sudha, T.; Mousa, S.A. Nanoformulated Bioactive Compounds Derived from Different Natural Products Combat Pancreatic Cancer Cell Proliferation. Int. J. Nanomed. 2020, 15, 2259. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.Y.; Ryu, H.J.; Lee, S.H.; Kim, D.Y.; Kim, M.J.; Lee, E.J.; Ryu, Y.M.; Kim, S.Y.; Kim, K.P.; Choi, E.Y.; et al. SiRNA Nanoparticle Targeting PD-L1 Activates Tumor Immunity and Abrogates Pancreatic Cancer Growth in Humanized Preclinical Model. Cells 2021, 10, 2734. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Setua, S.; Kumari, S.; Dan, N.; Massey, A.; Hafeez, B.B.; Yallapu, M.M.; Stiles, Z.E.; Alabkaa, A.; Yue, J.; et al. Superparamagnetic Iron Oxide Nanoparticles of Curcumin Enhance Gemcitabine Therapeutic Response in Pancreatic Cancer. Biomaterials 2019, 208, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Albukhaty, S.; Al-Musawi, S.; Mahdi, S.A.; Sulaiman, G.M.; Alwahibi, M.S.; Dewir, Y.H.; Soliman, D.A.; Rizwana, H. Investigation of Dextran-Coated Superparamagnetic Nanoparticles for Targeted Vinblastine Controlled Release, Delivery, Apoptosis Induction, and Gene Expression in Pancreatic Cancer Cells. Molecules 2020, 25, 4721. [Google Scholar] [CrossRef] [PubMed]
- Tarannum, M.; Hossain, A.; Holmes, B.; Yan, S.; Mukherjee, P.; Vivero-Escoto, J.L.; Vivero-Escoto, J.L.; Hossain, M.A.; Yan, S.; Mukherjee, P.; et al. Advanced Nanoengineering Approach for Target-Specific, Spatiotemporal, and Ratiometric Delivery of Gemcitabine—Cisplatin Combination for Improved Therapeutic Outcome in Pancreatic Cancer. Small 2022, 18, 2104449. [Google Scholar] [CrossRef] [PubMed]
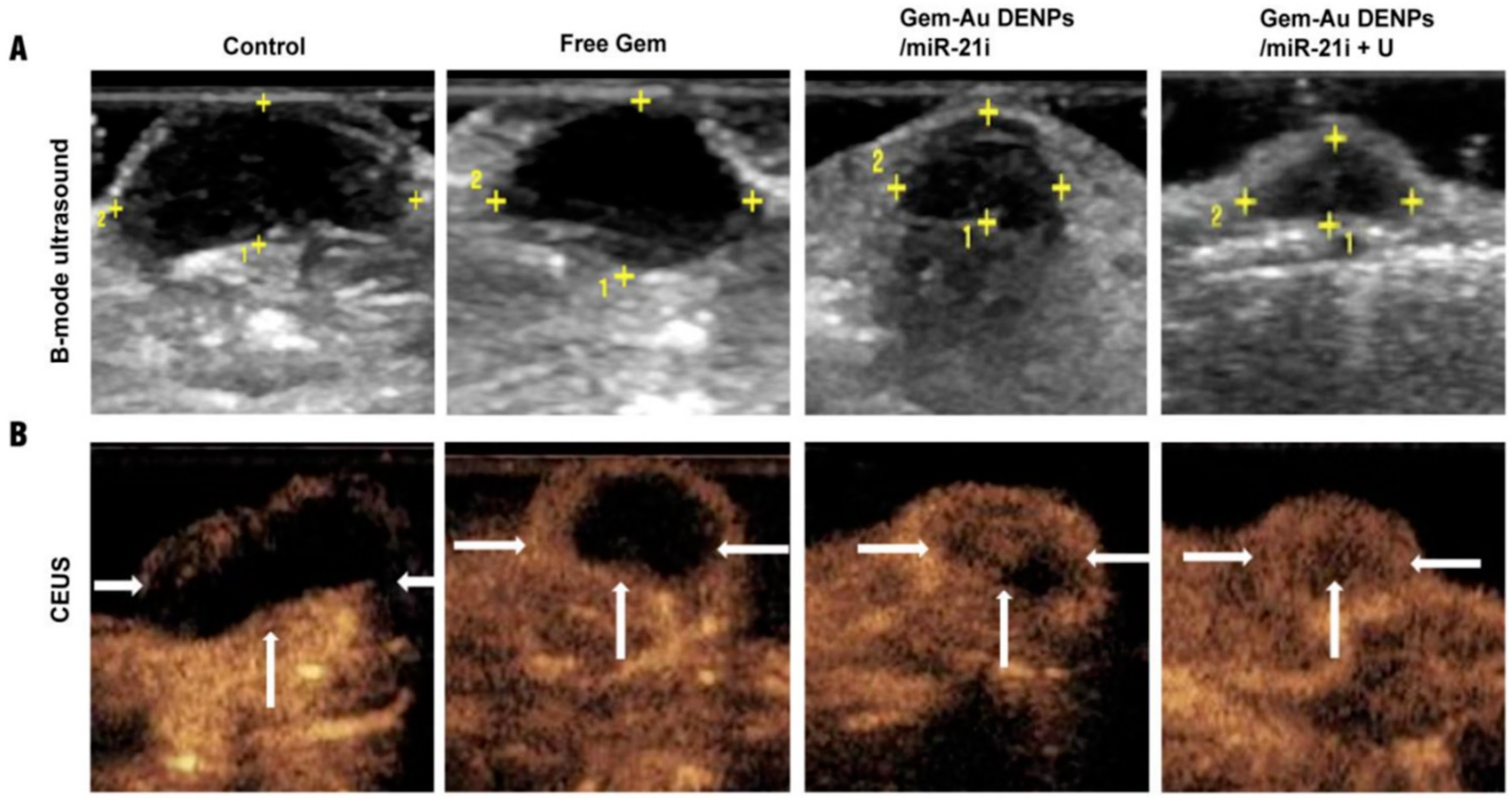
- Lin, L.; Fan, Y.; Gao, F.; Jin, L.; Li, D.; Sun, W.; Li, F.; Qin, P.; Shi, Q.; Shi, X.; et al. UTMD-Promoted Co-Delivery of Gemcitabine and MiR-21 Inhibitor by Dendrimer-Entrapped Gold Nanoparticles for Pancreatic Cancer Therapy. Theranostics 2018, 8, 1923–1939. [Google Scholar] [CrossRef] [PubMed]
- Coelho, S.C.; Reis, D.P.; Pereira, M.C.; Coelho, M.A.N. Doxorubicin and Varlitinib Delivery by Functionalized Gold Nanoparticles against Human Pancreatic Adenocarcinoma. Pharmaceutics 2019, 11, 551. [Google Scholar] [CrossRef]
- Barroso, L.; Viegas, C.; Vieira, J.; Ferreira-Pêgo, C.; Costa, J.; Fonte, P. Lipid-Based Carriers for Food Ingredients Delivery. J. Food Eng. 2021, 295, 110451. [Google Scholar] [CrossRef]
- Alavi, M.; Karimi, N.; Safaei, M. Application of Various Types of Liposomes in Drug Delivery Systems. Adv. Pharm. Bull. 2017, 7, 3–9. [Google Scholar] [CrossRef] [PubMed]
- George, P.S.; Teni, B.; Maria, V.; Sotirios, K.R.; John, G.S. Liposomal Cisplatin Combined with Gemcitabine in Pretreated Advanced Pancreatic Cancer Patients: A Phase I-II Study. Oncol. Rep. 2006, 15, 1201–1204. [Google Scholar]
- Allen, T.M. Ligand-Targeted Therapeutics in Anticancer Therapy. Nat. Rev. Cancer 2002, 2, 750–763. [Google Scholar] [CrossRef]
- Daniels, T.R.; Delgado, T.; Helguera, G.; Penichet, M.L. The Transferrin Receptor Part II: Targeted Delivery of Therapeutic Agents into Cancer Cells. Clin. Immunol. 2006, 121, 159–176. [Google Scholar] [CrossRef]
- Zinger, A.; Koren, L.; Adir, O.; Poley, M.; Alyan, M.; Yaari, Z.; Noor, N.; Krinsky, N.; Simon, A.; Gibori, H.; et al. Collagenase Nanoparticles Enhance the Penetration of Drugs into Pancreatic Tumors. ACS Nano 2019, 13, 11008–11021. [Google Scholar] [CrossRef]
- Viegas, C.; Seck, F.; Fonte, P. An Insight on Lipid Nanoparticles for Therapeutic Proteins Delivery. J. Drug Deliv. Sci. Technol. 2022, 77, 103839. [Google Scholar] [CrossRef]
- Daraee, H.; Etemadi, A.; Kouhi, M.; Alimirzalu, S.; Akbarzadeh, A. Application of Liposomes in Medicine and Drug Delivery. Artif. Cells Nanomed. Biotechnol. 2014, 44, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Viegas, C.; Patrício, A.B.; Prata, J.M.; Nadhman, A.; Chintamaneni, P.K.; Fonte, P. Solid Lipid Nanoparticles vs. Nanostructured Lipid Carriers: A Comparative Review. Pharmaceutics 2023, 15, 1593. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.; Abdin, S.M.; Kamal, L.; Orive, G. Nanostructured Lipid Carriers for Delivery of Chemotherapeutics: A Review. Pharmaceutics 2020, 12, 288. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, S.; Jia, L.; Chu, F.; Zhou, Y.; He, Z.; Guo, M.; Chen, C.; Xu, L. Nanostructured Lipid Carriers for MicroRNA Delivery in Tumor Gene Therapy. Cancer Cell Int. 2018, 18, 101. [Google Scholar] [CrossRef]
- Müller, R.H.; Radtke, M.; Wissing, S.A. Nanostructured Lipid Matrices for Improved Microencapsulation of Drugs. Int. J. Pharm. 2002, 242, 121–128. [Google Scholar] [CrossRef]
- Borges, A.; de Freitas, V.; Mateus, N.; Fernandes, I.; Oliveira, J. Solid Lipid Nanoparticles as Carriers of Natural Phenolic Compounds. Antioxidants 2020, 9, 998. [Google Scholar] [CrossRef]
- Wang, T.; Liu, Z.; Zhang, Z.; Tang, S.; Yue, M.; Feng, S.; Hu, M.; Xuan, L.; Chen, Y. Evaluation of Antitumor Activity of Survivin Short Interfering RNA Delivered by Lipid Nanoparticles in Colon Cancer In Vitro and In Vivo. Oncol. Lett. 2017, 14, 2001–2008. [Google Scholar] [CrossRef]
- Aryani, N.L.D.; Siswandono; Soeratri, W.; Rahmasari, F.P.; Sari, D.R.K. Experimental Development and Molecular Docking: Nanostructured Lipid Carriers (NLCs) of Coenzyme Q10 Using Stearic Acid and Different Liquid Lipids as a Lipid Matrix. Int. J. Appl. Pharm. 2021, 13, 108–116. [Google Scholar] [CrossRef]
- Severino, P.; Andreani, T.; Macedo, A.S.; Fangueiro, J.F.; Santana, M.H.A.; Silva, A.M.; Souto, E.B. Current State-of-Art and New Trends on Lipid Nanoparticles (SLN and NLC) for Oral Drug Delivery. J. Drug Deliv. 2012, 2012, 750891. [Google Scholar] [CrossRef]
- Muchow, M.; Maincent, P.; Müller, R.H. Lipid Nanoparticles with a Solid Matrix (SLN, NLC, LDC) for Oral Drug Delivery. Drug Dev. Ind. Pharm. 2008, 34, 1394–1405. [Google Scholar] [CrossRef]
- Joshi, M.D.; Müller, R.H. Lipid Nanoparticles for Parenteral Delivery of Actives. Eur. J. Pharm. Biopharm. 2009, 71, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Judge, A.D.; Robbins, M.; Tavakoli, I.; Levi, J.; Hu, L.; Fronda, A.; Ambegia, E.; McClintock, K.; MacLachlan, I. Confirming the RNAi-Mediated Mechanism of Action of SiRNA-Based Cancer Therapeutics in Mice. J. Clin. Investig. 2009, 119, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Mahato, R.I. Lipid and Polymeric Carrier-Mediated Nucleic Acid Delivery. Expert. Opin. Drug Deliv. 2010, 7, 1209–1226. [Google Scholar] [CrossRef]
- Crucho, C.I.C.; Barros, M.T. Polymeric Nanoparticles: A Study on the Preparation Variables and Characterization Methods. Mater. Sci. Eng. 2017, 80, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Crucho, C.I.C. Stimuli-Responsive Polymeric Nanoparticles for Nanomedicine. ChemMedChem 2014, 14, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Macedo, A.S.; Castro, P.M.; Roque, L.; Thomé, N.G.; Reis, C.P.; Pintado, M.E.; Fonte, P. Novel and Revisited Approaches in Nanoparticle Systems for Buccal Drug Delivery. J. Control. Release 2020, 320, 125–141. [Google Scholar] [CrossRef]
- Viegas, C.; Pereira, D.S.M.; Fonte, P. Insights into Nanomedicine for Head and Neck Cancer Diagnosis and Treatment. Materials 2022, 15, 2086. [Google Scholar] [CrossRef]
- Tang, X.; Wang, G.; Shi, R.; Jiang, K.; Meng, L.; Ren, H.; Wu, J.; Hu, Y. Enhanced Tolerance and Antitumor Efficacy by Docetaxel-Loaded Albumin Nanoparticles. Drug Deliv. 2016, 23, 2686–2696. [Google Scholar] [CrossRef]
- Hung, S.W.; Mody, H.R.; Govindarajan, R. Overcoming Nucleoside Analog Chemoresistance of Pancreatic Cancer: A Therapeutic Challenge. Cancer Lett. 2012, 320, 138–149. [Google Scholar] [CrossRef]
- Yu, X.; Di, Y.; Xie, C.; Song, Y.; He, H.; Li, H.; Pu, X.; Lu, W.; Fu, D.; Jin, C. An In Vitro and In Vivo Study of Gemcitabine-Loaded Albumin Nanoparticles in a Pancreatic Cancer Cell Line. Int. J. Nanomed. 2015, 10, 6825–6834. [Google Scholar] [CrossRef]
- Rajeshkumar, N.V.; Yabuuchi, S.; Pai, S.G.; Tong, Z.; Hou, S.; Bateman, S.; Pierce, D.W.; Heise, C.; Von Hoff, D.D.; Maitra, A.; et al. Superior Therapeutic Efficacy of Nab-Paclitaxel over Cremophor-Based Paclitaxel in Locally Advanced and Metastatic Models of Human Pancreatic Cancer. Br. J. Cancer 2016, 115, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; An, Y.; Yuan, C.; Zhang, H.; Liang, C.; Ding, F.; Gao, Q.; Zhang, D. GEM-Loaded Magnetic Albumin Nanospheres Modified with Cetuximab for Simultaneous Targeting, Magnetic Resonance Imaging, and Double-Targeted Thermochemotherapy of Pancreatic Cancer Cells. Int. J. Nanomed. 2015, 10, 2507–2519. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, N.; Matsumura, Y.; Kataoka, K. Development of Polymeric Micelles for Targeting Intractable Cancers. Cancer Sci. 2016, 107, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Matsumura, Y.; Suzuki, M.; Shimizu, K.; Goda, R.; Nakamura, I.; Nakatomi, I.; Yokoyama, M.; Kataoka, K.; Kakizoe, T. NK105, a Paclitaxel-Incorporating Micellar Nanoparticle Formulation, Can Extend In Vivo Antitumour Activity and Reduce the Neurotoxicity of Paclitaxel. Br. J. Cancer 2005, 92, 1240–1246. [Google Scholar] [CrossRef]
- Tian, L.; Pei, R.; Zhong, L.; Ji, Y.; Zhou, D.; Zhou, S. Enhanced Targeting of 3D Pancreatic Cancer Spheroids by Aptamer-Conjugated Polymeric Micelles with Deep Tumor Penetration. Eur. J. Pharmacol. 2021, 894, 173814. [Google Scholar] [CrossRef]
- Salva, E.; Ekentok, C.; Turan, S.O.; Akbuga, J. Non-Viral SiRNA and ShRNA Delivery Systems in Cancer Therapy. In RNA Interference; InTech Open: London, UK, 2016; ISBN 9789533070940. [Google Scholar]
- Liu, T.; Liang, X.; Li, B.; Björkholm, M.; Jia, J.; Xu, D. Telomerase Reverse Transcriptase Inhibition Stimulates Cyclooxygenase 2 Expression in Cancer Cells and Synergizes with Celecoxib to Exert Anti-Cancer Effects. Br. J. Cancer 2013, 108, 2272–2280. [Google Scholar] [CrossRef]
- Rezvantalab, S.; Drude, N.I.; Moraveji, M.K.; Güvener, N.; Koons, E.K.; Shi, Y.; Lammers, T.; Kiessling, F. PLGA-Based Nanoparticles in Cancer Treatment. Front. Pharmacol. 2018, 9, 1260. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, J.; Zhao, M.; Tang, S.; Cheng, X.; Zhang, W.; Li, W.; Liu, X.; Peng, H.; Wang, Q. Effects of Polyethylene Glycol on the Surface of Nanoparticles for Targeted Drug Delivery. Nanoscale 2021, 13, 10748–10764. [Google Scholar] [CrossRef]
- Akhter, M.H.; Kumar, S.; Nomani, S. Sonication Tailored Enhance Cytotoxicity of Naringenin Nanoparticle in Pancreatic Cancer: Design, Optimization, and in Vitro Studies. Drug Dev. Ind. Pharm. 2020, 46, 659–672. [Google Scholar] [CrossRef]
- Jamil, A.; Aamir Mirza, M.; Anwer, M.K.; Thakur, P.S.; Alshahrani, S.M.; Alshetaili, A.S.; Telegaonkar, S.; Panda, A.K.; Iqbal, Z. Co-Delivery of Gemcitabine and Simvastatin through PLGA Polymeric Nanoparticles for the Treatment of Pancreatic Cancer: In-Vitro Characterization, Cellular Uptake, and Pharmacokinetic Studies. Drug Dev. Ind. Pharm. 2019, 45, 745–753. [Google Scholar] [CrossRef]
- Kudr, J.; Haddad, Y.; Richtera, L.; Heger, Z.; Cernak, M.; Adam, V.; Zitka, O. Magnetic Nanoparticles: From Design and Synthesis to Real World Applications. Nanomaterials 2017, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Samiei, M.; Davaran, S. Magnetic Nanoparticles: Preparation, Physical Properties, and Applications in Biomedicine. Nanoscale Res. Lett. 2012, 7, 144. [Google Scholar] [CrossRef]
- Hafiz, S.S.; Xavierselvan, M.; Gokalp, S.; Labadini, D.; Barros, S.; Duong, J.; Foster, M.; Mallidi, S. Eutectic Gallium-Indium Nanoparticles for Photodynamic Therapy of Pancreatic Cancer. ACS Appl. Nano Mater. 2022, 5, 6125–6139. [Google Scholar] [CrossRef]
- Sharma, A.; Madhunapantula, S.V.; Robertson, G.P. Toxicological Considerations When Creating Nanoparticle Based Drugs and Drug Delivery Systems? Expert. Opin. Drug Metab. Toxicol. 2012, 8, 47–69. [Google Scholar] [CrossRef] [PubMed]
- Bahadar, H.; Maqbool, F.; Niaz, K.; Abdollahi, M. Toxicity of Nanoparticles and an Overview of Current Experimental Models. Iran. Biomed. J. 2016, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of Cationic Lipids and Cationic Polymers in Gene Delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H. Onivyde for the Therapy of Multiple Solid Tumors. Onco Targets Ther. 2016, 9, 3001–3007. [Google Scholar] [CrossRef]
- Chang, C. The Immune Effects of Naturally Occurring and Synthetic Nanoparticles. J. Autoimmun. 2010, 34, J234–J246. [Google Scholar] [CrossRef]
- Keelan, J.A. Nanotoxicology: Nanoparticles versus the Placenta. Nat. Nanotechnol. 2011, 6, 263–264. [Google Scholar] [CrossRef]
- Kodiha, M.; Wang, Y.M.; Hutter, E.; Maysinger, D.; Stochaj, U. Off to the Organelles—Killing Cancer Cells with Targeted Gold Nanoparticles. Theranostics 2015, 5, 357. [Google Scholar] [CrossRef]
- Gu, Y.J.; Cheng, J.; Lin, C.C.; Lam, Y.W.; Cheng, S.H.; Wong, W.T. Nuclear Penetration of Surface Functionalized Gold Nanoparticles. Toxicol. Appl. Pharmacol. 2009, 237, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Lee, S.; Son, S.; Kim, S.H.; Leary, J.F.; Choi, K.; Kwon, I.C. Theranostic Nanoparticles for Future Personalized Medicine. J. Control. Release 2014, 190, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Situ, A.; Kang, Y.; Rose Villabroza, K.; Liao, Y.; Hyun Chang, C.; Donahue, T.; Nel, A.E.; Meng, H. Irinotecan Delivery by Lipid-Coated Mesoporous Silica Nanoparticles Shows Improved Efficacy and Safety over Liposomes for Pancreatic Cancer. ACS Nano 2016, 10, 2702–2715. [Google Scholar] [CrossRef]
- Miladi, I.; Aloy, M.T.; Armandy, E.; Mowat, P.; Kryza, D.; Magné, N.; Tillement, O.; Lux, F.; Billotey, C.; Janier, M.; et al. Combining Ultrasmall Gadolinium-Based Nanoparticles with Photon Irradiation Overcomes Radioresistance of Head and Neck Squamous Cell Carcinoma. Nanomedicine 2015, 11, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil®—The First FDA—Approved Nano-Drug: Lessons Learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Tang, F.; Li, L.; Chen, D.; Products, N.; For, A.; Or, U.S.E.; Roggers, R.; Kanvinde, S.; Boonsith, S.; Oupický, D.; et al. The Practicality of Mesoporous silica Nanoparticles as Drug Delivery Devices and Progress toward This Goal. Nano Today 2013, 46, 1278–1288. [Google Scholar]
- DiGiulio, S. FDA Approves Onivyde Combo Regimen for Advanced Pancreatic Cancer. Oncol. Times 2015, 37, 8. [Google Scholar] [CrossRef]
- Alphandéry, E.; Grand-Dewyse, P.; Lefèvre, R.; Mandawala, C.; Durand-Dubief, M. Cancer Therapy Using Nanoformulated Substances: Scientific, Regulatory and Financial Aspects. Expert. Rev. Anticancer Ther. 2015, 15, 1233–1255. [Google Scholar] [CrossRef]
- Gordon, E.M.; Hall, F.L. Rexin-G, a Targeted Genetic Medicine for Cancer. Expert. Opin. Biol. Ther. 2010, 10, 819–832. [Google Scholar] [CrossRef]
- Gordon, E.M.; Hall, F.L. Critical Stages in the Development of the First Targeted, Injectable Molecular-Genetic Medicine for Cancer. In Gene Therapy Applications; InTech Open: London, UK, 2011; pp. 461–492. [Google Scholar]
- Chawla, S.P.; Bruckner, H.; Morse, M.A.; Assudani, N.; Hall, F.L.; Gordon, E.M. A Phase I-II Study Using Rexin-G Tumor-Targeted Retrovector Encoding a Dominant-Negative Cyclin G1 Inhibitor for Advanced Pancreatic Cancer. Mol. Ther. J. Cell Press. 2019, 12, 56–67. [Google Scholar] [CrossRef]
- Pancreatic Ductal Adenocarcinoma, Pancreatic Cancer Stage IV, Pancreatic Cancer Non-Resectable Trial in Oxford. Available online: https://www.clincosm.com/trial/pancreatic-ductal-adenocarcinoma-cancer-stage-iv-non-resectable (accessed on 20 September 2022).
- PanDox: Targeted Doxorubicin in Pancreatic Tumours—Full Text View—Clinicaltrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04852367 (accessed on 26 October 2021).
- Focused Ultrasound Foundation. New Clinical Trial of Focused Ultrasound–Enhanced Chemotherapy for Pancreatic Cancer—Focused Ultrasound Foundation. Available online: https://www.fusfoundation.org/news/new-clinical-trial-of-focused-ultrasound-enhanced-chemotherapy-for-pancreatic-cancer (accessed on 26 October 2021).
- Kim, G. Nab-Paclitaxel for the Treatment of Pancreatic Cancer. Cancer Manag. Res. 2017, 9, 85–96. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Tumor (T) | Regional Lymph Nodes (N) | Distant Metastasis (M) | |||
|---|---|---|---|---|---|
| TX | Primary tumor cannot be assessed | NX | Regional lymph nodes cannot be assessed | M0 | No distant metastasis |
| T0 | No evidence of primary tumor | N0 | No regional lymph node metastasis | M1 | Distant metastasis |
| Tis | Carcinoma in situ | N1 | Regional lymph node metastasis | ||
| T1 | Tumor limited to the pancreas, 2 cm or less in greatest dimension | ||||
| T2 | Tumor limited to the pancreas, more than 2 cm in greatest dimension | ||||
| T3 | Tumor extends beyond the pancreas but without the involvement of the celiac axis or the superior mesenteric artery | ||||
| T4 | Tumor involves the celiac axis or the superior mesenteric artery; unresectable primary tumor | ||||
| Stage | Primary Tumor | Regional Lymph Nodes | Distant Metastasis |
|---|---|---|---|
| 0 | Carcinoma in situ | No regional lymph node metastasis | No distant metastasis |
| IA | Tumor limited to the pancreas, 2 cm or less in greatest dimension | No regional lymph node metastasis | No distant metastasis |
| IB | Tumor limited to the pancreas, more than 2 cm in greatest dimension | No regional lymph node metastasis | No distant metastasis |
| IIA | Tumor extends beyond the pancreas but without the involvement of the celiac axis or the superior mesenteric artery | No regional lymph node metastasis | No distant metastasis |
| IIB | Tumor limited to the pancreas; Tumor extends beyond the pancreas but without the involvement of the celiac axis or the superior mesenteric artery | Regional lymph node metastasis | No distant metastasis |
| III | Tumor involves the celiac axis or the superior mesenteric artery; unresectable primary tumor | No regional or regional lymph node metastasis | No distant metastasis |
| IV | Tumor limited to the pancreas; Tumor extends beyond the pancreas but without involvement of the celiac axis or the superior mesenteric artery; Tumor involves the celiac axis or the superior mesenteric artery; unresectable primary tumor | No regional or regional lymph node metastasis | Distant metastasis |
| Nanocarrier | Load | Characterization | Targeting Moiety | Targeting Cell/Tissue | In Vitro/Vivo Model | Application | Ref. |
|---|---|---|---|---|---|---|---|
| Liposomes | siRNA against HER-2 | Particle size = ~100 nm | TfR antibody receptor | Human pancreatic cancer cell line PANC-1 | Murine xenograft model | Pancreatic cancer | [113] |
| Nanoliposomal system (MM-398) | Irinotecan | Particle size = ~111 nm; PdI = 0.04 | Topoisomerase I | Metastatic pancreatic ductal adenocarcinoma (PDA) | Global, phase 3, randomized, open-label trial | 2nd line therapy in metastatic PDA | [114,115,116] |
| Liposome FF-10832 | Gemcitabine | Particle size = 79 ± 2 nm | DNA synthesis and ribonucleotide reductase | Pancreatic cancer cell lines | Mice with Capan-1, SUIT-2, and BxPC-3 tumors | Pancreatic cancer | [117] |
| Liposomes | Collagenase | - | Collagen | Extracellular collagen stroma matrix | Mice-bearing PDA tumors | Disassemble the collagen stroma matrix | [96] |
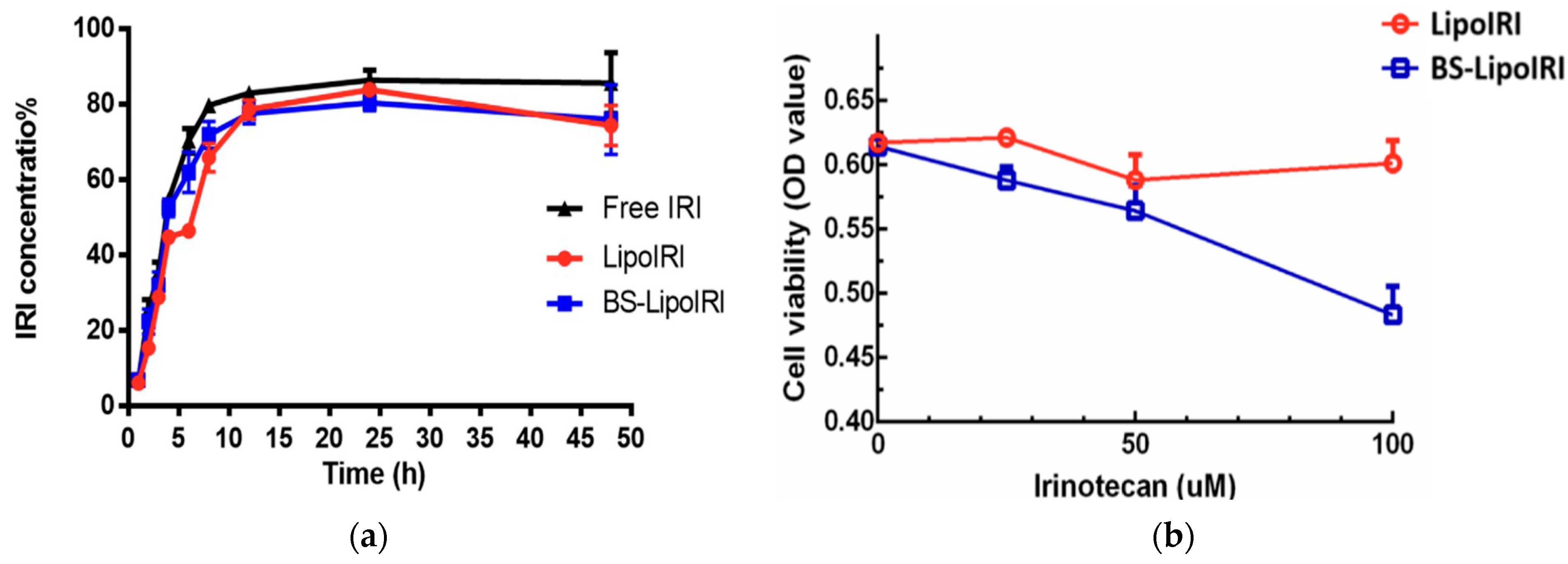
| Liposomes (BS-LipoIRI) | Irinotecan | Particle size = 125 nm; drug encapsulation efficiency = 80.95% | Epidermal growth factor receptor (EGFR) and fibroblast activation protein bispecific antibody | Pancreatic tumor cells and tumor-associated fibroblasts | Eight-week-old SCID mice | Human Pancreatic Tumor | [118] |
| Solid lipid nanoparticles (SLN) | Gemcitabine | Particle size = 603 ± 19 nm; entrapment efficiency = 68.3 ± 4.8% | DNA synthesis and ribonucleotide reductase | Patient-derived primary pancreatic cancer cell lines | MiaPaCa-2 and PPCL-46 cell lines | Human Pancreatic Tumor | [119] |
| SLN | Aspirin and Curcumin | Particle size of 150 and 250 nm; encapsulation efficiency of 85 and 69% | Cyclooxygenase-2; anti-inflammatory/anti-cancer effect | Pancreatic cancer cells | MIA Paca-2 and Panc-1 cell lines | Chemoprevention of pancreatic cancer | [120] |
| Cationic Nanostructured lipid carriers (NLC) | microRNA miR-34a and miR-143/145 | Nanovector size ~100 nm | SIRT1, CD44, aldehyde dehydrogenase, KRAS2 and Ras-responsive element binding protein-1 (RREB1) | Pancreatic cancer xenograft model | MiaPaCa-2 subcutaneous xenografts | Pancreatic cancer | [121] |
| Hyaluronic acid-coated NLCs | Gemcitabine and Baicalein (BCL) | - | DNA synthesis and ribonucleotide reductase | Human pancreatic adenocarcinoma cell lines | AsPC1 cells lines | Human pancreatic adenocarcinoma | [122] |
| Lipid-polymer hybrid nanoparticle | Gemcitabine and HIF1a siRNA | - | DNA synthesis and ribonucleotide reductase | - | Subcutaneous and orthotopic tumor models | Pancreatic cancer | [123] |
| Albumin nanoparticles encapsulated in modified thermosensitive liposomes | Paclitaxel | Particle size = 123.9 ± 1.9 nm | Mitotic arrest in the cell cycle at the mitotic phase | Tumor mouse models | Pan 02 subcutaneous and orthotopic tumor | PDA | [124] |
| Gelatin nanoparticles marked with a redox-responsive EGFR | Gemcitabine | - | DNA synthesis and ribonucleotide reductase | Orthotopic pancreatic cancer model | Panc-1 human pancreatic ductal adenocarcinoma cells | PDA | [125] |
| Nanobioconjugate chitosan-based | Gemcitabine and anti-EGFR antibodies | Encapsulation rate = 91.63% | DNA synthesis, ribonucleotide reductase and EGFR | Human pancreatic cancer cells | Human pancreatic cancer cell lines SW1990 | Pancreatic cancer | [16] |
| Chitosan nanoparticles | Quercetin and 5-fluorouracil | Particle size = 402 ± 52 nm; entrapment efficiency = 95 and 75% | Chromosome segregation and organization | Primary pancreatic cancer cell line and mouse cell line | MiaPaCa2 and primary mouse fibroblast cell line | Pancreatic cancer | [126] |
| Polymeric micelles with cellular membrane-disruptive molecules | Gemcitabine | Particle size from 107 ± 11.9 to 163.1 ± 13.1 nm | DNA synthesis and ribonucleotide reductase | Human pancreatic cancer cells | 3D spheroid, shell of fibroblast; NIH-3T3 cells over pancreatic BxPC-3 cells | Pancreatic cancer | [127] |
| Polymeric micelles | microRNA miR-34a and volasertib (BI6727) | Particle size = 100 nm; drug loading capacity = 10% | Suppression of Bcl-2 | Pancreatic cell lines | Orthotopic pancreatic tumor-bearing NSG mice, MIA PaCa-2R cell line | PDA | [128] |
| PAMAM dendrimers | Camptothecin | Particle size = ~20 nm | Topoisomerase inhibition | Mice tumor models | Patient-derived PDA xenograft and orthotopic PDA cell xenograft | PDA | [129] |
| Polymeric system onto PAMAM dendrimer | Gemcitabine | Particle size = ~120 nm | DNA synthesis and ribonucleotide reductase | Pancreatic cell lines and mice bearing Panc02 pancreatic tumor xenografts | Adherent Panc02 cells, 3D multicellular spheroids (MCSs) and ICR mice | PDA | [130] |
| Poly Lactic-co-Glycolic Acid (PLGA) nanoparticles | Naringenin (NARG) | Particle size = 150.45 ± 12.45 nm; PDI = 0.132 ± 0.026; Zeta potential = −20.5 ± 2.5 mV | Free radical scavenging activity | Pancreatic cell lines | - | Pancreatic adenocarcinoma | [124] |
| PLGA nanoparticles | Gemcitabine and simvastatin | Particle size = 258 ± 2.4 nm; PDI = 0.32 ± 0.052; zeta potential = −12.5 mV | DNA synthesis and ribonucleotide reductase; avoidance of translation of pancreatic intraepithelial neoplasia to PDA | Pancreatic cell lines | MCF-7 and MIA PaCa-2 cells; Wistar rats | PDA | |
| PLGA-PEG nanoparticles | 3, 3′-diindolylmethane (DIM), and ellagic acid (EA) | Particle size = 180–210 nm | Apoptosis induction | Human pancreatic cancer cell line | SUIT2 expressing firefly luciferase (SUIT2-Luc) | Pancreatic cancer | [131] |
| PLGA nanoparticle | siRNA | Particle size = 188.5 ± 1.2 nm | Programmed death-ligand 1 (PD-L1 | PDA tumor-bearing humanized mice | - | Pancreatic cancer | [132] |
| Superparamagnetic iron oxide nanoparticle (SPION) | Curcumin | Particle size = 120 to 140 nm; zeta potential = −17 to −20 mV | Anti-inflammatory/anti-cancer effect | Model pancreatic cancer mice | - | Pancreatic cancer | [133] |
| SPION coated with dextran and conjugated with folic acid | Vinblastin | Particle size = 74 ± 13 nm; zeta potential = −45 mV; polydispersity index = 0.080 | Mitotic arrest in the cell cycle at the mitotic phase | Pancreatic cell lines | PANC-1 pancreatic cancer cells | Pancreatic cancer | [134] |
| Mesoporous silica nanoparticles (MSNs) | Gemcitabine and cisplatin | Particle size between 120 to 1890 nm | DNA synthesis and reparation | PDA cell lines | PDA.MUC1 Mouse Model | PDA | [135] |
| Dendrimer-entrapped gold nanoparticles (AU-DNPs) | Gemcitabine and miR-21 inhibitors | Particle size between 154 to 276 nm | DNA synthesis | Xenografted pancreatic mouse tumors | SW1990 cells | Pancreatic cancer | [136] |
| Pegylated gold nanoparticles (PEGAuNPs) | Doxorubicin and varlitinib | Particle size = 24 ± 1 nm; zeta potential = −41 ± 2 mV | Topoisomerase-II-mediated DNA repair; tyrosine kinase | Pancreatic tumor | MIA PaCa-2, S2-013 cells, and hTERT immortalized human cell | Pancreatic cancer | [137] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viegas, C.; Patrício, A.B.; Prata, J.; Fonseca, L.; Macedo, A.S.; Duarte, S.O.D.; Fonte, P. Advances in Pancreatic Cancer Treatment by Nano-Based Drug Delivery Systems. Pharmaceutics 2023, 15, 2363. https://doi.org/10.3390/pharmaceutics15092363
Viegas C, Patrício AB, Prata J, Fonseca L, Macedo AS, Duarte SOD, Fonte P. Advances in Pancreatic Cancer Treatment by Nano-Based Drug Delivery Systems. Pharmaceutics. 2023; 15(9):2363. https://doi.org/10.3390/pharmaceutics15092363
Chicago/Turabian StyleViegas, Cláudia, Ana B. Patrício, João Prata, Leonor Fonseca, Ana S. Macedo, Sofia O. D. Duarte, and Pedro Fonte. 2023. "Advances in Pancreatic Cancer Treatment by Nano-Based Drug Delivery Systems" Pharmaceutics 15, no. 9: 2363. https://doi.org/10.3390/pharmaceutics15092363