
Antiviral Action against SARS-CoV-2 of a Synthetic Peptide Based on a Novel Defensin Present in the Transcriptome of the Fire Salamander (Salamandra salamandra)

 , , , , , ,
, , , , , ,  ,
,  , ,
, , 
 ,
,  ,
,  add
Show full author list
add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Transcriptome Assembly and Identification of Transcript Sequence by Homology
2.2. Sequence Alignment and Phylogenetic Analysis
2.3. In Silico Studies
2.3.1. Docking
2.3.2. Molecular Dynamics Simulations
2.3.3. Electronic Structure Calculations
2.4. Synthesis and Characterization of Peptide
2.5. In Vitro Synthetic SS-I Viability Evaluation
2.6. In Vitro Vero-CCL-81 Cell-Based SARS-CoV-2 Infection Assay
2.7. Hemolysis Assay
3. Results and Discussion
3.1. Identification and Characterization of the Defensin SS-I
3.2. Interactions between SS-I with ACE2 and SARS-CoV-2 Spike Protein (S1)
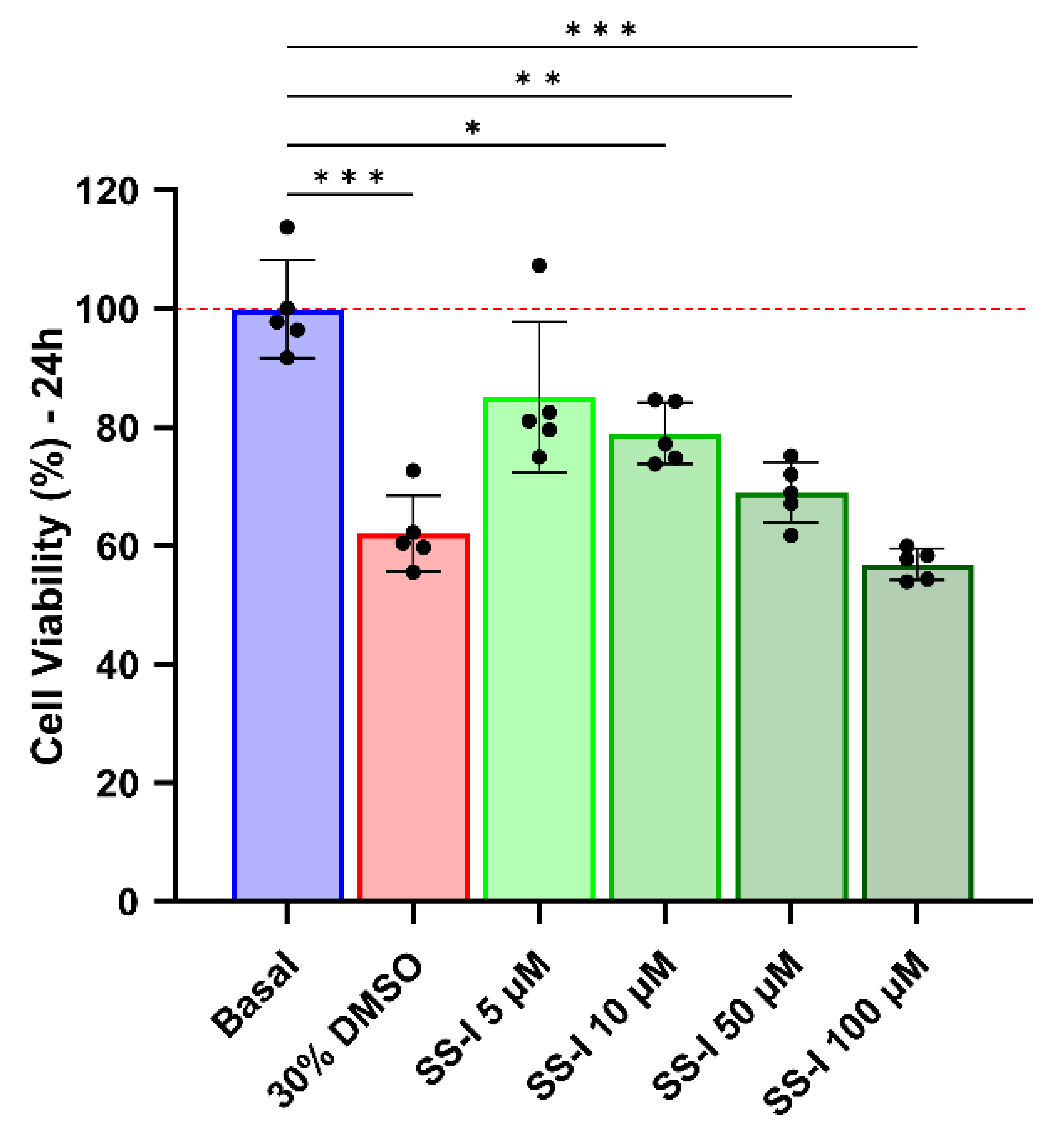
3.3. In Vitro Cytotoxic and Hemolytic Activity of Synthetic SS-I and SARS-CoV-2 Infection Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tamma, P.D.; Miller, M.A.; Dullabh, P.; Ahn, R.; Speck, K.; Gao, Y.; Scherpf, E.; Cosgrove, S.E. Association of a Safety Program for Improving Antibiotic Use with Antibiotic Use and Hospital-Onset Clostridioides Difficile Infection Rates among US Hospitals. JAMA Netw. Open 2021, 4, e210235. [Google Scholar] [CrossRef]
- Chafekar, A.; Fielding, B.C. MERS-CoV: Understanding the Latest Human Coronavirus Threat. Viruses 2018, 10, 93. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Boto, A.; Pérez de la Lastra, J.; González, C.C.; De La Lastra, J.M.P.; González, C.C. The Road from Host-Defense Peptides to a New Generation of Antimicrobial Drugs. Molecules 2018, 23, 311. [Google Scholar] [CrossRef] [PubMed]
- Di, L. Strategic Approaches to Optimizing Peptide ADME Properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Dai, W. Peptide Developability at the Discovery-to-Development Interface—Current State and Future Opportunities. AAPS J. 2015, 17, 777–779. [Google Scholar] [CrossRef]
- da Costa, J.P.; Cova, M.; Ferreira, R.; Vitorino, R. Antimicrobial Peptides: An Alternative for Innovative Medicines? Appl. Microbiol. Biotechnol. 2015, 99, 2023–2040. [Google Scholar] [CrossRef]
- de la Torre, B.G.; Albericio, F. Peptide Therapeutics 2.0. Molecules 2020, 25, 2293. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Björn, C.; Ekblom, J. Antimicrobial Peptides as Therapeutic Agents: Opportunities and Challenges. Crit. Rev. Biotechnol. 2020, 40, 978–992. [Google Scholar] [CrossRef]
- Zhu, S. Discovery of Six Families of Fungal Defensin-like Peptides Provides Insights into Origin and Evolution of the CSalphabeta Defensins. Mol. Immunol. 2008, 45, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, K.A.T.; Graham, M.A.; Paape, T.D.; Vandenbosch, K.A. Genome Organization of More Than 300 Defensin-Like Genes in Arabidopsis. Plant Physiol. 2005, 138, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Meng, P.; Yang, S.; Shen, C.; Jiang, K.; Rong, M.; Lai, R. The First Salamander Defensin Antimicrobial Peptide. PLoS ONE 2013, 8, e83044. [Google Scholar] [CrossRef] [PubMed]
- Jarczak, J.; Kościuczuk, E.M.; Lisowski, P.; Strzałkowska, N.; Jóźwik, A.; Horbańczuk, J.; Krzyzewski, J.; Zwierzchowski, L.; Bagnicka, E. Defensins: Natural Component of Human Innate Immunity. Hum. Immunol. 2013, 74, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Thevissen, K.; Kristensen, H.-H.; Thomma, B.P.H.J.; Cammue, B.P.A.; François, I.E.J.A. Therapeutic Potential of Antifungal Plant and Insect Defensins. Drug Discov. Today 2007, 12, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Blecha, F. Antimicrobial Peptides and Bacteriocins: Alternatives to Traditional Antibiotics. Anim. Health Res. Rev. 2008, 9, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Kong, Y.; Wang, H.; Yan, T.; Feng, F.; Bian, J.; Yang, Y.; Yu, H. A Defensin-like Antimicrobial Peptide from the Venoms of Spider, Ornithoctonus hainana. J. Pept. Sci. 2011, 17, 540–544. [Google Scholar] [CrossRef]
- Rodriguez, A.; Pedersen, M.; Villegas, E.; Rivas-Santiago, B.; Villegas-Moreno, J.; Amero, C.; Norton, R.S.; Corzo, G. Antimicrobial Activity and Structure of a Consensus Human β-Defensin and Its Comparison to a Novel Putative HBD10. Proteins Struct. Funct. Bioinform. 2020, 88, 175–186. [Google Scholar] [CrossRef]
- Patocka, J.; Nepovimova, E.; Klimova, B.; Wu, Q.; Kuca, K. Antimicrobial Peptides: Amphibian Host Defense Peptides. Curr. Med. Chem. 2019, 26, 5924–5946. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef]
- Marani, M.M.; Aguilar, S.; Cuzziol Boccioni, A.P.; Cancelarich, N.L.; Basso, N.G.; Albericio, F. Identification of New Ocellatin Antimicrobial Peptides by CDNA Precursor Cloning in the Frame of This Family of Intriguing Peptides. Antibiotics 2020, 9, 751. [Google Scholar] [CrossRef]
- Linnæus, C. Systema Naturae per Regna Tria Naturae, Secundum Classes, Ordines, Genera, Species, Cum Characteribus, Differentiis, Synonymis, Locis, 10th ed.; Salvii, L., Ed.; Stockholm, Sweden, 1758. [Google Scholar]
- Lüddecke, T.; Schulz, S.; Steinfartz, S.; Vences, M. A Salamander’s Toxic Arsenal: Review of Skin Poison Diversity and Function in True Salamanders, Genus Salamandra. Sci. Nat. 2018, 105, 56. [Google Scholar] [CrossRef] [PubMed]
- Mebs, D.; Pogoda, W. Variability of Alkaloids in the Skin Secretion of the European Fire Salamander (Salamandra Salamadra terrestris). Toxicon 2005, 45, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Knepper, J.; Lüddecke, T.; Preißler, K.; Vences, M.; Schulz, S. Isolation and Identification of Alkaloids from Poisons of Fire Salamanders (Salamandra salamandra). J. Nat. Prod. 2019, 82, 1319–1324. [Google Scholar] [CrossRef]
- Plácido, A.; Bueno, J.; Barbosa, E.A.; Moreira, D.C.; Dias, J.D.N.; Cabral, W.F.; Albuquerque, P.; Bessa, L.J.; Freitas, J.; Kuckelhaus, S.A.S.; et al. The Antioxidant Peptide Salamandrin-I: First Bioactive Peptide Identified from Skin Secretion of Salamandra Genus (Salamandra salamandra). Biomolecules 2020, 10, 512. [Google Scholar] [CrossRef] [PubMed]
- Goedbloed, D.J.; Czypionka, T.; Altmüller, J.; Rodriguez, A.; Küpfer, E.; Segev, O.; Blaustein, L.; Templeton, A.R.; Nolte, A.W.; Steinfartz, S. Parallel Habitat Acclimatization Is Realized by the Expression of Different Genes in Two Closely Related Salamander Species (Genus Salamandra). Heredity 2017, 119, 429–437. [Google Scholar] [CrossRef]
- Czypionka, T.; Krugman, T.; Altmüller, J.; Blaustein, L.; Steinfartz, S.; Templeton, A.R.; Nolte, A.W. Ecological Transcriptomics—A Non-Lethal Sampling Approach for Endangered Fire Salamanders. Methods Ecol. Evol. 2015, 6, 1417–1425. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2015. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 29 June 2022).
- Krueger, F.; James, F.; Ewels, P.; Afyounian, E.; Weinstein, M.; Schuster-Boeckler, B.; Hulselmans, G.; Sclamons. FelixKrueger/TrimGalore: V0.6.10-Add Default Decompression Path. Zenodo. 2023. Available online: https://zenodo.org/records/7598955 (accessed on 9 May 2023).
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. RnaSPAdes: A de Novo Transcriptome Assembler and Its Application to RNA-Seq Data. Gigascience 2019, 8, giz100. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and Sequence Analysis Tools Services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, B.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W.H. Evolview v3: A Webserver for Visualization, Annotation, and Management of Phylogenetic Trees. Nucleic Acids Res. 2019, 47, W270–W275. [Google Scholar] [CrossRef]
- He, Z.; Zhang, H.; Gao, S.; Lercher, M.J.; Chen, W.H.; Hu, S. Evolview v2: An Online Visualization and Management Tool for Customized and Annotated Phylogenetic Trees. Nucleic Acids Res. 2016, 44, W236–W241. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W.H. EvolView, an Online Tool for Visualizing, Annotating and Managing Phylogenetic Trees. Nucleic Acids Res. 2012, 40, W569–W572. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at Its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.; Till, S.; Mirdita, M.; Steinegger, M.; Söding, J.; Lupas, A.N.; Alva, V. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.; DeLano, W. PyMOL 2020. Available online: http://www.pymol.org/pymol (accessed on 30 June 2022).
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An Automated Pipeline for the Setup of Poisson-Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Honorato, R.V.; Koukos, P.I.; Jiménez-García, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A.M.J.J. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021, 8, 729513. [Google Scholar] [CrossRef]
- RCSB PDB-6LZG: Structure of Novel Coronavirus Spike Receptor-Binding Domain Complexed with Its Receptor ACE2. Available online: https://www.rcsb.org/structure/6LZG (accessed on 29 June 2022).
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Ban, X.; Lahiri, P.; Dhoble, A.S.; Li, D.; Gu, Z.; Li, C.; Cheng, L.; Hong, Y.; Li, Z.; Kaustubh, B. Evolutionary Stability of Salt Bridges Hints Its Contribution to Stability of Proteins. Comput. Struct. Biotechnol. J. 2019, 17, 895–903. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab Initio Study of Solvated Molecules: A New Implementation of the Polarizable Continuum Model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Yang, W.; Mortier, W.J. The Use of Global and Local Molecular Parameters for the Analysis of the Gas-Phase Basicity of Amines. J. Am. Chem. Soc. 1986, 108, 5708–5711. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, E.A.; Plácido, A.; Moreira, D.C.; Albuquerque, L.; Dematei, A.; Silva-Carvalho, A.É.; Cabral, W.F.; Báo, S.N.; Saldanha-Araújo, F.; Kuckelhaus, S.A.S.; et al. The Peptide Secreted at the Water to Land Transition in a Model Amphibian Has Antioxidant Effects. Proc. R. Soc. B Biol. Sci. 2021, 288, 20211531. [Google Scholar] [CrossRef] [PubMed]
- Plácido, A.; do Pais do Amaral, C.; Teixeira, C.; Nogueira, A.; Brango-Vanegas, J.; Alves Barbosa, E.; Moreira, D.C.; Silva-Carvalho, A.É.; da Silva, M.d.G.; do Nascimento Dias, J.; et al. Neuroprotective Effects on Microglia and Insights into the Structure–Activity Relationship of an Antioxidant Peptide Isolated from Pelophylax perezi. J. Cell Mol. Med. 2022, 26, 2793–2807. [Google Scholar] [CrossRef] [PubMed]
- Alves, G.G.B.; Lavarda, F.C.; Graeff, C.F.O.; Batagin-Neto, A. Reactivity of Eumelanin Building Blocks: A DFT Study of Monomers and Dimers. J. Mol. Graph. Model. 2020, 98, 107609. [Google Scholar] [CrossRef] [PubMed]
- Maia, R.A.; Ventorim, G.; Batagin-Neto, A. Reactivity of Lignin Subunits: The Influence of Dehydrogenation and Formation of Dimeric Structures. J. Mol. Model. 2019, 25, 228. [Google Scholar] [CrossRef]
- Chan, W.C.; White, P.D. (Eds.) Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Practical Approach Series; Oxford University Press: Oxford, UK, 2000; ISBN 0199637253. [Google Scholar]
- Subirós-Funosas, R.; Prohens, R.; Barbas, R.; El-Faham, A.; Albericio, F. Oxyma: An Efficient Additive for Peptide Synthesis to Replace the Benzotriazole-Based HOBt and HOAt with a Lower Risk of Explosion. Chemistry 2009, 15, 9394–9403. [Google Scholar] [CrossRef]
- Dematei, A.; Nunes, J.B.; Moreira, D.C.; Jesus, J.A.; Laurenti, M.D.; Mengarda, A.C.A.; Vieira, M.S.; Do Amaral, C.P.; Domingues, M.M.; De Moraes, J.; et al. Mechanistic Insights into the Leishmanicidal and Bactericidal Activities of Batroxicidin, a Cathelicidin-Related Peptide from a South American Viper (Bothrops atrox). J. Nat. Prod. 2021, 84, 1787–1798. [Google Scholar] [CrossRef]
- Shi, A.; Fan, F.; Broach, J.R. Microbial Adaptive Evolution. J. Ind. Microbiol. Biotechnol. 2022, 49, 76. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Y.; Zhang, Y.; Lee, W.H.; Zhang, Y. Rich Diversity and Potency of Skin Antioxidant Peptides Revealed a Novel Molecular Basis for High-Altitude Adaptation of Amphibians. Sci. Rep. 2016, 6, 19866. [Google Scholar] [CrossRef] [PubMed]
- ProteinProspector. Available online: https://prospector.ucsf.edu/prospector/mshome.htm (accessed on 9 May 2023).
- Xiong, Q.; Cao, L.; Ma, C.; Tortorici, M.A.; Liu, C.; Si, J.; Liu, P.; Gu, M.; Walls, A.C.; Wang, C.; et al. Close Relatives of MERS-CoV in Bats Use ACE2 as Their Functional Receptors. Nature 2022, 612, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Nazario de Moraes, L.; Tommasini Grotto, R.M.; Targino Valente, G.; de Carvalho Sampaio, H.; Magro, A.J.; Fogaça, L.; Wolf, I.R.; Perahia, D.; Faria Silva, G.; Plana Simões, R. A Novel Molecular Mechanism to Explain Mutations of the HCV Protease Associated with Resistance against Covalently Bound Inhibitors. Virus Res. 2019, 274, 197778. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Kokubo, H. Exploring the Stability of Ligand Binding Modes to Proteins by Molecular Dynamics Simulations: A Cross-Docking Study. J. Chem. Inf. Model. 2017, 57, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- ISO 10993-5; Biological Evaluation of Medical Devices. International Organization for Standardization: Geneva, Switzerland, 2009.
- Hwang, P.M.; Vogel, H.J. Structure-Function Relationships of Antimicrobial Peptides. Biochem. Cell Biol. 1998, 76, 235–246. [Google Scholar] [CrossRef]
- Klüver, E.; Schulz-Maronde, S.; Scheid, S.; Meyer, B.; Forssmann, W.G.; Adermann, K. Structure-Activity Relation of Human β-Defensin 3: Influence of Disulfide Bonds and Cysteine Substitution on Antimicrobial Activity and Cytotoxicity. Biochemistry 2005, 44, 9804–9816. [Google Scholar] [CrossRef]
- Greco, I.; Molchanova, N.; Holmedal, E.; Jenssen, H.; Hummel, B.D.; Watts, J.L.; Håkansson, J.; Hansen, P.R.; Svenson, J. Correlation between Hemolytic Activity, Cytotoxicity and Systemic in Vivo Toxicity of Synthetic Antimicrobial Peptides. Sci. Rep. 2020, 10, 13206. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, L.; Li, J.; Suresh, A.; Verma, C.; Foo, Y.H.; Yap, E.P.H.; Tan, D.T.H.; Beuerman, R.W. Linear Analogues of Human β-Defensin 3: Concepts for Design of Antimicrobial Peptides with Reduced Cytotoxicity to Mammalian Cells. ChemBioChem 2008, 9, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Ilker, M.F.; Nüsslein, K.; Tew, G.N.; Coughlin, E.B. Tuning the Hemolytic and Antibacterial Activities of Amphiphilic Polynorbornene Derivatives. J. Am. Chem. Soc. 2004, 126, 15870–15875. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Mechanism of the Binding, Insertion and Destabilization of Phospholipid Bilayer Membranes by α-Helical Antimicrobial and Cell Non-Selective Membrane-Lytic Peptides. Biochim. Biophys. Acta (BBA) Biomembr. 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Maturana, P.; Martinez, M.; Noguera, M.E.; Santos, N.C.; Disalvo, E.A.; Semorile, L.; Maffia, P.C.; Hollmann, A. Lipid Selectivity in Novel Antimicrobial Peptides: Implication on Antimicrobial and Hemolytic Activity. Colloids Surf. B Biointerfaces 2017, 153, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Manzini, M.C.; Perez, K.R.; Riske, K.A.; Bozelli, J.C.; Santos, T.L.; Da Silva, M.A.; Saraiva, G.K.V.; Politi, M.J.; Valente, A.P.; Almeida, F.C.L.; et al. Peptide:Lipid Ratio and Membrane Surface Charge Determine the Mechanism of Action of the Antimicrobial Peptide BP100. Conformational and Functional Studies. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 1985–1999. [Google Scholar] [CrossRef] [PubMed]
- Malanovic, N.; Leber, R.; Schmuck, M.; Kriechbaum, M.; Cordfunke, R.A.; Drijfhout, J.W.; De Breij, A.; Nibbering, P.H.; Kolb, D.; Lohner, K. Phospholipid-Driven Differences Determine the Action of the Synthetic Antimicrobial Peptide OP-145 on Gram-Positive Bacterial and Mammalian Membrane Model Systems. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1848, 2437–2447. [Google Scholar] [CrossRef] [PubMed]
- Belokoneva, O.S.; Villegas, E.; Corzo, G.; Dai, L.; Nakajima, T. The Hemolytic Activity of Six Arachnid Cationic Peptides Is Affected by the Phosphatidylcholine-to-Sphingomyelin Ratio in Lipid Bilayers. Biochim. Biophys. Acta (BBA) Biomembr. 2003, 1617, 22–30. [Google Scholar] [CrossRef]
- Virtanen, J.A.; Cheng, K.H.; Somerharju, P. Phospholipid Composition of the Mammalian Red Cell Membrane Can Be Rationalized by a Superlattice Model. Proc. Natl. Acad. Sci. USA 1998, 95, 4964. [Google Scholar] [CrossRef]
- Reynolds, N.L.; De Cecco, M.; Taylor, K.; Stanton, C.; Kilanowski, F.; Kalapothakis, J.; Seo, E.; Uhrin, D.; Campopiano, D.; Govan, J.; et al. Peptide Fragments of a β-Defensin Derivative with Potent Bactericidal Activity. Antimicrob. Agents Chemother. 2010, 54, 1922. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACE2-PEP-S2 | SPIKE-PEP-S2 | ||||
|---|---|---|---|---|---|
| H-Bonds | |||||
| Donor | Acceptor | Occupancy | Donor | Acceptor | Occupancy |
| Ala16-PEP | Asp30-ACE2 | 93.72% | Arg42-PEP | Glu484-SPIKE | 97.74% |
| Thr15-PEP | Asp30-ACE2 | 93.28% | Thr500-SPIKE | Asp31-PEP | 30.33% |
| Lys26-ACE2 | Asp31-PEP | 65.47% | Phe1-PEP | Asn501-SPIKE | 22.97% |
| Gly5-PEP | Ala387-ACE2 | 64.17% | Arg42-PEP | Gly482-SPIKE | 20.09% |
| Arg42-PEP | Glu37-ACE2 | 62.71% | Phe1-PEP | Gln498-SPIKE | 19.83% |
| Arg14-PEP | Asp30-ACE2 | 62.67% | Asn450-SPIKE | Met44-PEP | 19.13% |
| Trp4-PEP | Gln388-ACE2 | 36.05% | Asn39-PEP | Glu484-SPIKE | 17.19% |
| Lys353-ACE2 | Leu43-PEP | 27.77% | Gln498-SPIKE | Asp31-PEP | 14.91% |
| Tyr33-PEP | Asp30-ACE2 | 15.81% | Asn501-SPIKE | Phe1-PEP | 14.73% |
| Trp4-PEP | Ala387-ACE2 | 15.35% | Tyr33-PEP | Gln498-SPIKE | 12.20% |
| Thr15-PEP | Gly446-SPIKE | 11.32% | |||
| Asn39-PEP | Glu484-SPIKE | 10.58% | |||
| Salt Bridges | |||||
| Residues | Occupancy | Residues | Occupancy | ||
| Asp31-PEP/Lys26-ACE2 | 82.18% | Glu484-SPIKE/Arg42-PEP | 93.90% | ||
| Asp30-ACE2/Arg14-PEP | 68.21% | ||||
| Glu37-ACE2/Arg42-PEP | 61.13% | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barros, A.L.A.N.; Silva, V.C.; Ribeiro-Junior, A.F.; Cardoso, M.G.; Costa, S.R.; Moraes, C.B.; Barbosa, C.G.; Coleone, A.P.; Simões, R.P.; Cabral, W.F.; et al. Antiviral Action against SARS-CoV-2 of a Synthetic Peptide Based on a Novel Defensin Present in the Transcriptome of the Fire Salamander (Salamandra salamandra). Pharmaceutics 2024, 16, 190. https://doi.org/10.3390/pharmaceutics16020190
Barros ALAN, Silva VC, Ribeiro-Junior AF, Cardoso MG, Costa SR, Moraes CB, Barbosa CG, Coleone AP, Simões RP, Cabral WF, et al. Antiviral Action against SARS-CoV-2 of a Synthetic Peptide Based on a Novel Defensin Present in the Transcriptome of the Fire Salamander (Salamandra salamandra). Pharmaceutics. 2024; 16(2):190. https://doi.org/10.3390/pharmaceutics16020190
Chicago/Turabian StyleBarros, Ana Luisa A. N., Vladimir C. Silva, Atvaldo F. Ribeiro-Junior, Miguel G. Cardoso, Samuel R. Costa, Carolina B. Moraes, Cecília G. Barbosa, Alex P. Coleone, Rafael P. Simões, Wanessa F. Cabral, and et al. 2024. "Antiviral Action against SARS-CoV-2 of a Synthetic Peptide Based on a Novel Defensin Present in the Transcriptome of the Fire Salamander (Salamandra salamandra)" Pharmaceutics 16, no. 2: 190. https://doi.org/10.3390/pharmaceutics16020190