Antiretroviral Drug Interactions: Overview of Interactions Involving New and Investigational Agents and the Role of Therapeutic Drug Monitoring for Management

Abstract
: Antiretrovirals are prone to drug-drug and drug-food interactions that can result in subtherapeutic or supratherapeutic concentrations. Interactions between antiretrovirals and medications for other diseases are common due to shared metabolism through cytochrome P450 (CYP450) and uridine diphosphate glucuronosyltransferase (UGT) enzymes and transport by membrane proteins (e.g., p-glycoprotein, organic anion-transporting polypeptide). The clinical significance of antiretroviral drug interactions is reviewed, with a focus on new and investigational agents. An overview of the mechanistic basis for drug interactions and the effect of individual antiretrovirals on CYP450 and UGT isoforms are provided. Interactions between antiretrovirals and medications for other co-morbidities are summarized. The role of therapeutic drug monitoring in the detection and management of antiretroviral drug interactions is also briefly discussed.1. Introduction
The introduction of triple combination antiretroviral therapy has led to dramatic reductions in HIV-related morbidity and mortality [1]. Despite improvements in patient outcomes, selection of antiretroviral therapy remains challenging for clinicians due to resistance considerations, overlapping drug toxicities, and drug-drug and drug-food interactions. Drug-drug interactions among antiretrovirals are common and often require dose modification to mitigate unwanted adverse events and to sustain therapeutic concentrations. As HIV management has migrated from treatment of an acute infection to a chronic disease, the potential for antiretroviral drug interactions with medications for other chronic diseases has increased. It is important for clinicians to carefully consider the consequences of combining antiretrovirals with some drugs where the risk of adverse events or treatment failure may be increased due to unfavorable drug interactions.
This article provides an overview of common antiretroviral pharmacokinetic drug interactions, with an emphasis on newer antiretroviral agents introduced in the past five years and promising investigational agents. The mechanistic basis for drug interactions and metabolism of antiretroviral classes is briefly reviewed, followed by detailed discussion of the drug interaction potential between different antiretrovirals and with various drugs and drug classes. Recommendations for clinical management are also provided. The role of therapeutic drug monitoring in the detection and management of drug interactions is also discussed.
2. Mechanisms of Interaction
Drug interactions involving antiretrovirals can be classified as either pharmacokinetic or pharmacodynamic based on the mechanism of interaction. Pharmacokinetic interactions impact the absorption, distribution, metabolism, or excretion of antiretrovirals, whereas pharmacodynamic interactions result in synergistic, additive, or antagonistic drug response when they occur. Many antiretrovirals are substrates for transport proteins (e.g., organic anion-transporting polypeptide [OATP] 1B1, OATP1B3, and organic cation protein 1 [OCT1] in the liver; MDR1, breast cancer resistance protein [BCRP] and multidrug resistance protein 2 [MRP2] in the gut and liver) and can exhibit altered absorption, distribution, or excretion when coadministered with drugs that affect these proteins [2].
Interactions involving altered metabolism occur as a result of induction or inhibition of specific metabolic enzymes. Inhibition of metabolism most commonly results from competitive, non-competitive, or mechanism-based inhibition [3]. Competitive inhibition occurs when concentration of the inhibiting agent is sufficiently high to block metabolic conversion of the affected drug at the respective isoenzyme. Non-competitive inhibition occurs from allosteric inhibition, where binding to a site proximal to the catalytic binding site results in a conformational change in the catalytic site. Mechanism-based inhibition occurs when reactive intermediates bind irreversibly to the catalytic binding site [3]. Several antiretrovirals (ritonavir, amprenavir, nelfinavir, delavirdine) have been identified as mechanism-based inhibitors and are associated with clinically significant interactions with other drugs [4].
Induction of metabolism can occur when binding of drugs to nuclear receptors (e.g., pregnane X receptor [PXR], constitutive androstane receptor [CAR], hydrocarbon receptor) causes transcriptional factor activation, resulting in increased production of metabolic enzyme [5]. Individual protease inhibitors and NNRTIs have been found to be ligands for PXR and CAR, resulting in induction of specific isoenzymes or transport proteins (e.g., MDR1) that can decrease systemic exposure and increase the risk for therapeutic failure [6]. The timing of drug coadministration during pharmacokinetic interaction studies must be considered since agents that function as metabolic inducers at steady-state concentrations can exhibit inhibition during the first several weeks of therapy, leading to incorrect conclusions regarding their metabolic effects.
Pharmacogenetics can also determine whether a particular metabolic interaction is clinically significant. Polymorphic metabolism in CYP450 isoforms can influence the magnitude of change in serum concentration that is observed by either attenuating or magnifying the extent of reduction or increase. Cytochrome P450 (CYP) 3A4, CYP2B6, CYP2C9, CYPC19 and UGT all exhibit polymorphic metabolism and, therefore, can influence the significance of drug-drug interactions in specific patient populations [7–9].
5. Therapeutic Drug Monitoring to Manage Drug-Drug Interactions
5.1. Rationale for Therapeutic Drug Monitoring
Therapeutic drug monitoring (TDM) is the practice of dosing medications in response to plasma drug concentrations with the goal of maintaining concentrations within a clinically determined therapeutic target range. The aim is to optimize the clinical efficacy of a medication while minimizing or eliminating its concentration-dependent toxicities. In order for TDM to be valuable, a relationship between plasma concentrations and efficacy and/or toxicity must be established with a therapeutic agent. The role of TDM in regard to antiretrovirals continues to be largely undefined. Various small prospective and retrospective studies have displayed benefits in achieving virologic outcomes and minimizing toxicity when TDM is used in routine practice [157–159]. However, experts agree that large, well-designed trials are still needed to clearly delineate the role of TDM and to identify which patients may benefit most from its use [160,161]. One of the proposed populations where TDM may be advantageous is for patients at risk for clinically significant drug interactions [160]. Given that patients receiving antiretrovirals may experience complex, often unpredictable interactions with concomitant medications, the use of TDM can potentially allow examination and correction of altered serum concentrations prior to the development of virologic failure or adverse events.
The rationale for TDM in patients treated with antiretrovirals is primarily based on the large inter-patient variation in serum concentration that some agents exhibit when the same dose is administered to different patients [157,162,163]. This variation may be due to many factors including alteration in absorption, membrane transport, hepatic metabolism, and drug interactions. It follows that, if plasma drug concentrations can be reliably maintained within established ranges for efficacy and safety, clinicians can improve treatment response and limit toxicity risks for individual patients. Antiretroviral resistance development is associated with subtherapeutic drug levels and intermittent antiretroviral administration [164]. If clinicians are able to determine which patients are at risk for resistance development earlier in treatment (e.g., suboptimal drug concentrations due to erratic adherence from poor drug tolerability), dose alteration could potentially ameliorate drug intolerance and avoid the acquisition of antiretroviral resistance altogether. Another theoretical benefit of TDM is optimization of antiretroviral regimens in patients who have limited treatment options secondary to either extensive resistance or economic constraints. Adjusting doses to increase virologic suppression and limit toxicity might allow providers to use individual antiretrovirals longer without needing to change therapy.
The role of TDM for different antiretroviral classes varies. Protease inhibitors and NNRTIs display greater than 100-fold variation in pharmacokinetic parameters (AUC, Cmax, Cmin) associated with efficacy and toxicity end points [157,162,163,165]. This characteristic, combined with data supporting concentration-effect and concentration-toxicity relationships, makes protease inhibitors and NNRTIs the most likely candidates for TDM [157,161,166]. The CCR5 receptor antagonist, maraviroc, is a substrate for CYP3A4 and is highly susceptible to serum concentration changes when coadministered with strong CYP3A4 inducers or inhibitors. However, limited data exist on an observed association between maraviroc trough concentrations and virologic response to support it as a candidate for TDM [64,109,167]. The fusion inhibitor, enfuvirtide, and the integrase inhibitor, raltegravir, have relatively few drug interactions with other antiretrovirals or medications and do not utilize the CYP450 system for metabolism; therefore, no compelling role for TDM exists for them [165]. The clinical effect of NRTIs is related to the intracellular concentration of their active triphosphate; therefore, plasma concentrations are less predictive of clinical response or toxicity [168]. These agents also are predominantly eliminated renally and, with the exception of tenofovir, exhibit minimal potential for metabolic interactions, making plasma concentration monitoring of limited utility for NRTIs in general.
5.2. Barriers to Therapeutic Drug Monitoring
Barriers to routine application of TDM in clinical practice remain fairly formidable. Significant debate continues about which pharmacokinetic parameter(s) provides the best measure for drug efficacy or toxicity. Researchers have used a combination of Cmin, Cmax, AUC, and concentration ratio to assess efficacy and toxicity outcomes [169]. At the present time, the Cmin provides the best measure of virologic effect for most clinical scenarios without requiring complex calculations and population parameters [165]. However, definitive target values remain to be defined. Consensus guidelines have identified target trough concentrations for TDM in antiretroviral-naïve patients with wild-type virus; these values are listed in Table 3 [109,160,165,170]. It is not clear if individual targets are applicable to both antiretroviral-naïve and antiretroviral-experienced patients. In the case of antiretroviral-experienced patients, resistance mutations may increase the target antiretroviral serum concentration required to achieve viral suppression. Several studies have attempted to define the antiretroviral inhibitory quotient, which represents a combination of both pharmacokinetic/pharmacodynamic properties of the medication as well as virus-specific resistance information from either genotypic or phenotypic testing. Several different IQs have been proposed, with the PIQ (phenotype-based) and GIQ (genotype-based) discussed most predominately. The PIQ represents the ratio between the Cmin and the IC50 or IC90 measured by phenotypic resistance testing. The GIQ is defined as the ratio between Cmin and the number of resistance-associated mutations [161,165,169]. At the present time, no agreement exists on which inhibitory quotient is most closely tied to positive therapeutic outcomes. Preliminary target cutoffs have been proposed but require further study to correlate antiretroviral dose with viral response in patients with underlying resistance. It may be possible to overcome low- to mid-level resistance by utilizing IQ targets to individualize antiretroviral doses, giving clinicians an effective tool to achieve virologic success in highly treatment-experienced patients with numerous resistance mutations [161]. It is possible that targets will also vary based on the extent of synergy in individual antiretroviral combinations. Additionally, most protease inhibitors and NNRTIs are highly bound to plasma proteins; however, current targets are expressed as total plasma concentrations and do not correct for altered protein binding.
Laboratory validity is also a potential barrier to the clinical application of TDM results in different countries. Various methodologies can be employed by different clinical laboratories to measure antiretroviral plasma concentrations [160,171]. There is currently no standardized procedure or regulated quality control for determination of antiretroviral serum concentrations in laboratories in most commercial settings, making it difficult to apply specific laboratory results to proposed serum concentration targets established in research studies. Within the United States, the availability of reputable laboratories to perform antiretroviral serum concentration measurements is also limited, necessitating transport of patient samples that can extend turnaround time and decrease the clinical utility of laboratory results for patient management.
5.3. Role of Therapeutic Drug Monitoring for Drug-Drug Interactions
Despite the barriers that exist to widespread acceptance, clinical application of TDM has increased in the past few years, particularly in some European countries. In the United States, TDM continues to be primarily used in a research capacity. Treatment guidelines in several countries offer some limited information and guidance in the use of TDM [109,172–174]. There is uniform agreement between guidelines that routine application of TDM in patients treated with antiretrovirals is currently not recommended based on clinical evidence [109,169,172–174]. Most guidelines, including the United States Department of Health and Human Services Antiretroviral Treatment Guidelines [109], state that TDM may be useful in specific patient populations that could be at risk for subtherapeutic or supratherapeutic antiretroviral concentrations. One of the populations identified is patients with potential drug-drug interactions [109,161,169,172–174]. Two recent studies have investigated which patients may be at greater risk for experiencing drug-drug interactions. The factors identified include older patients (>42 years old), presence of more than three co-morbid conditions, treatment with more than five non-antiretroviral agents, antiretroviral therapy consisting of more than three antiretroviral medications, and use of either a protease inhibitor- or NNRTI-containing regimen [175,176]. In patient populations such as these that may be at increased risk for clinically significant drug-drug interactions, TDM may be prudent at the initiation of therapy or with medication changes to assure successful attainment of treatment outcomes. In addition, TDM would be advised for any patients with a high potential for drug-drug interactions that are experiencing unexpected clinical outcomes (e.g., sluggish viral load response, excessive adverse events). Therapeutic drug monitoring may provide insight into the cumulative effect on individual drug concentrations for antiretroviral regimens that involve multiple drug-drug interactions and whether antiretroviral doses are appropriate to achieve desired therapeutic end points. Practitioners who wish to perform TDM should keep in mind the limitations discussed and utilize guideline targets if possible. Expert consultation is advised for management of complex cases where extensive antiretroviral drug resistance is involved.
6. Conclusions
Combination antiretroviral therapy is highly effective in reducing HIV-related morbidity and mortality; however, clinicians must balance treatment outcomes with the potential for complex drug-drug interactions between antiretroviral and with medications for other chronic diseases. Antiretrovirals are prone to drug-drug interactions as a result of shared metabolism through CYP450 and UGT isoforms and binding to membrane transporters. Protease inhibitors and NNRTIs, including the newer agents darunavir and etravirine, commonly alter the metabolism of other medications. The clinical significance of drug interactions between rilpivirine, the newest NNRTI, and other antiretrovirals remains to be determined. Maraviroc is the only CCR5 receptor antagonist presently available and frequently requires dose modification with other antiretrovirals. The integrase inhibitor, raltegravir, and investigational agent, dolutegravir, are less prone to clinically significant drug interactions; whereas the investigational agent, elvitegravir, is more likely to exhibit interaction with other drugs because of coadministration with a pharmacokinetic enhancer (e.g., ritonavir, cobicistat). Important drug interactions exist between antiretrovirals and acid suppressants, antilipidemics, and medications for other chronic diseases. Therapeutic drug monitoring may represent a viable mechanism to identify and manage drug interactions in individual patients, but important barriers exist to widespread application in clinical practice.

{kind=link}
| Predicted Enzyme Effect | ||||||||
|---|---|---|---|---|---|---|---|---|
| Antiretroviral | 3A4 | 2B6 | 2C9 | 2C19 | 2D6 | 1A2 | UGT | |
| PIs | Atazanavir | – | –a | – | ||||
| Darunavir/r | –a | |||||||
| Fosamprenavir | – | –a | – | – | – | |||
| Indinavir | – | – | – | –a | – | |||
| Lopinavir/r | –a | |||||||
| Nelfinavir | – | – | ||||||
| Ritonavir | b | |||||||
| Saquinavir | – | –a | – | – | – | – | ||
| Tipranavir/r | ||||||||
| NNRTIs | Delavirdine | – | –a | – | – | |||
| Efavirenz | ||||||||
| Etravirine | – | – | ||||||
| Nevirapine | – | – | – | |||||
| Rilpivirine | * | * | * | |||||
| INSTI | Raltegravir | – | – | – | – | – | – | |
| Elvitegravir/r | * | * | * | * | * | |||
| Dolutegravir | * | * | * | * | * | |||
| CRA | Maraviroc | – | – | – | – | – | – | |
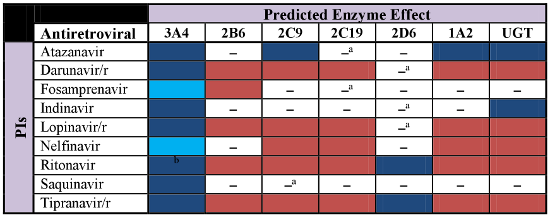
The predicted metabolic effects of antiretroviral agents on various cytochrome (CYP) P450 isoenzymes and uridine diphosphate glucuronosyltransferase (UGT) are illustrated according to the following:
 inhibition,
inhibition,
 induction,
induction,
 mixed induction/inhibition,
mixed induction/inhibition,
 substrate, [□] no significant effect, [*] not determined. The clinical significance of specific interactions between antiretrovirals and other drugs will be determined by the therapeutic and toxicity indices of the affected drug(s). The use of low-dose ritonavir for pharmacokinetic boosting is denoted by lowercase “/r” following individual antiretrovirals.aEnzyme not affected at clinically relevant antiretroviral concentrations.bAutoinduction of CYP3A4 by ritonavir is observed during the first 2 weeks of therapy, but CYP3A4 inhibition is most commonly evident with chronic therapy. PIs = protease inhibitors, NNRTIs = non-nucleoside reverse-transcriptase inhibitors, INSTIs = integrase strand transfer inhibitors, CRA = CCR5 receptor antagonist
substrate, [□] no significant effect, [*] not determined. The clinical significance of specific interactions between antiretrovirals and other drugs will be determined by the therapeutic and toxicity indices of the affected drug(s). The use of low-dose ritonavir for pharmacokinetic boosting is denoted by lowercase “/r” following individual antiretrovirals.aEnzyme not affected at clinically relevant antiretroviral concentrations.bAutoinduction of CYP3A4 by ritonavir is observed during the first 2 weeks of therapy, but CYP3A4 inhibition is most commonly evident with chronic therapy. PIs = protease inhibitors, NNRTIs = non-nucleoside reverse-transcriptase inhibitors, INSTIs = integrase strand transfer inhibitors, CRA = CCR5 receptor antagonist
| Medication | Antiretroviral | Predicted Effect | Management |
|---|---|---|---|
| Acid-Suppressants [106,107,109,110,115,149] | |||
| Antacids | Integrase Inhibitors | ⇩RAL, ETG, DTG |
|
| Atazanavir ± ritonavir | ⇩ATV | ||
| Fosamprenavir (unboosted) | ⇩APV | ||
| Tipranavir/ritonavir | ⇩TPV | ||
| H2-Receptor Antagonists (H2A) | Atazanavir/ritonavir | ⇩ATV |
|
| Atazanavir (unboosted) | ⇩ATV | ||
| Fosamprenavir (unboosted) | ⇩APV | ||
| Rilpivirine | ⇩RPV | ||
| Proton Pump Inhibitors (PPIs) | Atazanavir ± ritonavir | ⇩ATV |
|
| Indinavir (unboosted) | ⇩IDV | ||
| Nelfinavir | ⇩active metabolite (M8) | ||
| Saquinavir/ritonavir | ⇧SQV | ||
| Tipranavir/ritonavir | ⇩Omeprazole | ||
| Rilpivirine | ⇩RPV | ||
| Anticoagulants [13,109] | |||
| Clopidogrel | Etravirine | ⇩clopidogrel active metabolite |
|
| Warfarin | Boosted PIs, Nelfinavir | ⇩Warfarin |
|
| Unboosted PIs (except NFV) | ⇧/⇩Warfarin | ||
| Efavirenz, Etravirine, Delavirdine | ⇧Warfarin | ||
| Nevirapine | ⇩Warfarin | ||
| Anticonvulsants [109,150] | |||
| Carbamazepine (CBZ) | Boosted PIs (except DRV) | ⇩PI, ⇧CBZ |
|
| Darunavir/ritonavir | ⇧CBZ | ||
| Atazanavir, Fosamprenavir (unboosted) | ⇩PI | ||
| Efavirenz | ⇩EFV, ⇩CBZ | ||
| Lamotrigine | Boosted PIs | ⇩LMG |
|
| Phenytoin (PHT) | Boosted PIs | ⇩PHT, ⇩PI (⇧APV) |
|
| Atazanavir, Fosamprenavir (unboosted) | ⇩ATV, APV | ||
| Efavirenz, Etravirine | (⇧PHT) | ||
| Maraviroc | ⇩MVC | ||
| Valproic Acid (VPA) | Lopinavir/ritonavir | ⇩VPA, ⇧LPV |
|
| Zidovudine | ⇧ZDV | ||
| Corticosteroids [109,151] | |||
| Dexamethasone | PIs | ⇩PIs |
|
| Budesonide | Boosted PIs | ⇧Budesonide |
|
| Fluticasone (inhaled/intranasal) | PIs, Delavirdine | ⇧Fluticasone |
|
| Prednisone | Boosted PIs | ⇧Prednisolone |
|
| Triamcinolone | Boosted PIs | ⇧Prednisolone |
|
| Antifungals [68, 109,113,123–126,131,152] | |||
| Fluconazole | Tipranavir/ritonavir | ⇧TPV/r |
|
| Etravirine, Nevirapine | ⇧ETV, NVP | ||
| Itraconazole | PIs | (⇧ITZ, ⇧PI) |
|
| NNRTIs (except DLV) | ⇩ITZ, ⇧ETV, NVP | ||
| Maraviroc | (⇧MVC) | ||
| Posaconazole | Atazanavir ± ritonavir | ⇧ATV |
|
| Efavirenz | ⇩PCZ | ||
| Voriconazole | Boosted PIs | ⇩VCZ |
|
| Efavirenz | ⇩VCZ, ⇧EFV | ||
| Nevirapine, Rilpivirine | (⇩VCZ, ⇧NVP, RPV) | ||
| Ketoconazole | PIs | ⇧KTZ |
|
| Efavirenz, Nevirapine | ⇩KTZ, ⇧NVP | ||
| Etravirine, Rilpivirine | ⇩KTZ, ⇧ETV, RPV | ||
| Maraviroc | ⇧MVC | ||
| Caspofungin | Efavirenz, Nevirapine | ⇧Caspofungin |
|
| Antilipidemics [109,137–139] | |||
| Statins (Simvastatin, Lovastatin) | PIs | ⇧Statin |
|
| Efavirenz, Etravirine, Nevirapine | ⇩Statin | ||
| Rosuvastatin | Lopinavir/ritonavir | ⇧Rosuvastatin |
|
| Antimycobacterials [109,153–156] | |||
| Clarithromycin | Atazanavir ± ritonavir | ⇧Clarithromycin |
|
| Boosted PIs (except ATV) | ⇧Clarithromycin | ||
| NNRTIs (except DLV) | ⇩Clarithromycin | ||
| Maraviroc | ⇧MVC | ||
| Rifampin | PIs | ⇩PIs |
|
| Efavirenz | ⇩EFV | ||
| NNRTIs (except EFV) | ⇩NNRTIs | ||
| Raltegravir | ⇩RAL | ||
| Maraviroc | ⇩MVC | ||
| Rifabutin | Boosted PIs, Atazanavir (unboosted) | ⇧Rifabutin |
|
| Fosamprenavir, Indinavir (unboosted) | ⇧Rifabutin | ||
| Efavirenz | ⇩Rifabutin | ||
| Etravirine | ⇩Rifabutin, ⇩ETV | ||
| Rilpivirine, Delavirdine | ⇩RPV, DLV | ||
| Erectile Dysfunction Agents [109] | |||
| PDE5 Inhibitors (Sildenafil, Tadalafil, Vardenafil) | PIs, Delavirdine | ⇧PDE5 Inhibitor |
|
| Etravirine | ⇩PDE5 Inhibitor | ||
| Miscellaneous [18,109,141,142] | |||
| Levothyroxine | Boosted PIs (except ATV, IDV) | ⇩Levothyroxine |
|
| Atazanavir, Indinavir | ⇧Levothyroxine | ||
| Salmeterol | PIs ± ritonavir | ⇧Salmeterol |
|
APV = amprenavir, ART = antiretroviral therapy, ATV = atazanavir, CBZ = carbamazepine, DLV = delavirdine, DRV = darunavir, DTG = dolutegravir, FPV = fosamprenavir, EFV = efavirenz, ETG = elvitegravir, ETV = etravirine, FLU = fluconazole, IDV = indinavir, ITZ = itraconazole, KTZ = ketoconazole, LMG = lamotrigine, LPV = lopinavir, MVC = maraviroc, NNRTIs = non-nucleoside reverse-transcriptase inhibitors, NVP = nevirapine, PCZ = posaconazole, PDE5 = phosphodiesterase-5 inhibitors, PHT = phenytoin, PIs = protease inhibitors, RAL = raltegravir, RPV = rilpivirine, TPV = tipranavir, TSH = thyroid stimulating hormone, VCZ = voriconazole.
| Antiretroviral Drug | Minimum Trough Concentrations (Treatment-Naïve Patients) [ng/mL] | Minimum Trough Concentrations (Treatment-Experienced Patients) [ng/mL] |
|---|---|---|
| Atazanavir | 150 | |
| Darunavira | 3,300 | |
| Fosamprenavir | 400 | |
| Indinavir | 100 | |
| Lopinavir/ritonavir | 1,000 | |
| Nelfinavirb | 800 | |
| Ritonavir | ||
| Saquinavir | 100–250 | |
| Tipranavir | 20,500 | |
| Efavirenz | 1,000 | |
| Nevirapine | 3,000 | |
| Etravirinea | 275 | |
| Maraviroc | >50 | |
| Raltegravira | 72 |
aTarget serum concentrations represent median trough concentrations from clinical trials;bTarget serum concentrations represent the active metabolite (M8)
Conflict of Interest
The authors declare no conflict of interest.
References
- Palella, F.J., Jr.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N. Engl. J. Med. 1998, 338, 853–860. [Google Scholar]
- Zhang, L.; Huang, S.M.; Lesko, L.J. Transporter-mediated drug-drug interactions. Clin. Pharmacol. Ther. 2011, 89, 481–484. [Google Scholar]
- Pelkonen, O.; Turpeinen, M.; Hakkola, J.; Honkakoski, P.; Hukkanen, J.; Raunio, H. Inhibition and induction of human cytochrome P450 enzymes: current status. Arch. Toxicol. 2008, 82, 667–715. [Google Scholar]
- Zhou, S.; Yung Chan, S.; Cher Goh, B.; Chan, E.; Duan, W.; Huang, M.; McLeod, H.L. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin. Pharmacokinet. 2005, 44, 279–304. [Google Scholar]
- Tompkins, L.M.; Wallace, A.D. Mechanisms of cytochrome P450 induction. J. Biochem. Mol. Toxicol 2007, 21, 176–181. [Google Scholar]
- Svard, J.; Spiers, J.P.; Mulcahy, F.; Hennessy, M. Nuclear Receptor-Mediated Induction of CYP450 by Antiretrovirals: Functional Consequences of NR1I2 (PXR) Polymorphisms and Differential Prevalence in Whites and Sub-Saharan Africans. J. Acquir. Immune Defic. Syndr. 2010, 55, 536–539. [Google Scholar]
- Ingelman-Sundberg, M. Implications of polymorphic cytochrome p450-dependent drug metabolism for drug development. Drug Metab. Dispos. 2001, 29, 570–573. [Google Scholar]
- Gatanaga, H.; Hayashida, T.; Tsuchiya, K.; Yoshino, M.; Kuwahara, T.; Tsukada, H.; Fujimoto, K.; Sato, I.; Ueda, M.; Horiba, M.; et al. Successful efavirenz dose reduction in HIV type 1-infected individuals with cytochrome P450 2B6 *6 and *26. Clin. Infect. Dis. 2007, 45, 1230–1237. [Google Scholar]
- Bosma, P.J.; Chowdhury, J.R.; Bakker, C.; Gantla, S.; de Boer, A.; Oostra, B.A.; Lindhout, D.; Tytgat, G.N.; Jansen, P.L.; Oude Elferink, R.P.; et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert's syndrome. N. Engl. J. Med. 1995, 333, 1171–1175. [Google Scholar]
- Hsu, A.; Granneman, G.R.; Witt, G.; Locke, C.; Denissen, J.; Molla, A.; Valdes, J.; Smith, J.; Erdman, K.; Lyons, N.; Niu, P.; Decourt, J.P.; Fourtillan, J.B.; Girault, J.; Leonard, J.M. Multiple-dose pharmacokinetics of ritonavir in human immunodeficiency virus-infected subjects. Antimicrob. Agents Chemother. 1997, 41, 898–905. [Google Scholar]
- Kharasch, E.D.; Mitchell, D.; Coles, R.; Blanco, R. Rapid clinical induction of hepatic cytochrome P4502B6 activity by ritonavir. Antimicrob. Agents Chemother. 2008, 52, 1663–1669. [Google Scholar]
- Abbott Laboratories. Norvir (ritonavir) tablets and oral solution prescribing information. Available online: http://www.rxabbott.com/pdf/norvirtab_pi.pdf (accessed on 14 July 2011).
- Liedtke, M.D.; Rathbun, R.C. Drug interactions with antiretrovirals and warfarin. Expert Opin. Drug Saf. 2010, 9, 215–223. [Google Scholar]
- Kashuba, A.D.; Tierney, C.; Downey, G.F.; Acosta, E.P.; Vergis, E.N.; Klingman, K.; Mellors, J.W.; Eshleman, S.H.; Scott, T.R.; Collier, A.C. Combining fosamprenavir with lopinavir/ritonavir substantially reduces amprenavir and lopinavir exposure: ACTG protocol A5143 results. AIDS 2005, 19, 145–152. [Google Scholar]
- Corbett, A.H.; Patterson, K.B.; Tien, H.C.; Kalvass, L.A.; Eron, J.J.; Ngo, L.T.; Lim, M.L.; Kashuba, A.D. Dose separation does not overcome the pharmacokinetic interaction between fosamprenavir and lopinavir/ritonavir. Antimicrob. Agents Chemother. 2006, 50, 2756–2761. [Google Scholar]
- Hill, A.; van der Lugt, J.; Sawyer, W.; Boffito, M. How much ritonavir is needed to boost protease inhibitors? Systematic review of 17 dose-ranging pharmacokinetic trials. AIDS 2009, 23, 2237–2245. [Google Scholar]
- Zeldin, R.K.; Petruschke, R.A. Pharmacological and therapeutic properties of ritonavir-boosted protease inhibitor therapy in HIV-infected patients. J. Antimicrob. Chemother. 2004, 53, 4–9. [Google Scholar]
- Bristol-Myers Squibb. Reyataz (atazanavir sulfate) capsules prescribing information. Available online: http://us.gsk.com/products./us_viracept.pdf (accesssed on 14 July 2011).
- Sekar, V.; Spinosa-Guzman, S.; Meyvisch, P.; Stevens, T.; De Pauw, M.; Vangeneugden, T.; Hoetelmans, R. Cocktail study to investigate the in-vitro drug interaction potential of darunavir coadministered with low-dose ritonavir (DRV/r; RTV) on cytochrome P450 enzymes 2D6, 2C9 and 2C19. Presented at the 9th International Workshop on Clinical Pharmacology of HIV, New Orleans, LA, USA, 7–9 April 2008. Abstract P23.
- Veronese, L.; Rautaureau, J.; Sadler, B.M.; Gillotin, C.; Petite, J.P.; Pillegand, B.; Delvaux, M.; Masliah, C.; Fosse, S.; Lou, Y.; Stein, D.S. Single-dose pharmacokinetics of amprenavir, a human immunodeficiency virus type 1 protease inhibitor, in subjects with normal or impaired hepatic function. Antimicrob. Agents Chemother. 2000, 44, 821–826. [Google Scholar]
- Merck & Co., Inc. Crixivan (indinavir sulfate) capsules prescribing information. Available online: http://www.merck.com/product/usa/pi_circulars/c/crixivan/crixivan_pi.pdf (accessed on 19 July 2011).
- Yeh, R.F.; Gaver, V.E.; Patterson, K.B.; Rezk, N.L.; Baxter-Meheux, F.; Blake, M.J.; Eron, J.J., Jr.; Klein, C.E.; Rublein, J.C.; Kashuba, A.D. Lopinavir/ritonavir induces the hepatic activity of cytochrome P450 enzymes CYP2C9, CYP2C19, and CYP1A2 but inhibits the hepatic and intestinal activity of CYP3A as measured by a phenotyping drug cocktail in healthy volunteers. J. Acquir. Immune Defic. Syndr. 2006, 42, 52–60. [Google Scholar]
- Dixit, V.; Hariparsad, N.; Li, F.; Desai, P.; Thummel, K.E.; Unadkat, J.D. Cytochrome P450 enzymes and transporters induced by anti-human immunodeficiency virus protease inhibitors in human hepatocytes: implications for predicting clinical drug interactions. Drug Metab. Dispos. 2007, 35, 1853–1859. [Google Scholar]
- Agouron Pharmaceuticals Inc. Viracept [package insert]. Available online: http://us.gsk.com/products./us_viracept.pdf (accessed on 21 July 2011).
- Eagling, V.A.; Back, D.J.; Barry, M.G. Differential inhibition of cytochrome P450 isoforms by the protease inhibitors, ritonavir, saquinavir and indinavir. Br. J. Clin. Pharmacol. 1997, 44, 190–194. [Google Scholar]
- Dumond, J.B.; Vourvahis, M.; Rezk, N.L.; Patterson, K.B.; Tien, H.C.; White, N.; Jennings, S.H.; Choi, S.O.; Li, J.; Wagner, M.J.; et al. A phenotype-genotype approach to predicting CYP450 and P-glycoprotein drug interactions with the mixed inhibitor/inducer tipranavir/ritonavir. Clin. Pharmacol. Ther. 2010, 87, 735–742. [Google Scholar]
- Voorman, R.L.; Payne, N.A.; Wienkers, L.C.; Hauer, M.J.; Sanders, P.E. Interaction of delavirdine with human liver microsomal cytochrome P450: inhibition of CYP2C9, CYP2C19, and CYP2D6. Drug Metab. Dispos. 2001, 29, 41–47. [Google Scholar]
- Liu, P.; Foster, G.; LaBadie, R.R.; Gutierrez, M.J.; Sharma, A. Pharmacokinetic interaction between voriconazole and efavirenz at steady state in healthy male subjects. J. Clin. Pharmacol. 2008, 48, 73–84. [Google Scholar]
- Scholler-Gyure, M.; Kakuda, T.N.; Van Solingen-Ristea, R.; Aharchi, F.; De Smedt, G.; Witek, J.; Nijs, S.; Vyncke, V.; Hoetelmans, R.M.W. Pharmacokinetic interaction between etravirine and fluconazole or voriconazole in HIV-negative volunteers, Presented at the 49th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, CA, USA, 12–15 September, 2009. Abstract A1-1299.
- Ma, Q.; Okusanya, O.O.; Smith, P.F.; Dicenzo, R.; Slish, J.C.; Catanzaro, L.M.; Forrest, A.; Morse, G.D. Pharmacokinetic drug interactions with non-nucleoside reverse transcriptase inhibitors. Expert Opin. Drug Metab. Toxicol. 2005, 1, 473–485. [Google Scholar]
- Back, D.; Gibbons, S.; Khoo, S. Pharmacokinetic drug interactions with nevirapine. J. Acquir. Immune Defic. Syndr. 2003, 34, S8–S14. [Google Scholar]
- Crauwels, H.M.; van Heeswijk, R.P.G.; Stevens, T.; Stevens, M.; Buelens, A.; Boven, K.; Hoetelmans, R.M.W. The effect of TMC278, a next-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) on CYP3A activity in vivo. Presented at the 10th International Workshop on Clinical Pharmacology of HIV Therapy, Amsterdam, The Netherlands, 15–17 April 2009. Abstract P28.
- Merck Sharp & Dohme Corp. Isentress (raltegravir) tablets prescribing information. Available online: http://www.merck.com/product/usa/pi_circulars/i/isentress/isentress_pi.pdf (accessed on 21 July 2011).
- German, P.; Wang, M.; Warren, D.; Kearney, B.P. Pharmacokinetic interaction between norgestimate/ethinyl estradiol and elvitegravir/cobicistat/emtricitabine/tenofovir single tablet regimen. Presented at the 12th International Workshop on Clinical Pharmacology of HIV Therapy, Miami, FL, USA, 13–15 April 2011. Abstract O_17.
- Min, S.; Song, I.; Borland, J.; Chen, S.; Lou, Y.; Fujiwara, T.; Piscitelli, S.C. Pharmacokinetics and safety of S/GSK1349572, a next-generation HIV integrase inhibitor, in healthy volunteers. Antimicrob. Agents Chemother. 2010, 54, 254–258. [Google Scholar]
- Hyland, R.; Dickins, M.; Collins, C.; Jones, H.; Jones, B. Maraviroc: in vitro assessment of drug-drug interaction potential. Br. J. Clin. Pharmacol. 2008, 66, 498–507. [Google Scholar]
- Sekar, V.; Guzman, S.; Stevens, T.; Depaepe, E.; Lefebvre, E.; Hoetelmans, R. Absolute bioavailability of TMC114, administered in the absence and presence of low-dose ritonavir. Presented at the 7th International Workshop on Clinical Pharmacology of HIV Therapy, Lisbon, Portugal, 20–22 April 2006. Abstract P86.
- Sekar, V.; Spinosa-Guzman, S.; Lefebvre, E.; Hoetelmans, R. Clinical pharmacology of TMC114 a new HIV protease inhibitor. Presented at the 16th International AIDS Conference, Toronto, Canada, 13–18 August 2006. Abstract TUPE0083.
- Sekar, V.; Kestens, D.; Spinosa-Guzman, S.; De Pauw, M.; De Paepe, E.; Vangeneugden, T.; Lefebvre, E.; Hoetelmans, R.M. The effect of different meal types on the pharmacokinetics of darunavir (TMC114)/ritonavir in HIV-negative healthy volunteers. J. Clin. Pharmacol. 2007, 47, 479–484. [Google Scholar]
- Sekar, V.J.; De Pauw, M.; Marien, K.; Peeters, M.; Lefebvre, E.; Hoetelmans, R.M. Pharmacokinetic interaction between TMC114/r and efavirenz in healthy volunteers. Antivir. Ther. 2007, 12, 509–514. [Google Scholar]
- Sekar, V.; Lefebvre, E.; Marien, K.; De Pauw, M.; Vangeneugden, T.; Pozniak, A.; Hoetelmans, R.M. Pharmacokinetic interaction between nevirapine and darunavir with low-dose ritonavir in HIV-1-infected patients. Br. J. Clin. Pharmacol. 2009, 68, 116–119. [Google Scholar]
- Scholler-Gyure, M.; Kakuda, T.N.; Sekar, V.; Woodfall, B.; De Smedt, G.; Lefebvre, E.; Peeters, M.; Hoetelmans, R.M. Pharmacokinetics of darunavir/ritonavir and TMC125 alone and coadministered in HIV-negative volunteers. Antivir. Ther. 2007, 12, 789–796. [Google Scholar]
- Sekar, V.; Lefebvre, E.; Spinosa-Guzman, S.; Boogaerts, G.; Kestens, D.; De Pauw, M.; De Paepe, E.; Workman, C.; Hoetelmans, R. Pharmacokinetic interaction between the protease inhibitors TMC114 and lopinavir/ritonavir, Presented at the 46th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, CA, USA, 27–30 September 2006. Abstract A-367.
- Sekar, V.J.; Lefebvre, E.; Marien, K.; De Pauw, M.; Vangeneugden, T.; Hoetelmans, R.M. Pharmacokinetic interaction between darunavir and saquinavir in HIV-negative volunteers. Ther. Drug Monit. 2007, 29, 795–801. [Google Scholar]
- Sekar, V.J.; Lefebvre, E.; De Marez, T.; Spinosa-Guzman, S.; De Pauw, M.; De Paepe, E.; Vangeneugden, T.; Hoetelmans, R.M. Pharmacokinetics of darunavir (TMC114) and atazanavir during coadministration in HIV-negative, healthy volunteers. Drugs R. D. 2007, 8, 241–248. [Google Scholar]
- Sekar, V.; Lefebvre, E.; De Marez, T.; De Pauw, M.; De Paepe, E.; Vangeneugden, T.; Hoetelmans, R.M. Pharmacokinetic interaction between indinavir and darunavir with low-dose ritonavir in healthy volunteers. Intervirology 2010, 53, 176–182. [Google Scholar]
- Bristol-Myers Squibb. Sustiva (efavirenz) capsules and tablets prescribing information. Available online: http://packageinserts.bms.com/pi/pi_sustiva.pdf (accessed on 21 July 2011).
- Scholler-Gyure, M.; Boffito, M.; Pozniak, A.L.; Leemans, R.; Kakuda, T.N.; Woodfall, B.; Vyncke, V.; Peeters, M.; Vandermeulen, K.; Hoetelmans, R.M. Effects of different meal compositions and fasted state on the oral bioavailability of etravirine. Pharmacotherapy 2008, 28, 1215–1222. [Google Scholar]
- Crauwels, H.; van Heeswijk, R.P.; Bollen, A.; Stevens, M.; Buelens, A.; Boven, K.; Hoetelmans, R.M.W. The effect of different types of food on the bioavailability of TMC278, an investigational non-nucleoside reverse transcriptase inhibitor (NNRTI). Presented at the 9th International Workshop on Clinical Pharmacology of HIV Therapy, New Orleans, LA, USA, 7–9 April 2008. Abstract P32.
- von Moltke, L.L.; Greenblatt, D.J.; Granda, B.W.; Giancarlo, G.M.; Duan, S.X.; Daily, J.P.; Harmatz, J.S.; Shader, R.I. Inhibition of human cytochrome P450 isoforms by nonnucleoside reverse transcriptase inhibitors. J. Clin. Pharmacol. 2001, 41, 85–91. [Google Scholar]
- European Medicines Agency. CHMP Assessment Report for Intelence. 2008. Available online: http://www.emea.europa.eu/humandocs/PDFs/EPAR/intelence/H-900-en6.pdf (accessed on 1 March 2010). [Google Scholar]
- Hughes, C.A.; Robinson, L.; Tseng, A.; MacArthur, R.D. New antiretroviral drugs: a review of the efficacy, safety, pharmacokinetics, and resistance profile of tipranavir, darunavir, etravirine, rilpivirine, maraviroc, and raltegravir. Expert Opin. Pharmacother. 2009, 10, 2445–2466. [Google Scholar]
- Scholler-Gyure, M.; Kakuda, T.N.; Raoof, A.; De Smedt, G.; Hoetelmans, R.M. Clinical pharmacokinetics and pharmacodynamics of etravirine. Clin. Pharmacokinet. 2009, 48, 561–574. [Google Scholar]
- Scholler-Gyure, M.; Woodfall, B.; De Marez, T.; De Smedt, G.; Peeters, M.; Vandermeulen, K.; Hoetelmans, R. Pharmacokinetics of TMC125 with atazanavir and atazanavir/ritonavir. Presented at the 8th International Congress on Drug Therapy in HIV Infection, Glasgow, UK, 12–16 November 2006. Abstract P278.
- Scholler-Gyure, M.; Woodfall, B.; Bollen, A.; Peeters, M.; Vandermeulen, K.; Hoetelmans, R. Pharmacokinetics (pk) of amprenavir (APV) and TMC125 in HIV-infected volunteers receiving TMC125 with fosamprenavir/ritonavir (fosAPV/RTV), Presented at the 46th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, CA, USA, 27–30 September 2006. Abstract A-370.
- Mills, A.; Cahn, P.; Molina, J.M.; Nijs, S.; Vingerhoets, J.; Witek, J. Etravirine demonstrates durable efficacy in treatment-experienced patients in the DUET trials: pooled 96-week results. Presented at the 5th International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention Capetown, South Africa, 19–22 July 2009. Abstract MOPEB036.
- Baede, P.; Piscitelli, S.; Graham, N.; Van't Klooster, G. Drug interactions with TMC125, a potent next generation NNRTI. Presented at the 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy, San Diego, CA, USA, 27–30 September 2002. Abstract A-1827.
- Scholler-Gyure, M.; Kraft, M.; Hoetelmans, R.; Vyncke, V.; Vandermeulen, K.; Peeters, M.; Bastiaanse, L.; Debroye, C.; Woodfall, B.; Baeten, B. Significant decrease in TMC125 exposures when co-administered with tipranavir boosted with ritonavir in healthy subjects. Presented at the 13th Conference on Retroviruses and Opportunistic Infections, Denver, CO, USA, 5–8 February 2006. Abstract 583.
- Hoetelmans, R.; van Heeswijk, R.; Kestens, D.; Stevens, M.; Peeters, M.; Williams, P.; Bastlaanse, L.; Buffels, R.; Woodfall, B. Pharmacokinetic interaction between the novel non-nucleoside reverse transcriptase inhibitor (NNRTI) TMC278 and tenofovir disoproxil fumarate (TDF) in healthy volunteers. Presented at the 3rd International AIDS Society Conference on HIV Pathogenesis and Treatment, Rio de Janeiro, Brazil, 24–27 July 2005. Abstract WePe3.3C15.
- van Heeswijk, R.P.G.; Hoetelmans, R.M.W.; Kestens, D.; Stevens, M.; Peeters, M.; Williams, P.; Woodfall, B.; Boven, K. The pharmacokinetic (PK) interaction between TMC278, a next generation non-nucleoside reverse transcriptase inhibitor (NNRTI), and once daily darunavir/ritonavir (DRV/r) in HIV-negative volunteers. Presented at the 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 17–20 September 2007. Abstract H-1042.
- Hoetelmans, R.; van Heeswijk, R.; Kestens, D.; et al. Pharmacokinetic interaction between TMC278, and investigational NNRTI and lopinavir/r in healthy volunteers. Presented at the 10th European AIDS Conference, Dublin, Ireland, 17–20 November 2005. abstract PE4.3/1.
- Tibotec Therapeutics. Edurant (rilpivirine) tablets prescribing information. Available online: http://www.edurant-info.com/sites/default/files/EDURANT-PI.pdf (accessed on 21 July 2011).
- Pfizer, Inc. Maraviroc tablets NDA 22-128: Antiviral Drugs Advisory Committee briefing document. Available online: http://www.fda.gov/OHRMS/DOCKTES/AC/07/briefing/2007-4283b-01-Pfizer.pdf (accessed on 9 July 2011).
- ViiV Healthcare. Selzentry (maraviroc) tablets prescribing information. Available online: http://www.viivhealthcare.com/products/∼/media/Files/G/GlaxoSmithKline-Plc/Attachments/pdfs/products/us_selzentry_jul2011.pdf (accessed on 21 July 2011).
- Abel, S.; Jenkins, T.M.; Whitlock, L.A.; Ridgway, C.E.; Muirhead, G.J. Effects of CYP3A4 inducers with and without CYP3A4 inhibitors on the pharmacokinetics of maraviroc in healthy volunteers. Br. J. Clin. Pharmacol. 2008, 65, 38–46. [Google Scholar]
- Pozniak, A.L.; Boffito, M.; Russell, D.; Ridgway, C.E.; Muirhead, G.J. A novel probe drug interaction study to investigate the effect of selected antiretroviral combinations on the pharmacokinetics of a single oral dose of maraviroc in HIV-positive subjects. Br. J. Clin. Pharmacol. 2008, 65, 54–59. [Google Scholar]
- Kakuda, T.N.; Abel, S.; Davis, J.; Hamlin, J.; Scholler-Gyure, M.; Mack, R.; Ndongo, N.; Petit, W.; Ridgway, C.; Sekar, V.; Tweedy, S.; Hoetelmans, R.M. Pharmacokinetic interactions of maraviroc with darunavir-ritonavir, etravirine, and etravirine-darunavir-ritonavir in healthy volunteers: results of two drug interaction trials. Antimicrob. Agents Chemother. 2011, 55, 2290–2296. [Google Scholar]
- Abel, S.; Russell, D.; Taylor-Worth, R.J.; Ridgway, C.E.; Muirhead, G.J. Effects of CYP3A4 inhibitors on the pharmacokinetics of maraviroc in healthy volunteers. Br. J. Clin. Pharmacol. 2008, 65, 27–37. [Google Scholar]
- Ramanathan, S.; Abel, S.; Tweedy, S.; West, S.; Hui, J.; Kearney, B.P. Pharmacokinetic interaction of ritonavir-boosted elvitegravir and maraviroc. J. Acquir. Immune Defic. Syndr. 2010, 53, 209–214. [Google Scholar]
- Andrews, E.; Glue, P.; Fang, J.; Crownover, P.; Tressler, R.; Damle, B. Assessment of the pharmacokinetics of co-administered maraviroc and raltegravir. Br. J. Clin. Pharmacol. 2010, 69, 51–57. [Google Scholar]
- Kobayashi, M.; Yoshinaga, T.; Seki, T.; Wakasa-Morimoto, C.; Brown, K.W.; Ferris, R.; Foster, S.A.; Hazen, R.J.; Miki, S.; Suyama-Kagitani, A.; et al. In Vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob. Agents Chemother. 2011, 55, 813–821. [Google Scholar]
- Kassahun, K.; McIntosh, I.; Cui, D.; Hreniuk, D.; Merschman, S.; Lasseter, K.; Azrolan, N.; Iwamoto, M.; Wagner, J.A.; Wenning, L.A. Metabolism and disposition in humans of raltegravir (MK-0518), an anti-AIDS drug targeting the human immunodeficiency virus 1 integrase enzyme. Drug Metab. Dispos. 2007, 35, 1657–1663. [Google Scholar]
- Zhang, D.; Chando, T.J.; Everett, D.W.; Patten, C.J.; Dehal, S.S.; Humphreys, W.G. In vitro inhibition of UDP glucuronosyltransferases by atazanavir and other HIV protease inhibitors and the relationship of this property to in vivo bilirubin glucuronidation. Drug Metab. Dispos. 2005, 33, 1729–1739. [Google Scholar]
- Iwamoto, M.; Wenning, L.A.; Mistry, G.C.; Petry, A.S.; Liou, S.Y.; Ghosh, K.; Breidinger, S.; Azrolan, N.; Gutierrez, M.J.; Bridson, W.E.; et al. Atazanavir modestly increases plasma levels of raltegravir in healthy subjects. Clin. Infect. Dis. 2008, 47, 137–140. [Google Scholar]
- Cattaneo, D.; Ripamonti, D.; Baldelli, S.; Cozzi, V.; Conti, F.; Clementi, E. Exposure-related effects of atazanavir on the pharmacokinetics of raltegravir in HIV-1-infected patients. Ther. Drug Monit. 2010, 32, 782–786. [Google Scholar]
- Hanley, W.D.; Wenning, L.A.; Moreau, A.; Kost, J.T.; Mangin, E.; Shamp, T.; Stone, J.A.; Gottesdiener, K.M.; Wagner, J.A.; Iwamoto, M. Effect of tipranavir-ritonavir on pharmacokinetics of raltegravir. Antimicrob. Agents Chemother. 2009, 53, 2752–2755. [Google Scholar]
- Cooper, D.A.; Steigbigel, R.T.; Gatell, J.M.; Rockstroh, J.K.; Katlama, C.; Yeni, P.; Lazzarin, A.; Clotet, B.; Kumar, P.N.; Eron, J.E.; et al. Subgroup and resistance analyses of raltegravir for resistant HIV-1 infection. N. Engl. J. Med. 2008, 359, 355–365. [Google Scholar]
- Anderson, M.S.; Kakuda, T.N.; Hanley, W.; Miller, J.; Kost, J.T.; Stoltz, R.; Wenning, L.A.; Stone, J.A.; Hoetelmans, R.M.; Wagner, J.A.; Iwamoto, M. Minimal pharmacokinetic interaction between the human immunodeficiency virus nonnucleoside reverse transcriptase inhibitor etravirine and the integrase inhibitor raltegravir in healthy subjects. Antimicrob. Agents Chemother. 2008, 52, 4228–4232. [Google Scholar]
- Menard, A.; Solas, C.; Mokthari, S.; Bregigeon, S.; Drogoul, M.P.; Tamalet, C.; Lacarelle, B.; Martin, I.P. Etravirine-raltegravir, a marked interaction in HIV-1-infected patients: about four cases. AIDS 2009, 23, 869–871. [Google Scholar]
- Barrail-Tran, A.; Yazdanpanah, Y.; Fagard, C.; Colin, C.; Piketty, C.; Descamps, D.; Molina, J.M.; Chene, G.; Taburet, A.M. Lack of Interaction between Etravirine and Raltegravir plus Darunavir/Ritonavir When Combined in Treatment-experienced Patients: A Substudy of the ANRS 139 TRIO Trial. Presented at the 17th Conference on Retroviruses and Opportunistic Infections, San Francisco, CA, USA, 16–19 February 2010. Abstract 606.
- Wenning, L.; Anderson, M.; Petry, A.; Friedman, E.; Kost, J.; James, S.; Strohmaier, K.M.; Miller, D.; Merschman, S.; Bieberdorf, F.; et al. Raltegravir (RAL) dose proportionality and effect of food. Presented at the 47th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, CA, USA, 17–20 September 2007. Abstract H-1046.
- Ramanathan, S.; Mathias, A.A.; German, P.; Kearney, B.P. Clinical pharmacokinetic and pharmacodynamic profile of the HIV integrase inhibitor elvitegravir. Clin. Pharmacokinet. 2011, 50, 229–244. [Google Scholar]
- Kawaguchi, I.; Ishikawa, T.; Ishibashi, M.; Irie, S.; Kakee, A. Safety and pharmacokinetics of single oral dose of JTK-303/GS-9137, a novel HIV integrase inhibitor, in healthy volunteers. Presented at the 13th Conference on Retroviruses and Opportunistic Infections, Denver, CO, USA, 5–9 February 2006. Abstract 580.
- Ramanathan, S.; Shen, G.; Hinkle, J.; Enejosa, J.; Kearney, B.P. Pharmacokinetics of coadministered ritonavir-boosted elvitegravir and zidovudine, didanosine, stavudine, or abacavir. J. Acquir. Immune Defic. Syndr. 2007, 46, 160–166. [Google Scholar]
- Ramanathan, S.; Shen, G.; Cheng, A.; Kearney, B.P. Pharmacokinetics of emtricitabine, tenofovir, and GS-9137 following coadministration of emtricitabine/tenofovir disoproxil fumarate and ritonavir-boosted GS-9137. J. Acquir. Immune Defic. Syndr. 2007, 45, 274–279. [Google Scholar]
- Mathias, A.A.; Hinkle, J.; Shen, G.; Enejosa, J.; Piliero, P.J.; Sekar, V.; Mack, R.; Tomaka, F.; Kearney, B.P. Effect of ritonavir-boosted tipranavir or darunavir on the steady-state pharmacokinetics of elvitegravir. J. Acquir. Immune Defic. Syndr. 2008, 49, 156–162. [Google Scholar]
- Ramanathan, S.; Kakuda, T.N.; Mack, R.; West, S.; Kearney, B.P. Pharmacokinetics of elvitegravir and etravirine following coadministration of ritonavir-boosted elvitegravir and etravirine. Antivir. Ther. 2008, 13, 1011–1017. [Google Scholar]
- Ramanathan, S.; Mathias, A.A.; Shen, G.; Holmes, C.; Kearney, B.P. Lack of clinically relevant drug-drug interaction between ritonavir-boosted GS-9137 (elvitegravir) and fosamprenavir/r. Presented at the 4th IAS Conference on HIV Pathogenesis, Treatment and Prevention, Sydney, Australia, 22–25 July 2007. Abstract WEPEB014.
- Mathias, A.; West, S.; Enejosa, J.; Kearney, B. A pharmacokinetic interaction between lopinavir/r and elvitegravir. Presented at the 47th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 17–20 September 2007. Abstract A-1418.
- Mathias, A.; Ramanathan, S.; Hinkle, J.; West, S.; Enejosa, J.; Kearney, B. Effect of atazanavir/r on the steady-state pharmacokinetics of elvitegravir. Presented at the 47th Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 17–20 September 2007. Abstract A-1417.
- Ramanathan, S.; West, S.; Hui, J.; Chuck, S.L.; Kearney, B.P. Clinical pharmacokinetics of once-daily elvitegravir boosted by atazanavir versus ritonavir. Presented at the 9th International Workshop on Clinical Pharmacology of HIV Therapy, New Orleans, LA, USA, 7–9 April 2008. Abstract O18.
- Song, I.; Min, S.S.; Borland, J.; Lou, Y.; Chen, S.; Patel, P.; Ishibashi, T.; Piscitelli, S.C. The effect of lopinavir/ritonavir and darunavir/ritonavir on the HIV integrase inhibitor S/GSK1349572 in healthy participants. J. Clin. Pharmacol. 2011, 51, 237–242. [Google Scholar]
- Song, I.; Borland, J.; Chen, S.; Patel, P.; Wajima, T.; Peppercorn, A.; Piscitelli, S. Effect of food on the pharmacokinetics of the integrase inhibitor, dolutegravir (S/GSK1349572). Presented at the 12th International Workshop on Clinical Pharmacology of HIV Therapy, Miami, FL, USA, 13–15 May 2011. Abstract P_12.
- Song, I.; Borland, J.; Lou, Y.; Chen, S.; Patel, P.; Guta, P.; Wajima, T.; Peppercorn, A.; Piscitelli, S. Effects of enzyme inducers, tipranavir and efavirenz, on the pharmacokinetics of the integrase inhibitor, dolutegravir (S/GSK1349572). Presented at the 12th International Workshop on Clinical Pharmacology of HIV Therapy, Miami, FL, USA, 13–15 May 2011. Abstract O_02.
- Song, I.; Borland, J.; Chen, S.; Lou, Y.; Peppercorn, A.; Wajima, T.; Min, S.; Piscitelli, S.C. Effect of atazanavir and atazanavir/ritonavir on the pharmacokinetics of the next-generation HIV integrase inhibitor, S/GSK1349572. Br. J. Clin. Pharmacol. 2011, 72, 103–108. [Google Scholar]
- Song, I.; Borland, J.; Min, S.; Lou, Y.; Chen, S.; Patel, P.; Wajima, T.; Piscitelli, S.C. Effects of etravirine alone and with ritonavir-boosted protease inhibitors on the pharmacokinetics of dolutegravir. Antimicrob. Agents Chemother. 2011, 55, 3517–3521. [Google Scholar]
- Song, I.; Min, S.S.; Borland, J.; Lou, Y.; Chen, S.; Ishibashi, T.; Wajima, T.; Piscitelli, S.C. Lack of interaction between the HIV integrase inhibitor S/GSK1349572 and tenofovir in healthy subjects. J. Acquir. Immune Defic. Syndr. 2010, 55, 365–367. [Google Scholar]
- Hazuda, D.J.; Felock, P.; Witmer, M.; Wolfe, A.; Stillmock, K.; Grobler, J.A.; Espeseth, A.; Gabryelski, L.; Schleif, W.; Blau, C.; Miller, M.D. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 2000, 287, 646–650. [Google Scholar]
- Kiser, J.J.; Bumpass, J.B.; Meditz, A.L.; Anderson, P.L.; Bushman, L.; Ray, M.; Predhomme, J.A.; Rower, J.; Mawhinney, S.; Brundage, R. Effect of antacids on the pharmacokinetics of raltegravir in human immunodeficiency virus-seronegative volunteers. Antimicrob. Agents Chemother. 2010, 54, 4999–5003. [Google Scholar]
- Ramanathan, S.; Shen, G.; Hinkle, J.; Enejosa, J.; Kearney, B. Pharmacokinetic evaluation of drug interactions with ritonavir-boosted HIV integrase inhibitor GS-9137 (elvitegravir) and acid-reducing agents. Presented at the 8th International Workshop on Clinical Pharmacology of HIV Therapy, Budapest, Hungary, 16–18 April 2007. Abstract 69.
- Patel, P.; Song, I.; Borland, J.; Patel, A.; Lou, Y.; Chen, S.; Wajima, T.; Peppercorn, A.; Min, S.S.; Piscitelli, S.C. Pharmacokinetics of the HIV integrase inhibitor S/GSK1349572 co-administered with acid-reducing agents and multivitamins in healthy volunteers. J. Antimicrob. Chemother. 2011, 66, 1567–1572. [Google Scholar]
- van Heeswijk, R.; Sabo, J.P.; Copper, C.; Cameron, W.; MacGregor, T.R.; Elgadi, M.; Harris, F.; McCallister, S.; Mayers, D. The pharmacokinetic interactions between tipranavir/ritonavir 500 mg/200 mg bid (TPV/r) and atorvastatin, antacid and CYP3A4 in healthy volunteers. In Abstract 52, Presented at the 5th International workshop on Clinical Pharmacology in HIV Therapy, Rome, Italy, 1–3 April 2004.
- Kiser, J.J.; Lichtenstein, K.A.; Anderson, P.L.; Fletcher, C.V. Effects of esomeprazole on the pharmacokinetics of atazanavir and fosamprenavir in a patient with human immunodeficiency virus infection. Pharmacotherapy 2006, 26, 511–514. [Google Scholar]
- Ford, S.L.; Wire, M.B.; Lou, Y.; Baker, K.L.; Stein, D.S. Effect of antacids and ranitidine on the single-dose pharmacokinetics of fosamprenavir. Antimicrob. Agents Chemother. 2005, 49, 467–469. [Google Scholar]
- Lin, J.H.; Chen, I.W.; Vastag, K.J.; Ostovic, D. pH-dependent oral absorption of L-735,524, a potent HIV protease inhibitor, in rats and dogs. Drug Metab. Dispos. 1995, 23, 730–735. [Google Scholar]
- van Heeswijk, R.P.; Hoetelmans, R.M.; Kestens, D.; Stevens, M.; Peeters, M.; Williams, P.; Woodfall, B.; Boven, K. The pharmacokinetic (PK) interaction between famotidine and TMC278, a next generation non-nucleoside reverse transcriptase inhibitor (NNRTI), in HIV-negative volunteers, Presented at the 4th International AIDS Society Conference on HIV Pathogenesis, Treatment, and Prevention, Sydney, Australia,, 22–25 July 2007. Abstract TUPDB01.
- Crauwels, H.M.; van Heeswijk, R.P.G.; Kestens, D.; Stevens, M.; Buelens, A.; Boven, K.; Hoetelmans, R.M.W. The pharmacokinetic (PK) interaction between omeprazole and TMC278, an investigational non-nucleoside reverse transcriptase inhibitor (NNRTI). Presented at the 9th International Congress on Drug Therapy in HIV Infection, Glasgow, UK, 9– 13 November 2008. Abstract P239.
- Shelton, M.J.; Ford, S.L.; Borland, J.; Lou, Y.; Wire, M.B.; Min, S.S.; Xue, Z.G.; Yuen, G. Coadministration of esomeprazole with fosamprenavir has no impact on steady-state plasma amprenavir pharmacokinetics. J. Acquir. Immune Defic. Syndr. 2006, 42, 61–67. [Google Scholar]
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. Available online: http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf (accessed on 1 July 2011).
- Tappouni, H.L.; Rublein, J.C.; Donovan, B.J.; Hollowell, S.B.; Tien, H.C.; Min, S.S.; Theodore, D.; Rezk, N.L.; Smith, P.C.; Tallman, M.N.; Raasch, R.H.; Kashuba, A.D. Effect of omeprazole on the plasma concentrations of indinavir when administered alone and in combination with ritonavir. Am. J. Health Syst. Pharm. 2008, 65, 422–428. [Google Scholar]
- Iwamoto, M.; Wenning, L.A.; Nguyen, B.Y.; Teppler, H.; Moreau, A.R.; Rhodes, R.R.; Hanley, W.D.; Jin, B.; Harvey, C.M.; Breidinger, S.A.; Azrolan, N.; Farmer, H.F., Jr.; Isaacs, R.D.; Chodakewitz, J.A.; Stone, J.A.; Wagner, J.A. Effects of omeprazole on plasma levels of raltegravir. Clin. Infect. Dis. 2009, 48, 489–492. [Google Scholar]
- Rhame, F.; Matson, M.; Wood, D.; Comisar, W.; Petry, A.; Liu, C.; Wenning, L.; Wagner, J.; Iwamoto, M.; Brainard, D. Effects of famotidine and omeprazole on raltegravir pharmacokinetics in HIV-infected individuals. Presented at the 12th European AIDS Conference, 11–14 November 2009, Abstract PE4.1/1. Cologne, Germany.
- Boehringer Ingelheim Pharmaceuticals, Inc. Aptivurs (tipranavir) capsules prescribing information. Available online: http://bidocs.boehringer-ingelheim.com/BIWebAccess/ViewServlet.ser?docBase=renetnt&folderPath=/Prescribing+Information/PIs/Aptivus/10003515+US+01.pdf (accessed on 28 July 2011).
- Saberi, P.; Ranatunga, D.K.; Quesenberry, C.P.; Silverberg, M.J. Clinical implications of the nelfinavir-proton pump inhibitor drug interaction in patients with human immunodeficiency virus. Pharmacotherapy 2011, 31, 253–261. [Google Scholar]
- Fang, A.F.; Damle, B.D.; LaBadie, R.R.; Crownover, P.H.; Hewlett, D., Jr.; Glue, P.W. Significant decrease in nelfinavir systemic exposure after omeprazole coadministration in healthy subjects. Pharmacotherapy 2008, 28, 42–50. [Google Scholar]
- Sekar, V.J.; Lefebvre, E.; De Pauw, M.; Vangeneugden, T.; Hoetelmans, R.M. Pharmacokinetics of darunavir/ritonavir and ketoconazole following co-administration in HIV-healthy volunteers. Br. J. Clin. Pharmacol. 2008, 66, 215–221. [Google Scholar]
- Abbott Laboratories. Kaletra (lopinavir/ritonavir) tablets and oral solution prescribing information. Available online: http://www.rxabbott.com/pdf/kaletratabpi.pdf (accessed on 28 July 2011).
- Bertz, R.; Wong, C.; Carothers, L.; Lauva, I.; Dennis, S.; Valdes, J. Evaluation of the pharmacokinetics of multiple dose ritonavir and ketoconazole in combination. Clin. Pharmacol. Ther. 1998, 63, 230. [Google Scholar]
- Viiv Healthcare. Lexiva (fosamprenavir calcium) tablets and oral suspension prescribing information. Available online: http://us.gsk.com/products./us_lexiva.pdf (accessed on 8 September 2011).
- Crommentuyn, K.M.; Mulder, J.W.; Sparidans, R.W.; Huitema, A.D.; Schellens, J.H.; Beijnen, J.H. Drug-drug interaction between itraconazole and the antiretroviral drug lopinavir/ritonavir in an HIV-1-infected patient with disseminated histoplasmosis. Clin. Infect. Dis. 2004, 38, e73–e75. [Google Scholar]
- Pfizer-Roerig. Vfend (voriconazole) tablets, oral suspension, and IV prescribing information. Available online: http://labeling.pfizer.com/ShowLabeling.aspx?id=618 (accessed on 8 September 2011).
- Damle, B.; LaBadie, R.; Crownover, P.; Glue, P. Pharmacokinetic interactions of efavirenz and voriconazole in healthy volunteers. Br. J. Clin. Pharmacol. 2008, 65, 523–530. [Google Scholar]
- Kaul, S.; Ji, P.; Dudley, J.; Wu, W.; Whigan, D.; Olszyk, C.; Nandy, P.; Hughes, E.; Grasela, D. Pharmacokinetic interaction between efavirenz and diltiazem or itraconazole after multiple-dose administration in adult healthy subjects. Presented at the 14th Conference on Retroviruses and Opportunistic Infections, Los Angeles, CA, USA, 25–28 February 2007. Abstract 561.
- Koo, H.L.; Hamill, R.J.; Andrade, R.A. Drug-drug interaction between itraconazole and efavirenz in a patient with AIDS and disseminated histoplasmosis. Clin. Infect. Dis. 2007, 45, e77–e79. [Google Scholar]
- Huet, E.; Hadji, C.; Hulin, A.; Botterel, F.; Bretagne, S.; Levy, Y. Therapeutic monitoring is necessary for the association itraconazole and efavirenz in a patient with AIDS and disseminated histoplasmosis. AIDS 2008, 22, 1885–1886. [Google Scholar]
- Sriwiriyajan, S.; Mahatthanatrakul, W.; Ridtitid, W.; Jaruratanasirikul, S. Effect of efavirenz on the pharmacokinetics of ketoconazole in HIV-infected patients. Eur. J. Clin. Pharmacol. 2007, 63, 479–483. [Google Scholar]
- Krishna, G.; Moton, A.; Ma, L.; Martinho, M.; Seiberling, M.; McLeod, J. Effects of oral posaconazole on the pharmacokinetics of atazanavir alone and with ritonavir or with efavirenz in healthy adult volunteers. J. Acquir. Immune. Defic. Syndr. 2009, 51, 437–444. [Google Scholar]
- la Porte, C.J.; Sabo, J.P.; Elgadi, M.; Cameron, D.W. Interaction studies of tipranavir-ritonavir with clarithromycin, fluconazole, and rifabutin in healthy volunteers. Antimicrob. Agents Chemother. 2009, 53, 162–173. [Google Scholar]
- Agarwala, S.; Gray, K.; Nettle, R.; et al. Lack of pharmacokinetic interaction between atazanavir, ritonavir and fluconazole dosed to steady state in healthy volunteers; Presented at the 46th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, CA, USA, 27–30 September 2006, Abstract A-382.
- Geel, J.; Pitt, J.; Orrell, C.; van Dyk, M.; Wood, R. Effect of fluconazole on nevirapine pharmacokinetics. Presented at the 11th International AIDS Conference, Bangkok, Thailand, 11–16 July 2004. Abstract TuPeB4606.
- Stone, J.A.; Li, S.; Winchell, G.; Bi, S.; Wickersham, P.; Schwartz, M.; Kartsonis, N.; Sable, C. Population Pharmacokinetics of Caspofungin in Candidiasis Patients. Presented at the 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 14–17 September 2003. Abstract A-1571.
- Dube, M.P.; Stein, J.H.; Aberg, J.A.; Fichtenbaum, C.J.; Gerber, J.G.; Tashima, K.T.; Henry, W.K.; Currier, J.S.; Sprecher, D.; Glesby, M.J. Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medical Association of the Infectious Disease Society of America and the Adult AIDS Clinical Trials Group. Clin. Infect. Dis. 2003, 37, 613–627. [Google Scholar]
- Fichtenbaum, C.J.; Gerber, J.G.; Rosenkranz, S.L.; Segal, Y.; Aberg, J.A.; Blaschke, T.; Alston, B.; Fang, F.; Kosel, B.; Aweeka, F. Pharmacokinetic interactions between protease inhibitors and statins in HIV seronegative volunteers: ACTG Study A5047. AIDS 2002, 16, 569–577. [Google Scholar]
- Busti, A.J.; Bain, A.M.; Hall, R.G., 2nd; Bedimo, R.G.; Leff, R.D.; Meek, C.; Mehvar, R. Effects of atazanavir/ritonavir or fosamprenavir/ritonavir on the pharmacokinetics of rosuvastatin. J. Cardiovasc. Pharmacol. 2008, 51, 605–610. [Google Scholar]
- Carr, R.A.; Andre, R.J.; Bertz, R.J.; Hsu, A.; Lam, W.; Chang, M.; Chen, P.; Williams, L.; Bernstein, B.; Sun, E. Concomitant administration of ABT 378/ritonavir results in clinically important pharmacokinetic interaction with atorvastatin but not pravastatin. Presented at the 40th Interscience Conference on Antimicrobial Agents and Chemotherapy, Toronto, Ontario, Canada, 17–20 September 2000. Abstract 334.
- Samineni, D.; Desai, P.B.; Sallans, L.; Fichtenbaum, C.J. Steady-State Pharmacokinetic Interactions of Darunavir/Ritonavir With Lipid-Lowering Agent Rosuvastatin. J. Clin. Pharmacol. 2011. in press. [Google Scholar]
- Kiser, J.J.; Gerber, J.G.; Predhomme, J.A.; Wolfe, P.; Flynn, D.M.; Hoody, D.W. Drug/Drug interaction between lopinavir/ritonavir and rosuvastatin in healthy volunteers. J. Acquir. Immune Defic. Syndr. 2008, 47, 570–578. [Google Scholar]
- Kowa Pharmaceuticals America, Inc. Livalo (pitavastatin) tablets prescribing information. Available online: http://www.kowapharma.com/documents/LIVALO_PI_CURRENT.pdf (accessed on 8 September 2011).
- Morgan, R.; Campbell, S.; Suehira, K.; Sponseller, C.; Yu, C.; Medlock, M. Effects of steady-state lopinavir/ritonavir on the pharmacokinetics of pitavastatin in healthy adult volunteers, Presented at the 6th International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention, Rome, Italy, 17–20 July 2011. Abstract MOPE 170.
- Gerber, J.G.; Rosenkranz, S.L.; Fichtenbaum, C.J.; Vega, J.M.; Yang, A.; Alston, B.L.; Brobst, S.W.; Segal, Y.; Aberg, J.A. Effect of efavirenz on the pharmacokinetics of simvastatin, atorvastatin, and pravastatin: results of AIDS Clinical Trials Group 5108 Study. J. Acquir. Immune Defic. Syndr. 2005, 39, 307–312. [Google Scholar]
- Touzot, M.; Beller, C.L.; Touzot, F.; Louet, A.L.; Piketty, C. Dramatic interaction between levothyroxine and lopinavir/ritonavir in a HIV-infected patient. AIDS 2006, 20, 1210–1212. [Google Scholar]
- Lanzafame, M.; Trevenzoli, M.; Faggian, F.; Marcati, P.; Gatti, F.; Carolo, G.; Concia, E. Interaction between levothyroxine and indinavir in a patient with HIV infection. Infection 2002, 30, 54–55. [Google Scholar]
- Gupta, S.K.; Dube, M.P. Exogenous cushing syndrome mimicking human immunodeficiency virus lipodystrophy. Clin. Infect. Dis. 2002, 35, E69–E71. [Google Scholar]
- Hillebrand-Haverkort, M.E.; Prummel, M.F.; ten Veen, J.H. Ritonavir-induced Cushing's syndrome in a patient treated with nasal fluticasone. AIDS 1999, 13, 1803. [Google Scholar]
- GlaxoSmithKline. Flonase (fluticasone propionate) nasal spray prescribing information. Research Available online: http://us.gsk.com/products./us_flonase.pdf (accessed on 28 July 2011).
- Kedem, E.; Shahar, E.; Hassoun, G.; Pollack, S. Iatrogenic Cushing's syndrome due to coadministration of ritonavir and inhaled budesonide in an asthmatic human immunodeficiency virus infected patient. J. Asthma 2010, 47, 830–831. [Google Scholar]
- Penzak, S.R.; Formentini, E.; Alfaro, R.M.; Long, M.; Natarajan, V.; Kovacs, J. Prednisolone pharmacokinetics in the presence and absence of ritonavir after oral prednisone administration to healthy volunteers. J. Acquir. Immune Defic. Syndr. 2005, 40, 573–580. [Google Scholar]
- GlaxoSmithKline. Serevent diskus (salmeterol xinafoate inhalation powder). Available online: http://us.gsk.com/products./us_serevent_diskus.pdf (accessed on 28 July 2011).
- Winston, A.; Back, D.; Fletcher, C.; Robinson, L.; Unsworth, J.; Tolowinska, I.; Schutz, M.; Pozniak, A.L.; Gazzard, B.; Boffito, M. Effect of omeprazole on the pharmacokinetics of saquinavir-500 mg formulation with ritonavir in healthy male and female volunteers. AIDS 2006, 20, 1401–1406. [Google Scholar]
- Lertora, J.J.; Rege, A.B.; Greenspan, D.L.; Akula, S.; George, W.J.; Hyslop, N.E., Jr.; Agrawal, K.C. Pharmacokinetic interaction between zidovudine and valproic acid in patients infected with human immunodeficiency virus. Clin. Pharmacol. Ther. 1994, 56, 272–278. [Google Scholar]
- Dort, K.; Padia, S.; Wispelwey, B.; Moore, C.C. Adrenal suppression due to an interaction between ritonavir and injected triamcinolone: a case report. AIDS Res. Ther. 2009, 6, 10. [Google Scholar]
- van Heeswijk, R.; Hoetelmans, R.; Kestens, D.; Stevens, M.; Peeters, M.; Williams, P.; Woodfall, B.; Boven, K. The pharmacokinetic interaction between ketoconazole and TMC278, an investigational non-nucleoside reverse transcriptase inhibitor (NNRTI), in healthy HIV-negative subjects. Presented at the XVI International AIDS Conference, Toronto, Ontario, Canada, 13–18 August 2006. Abstract TUPE0087.
- Hamzeh, F.M.; Benson, C.; Gerber, J.; Currier, J.; McCrea, J.; Deutsch, P.; Ruan, P.; Wu, H.; Lee, J.; Flexner, C. Steady-state pharmacokinetic interaction of modified-dose indinavir and rifabutin. Clin. Pharmacol. Ther. 2003, 73, 159–169. [Google Scholar]
- Borin, M.T.; Chambers, J.H.; Carel, B.J.; Gagnon, S.; Freimuth, W.W. Pharmacokinetic study of the interaction between rifampin and delavirdine mesylate. Clin. Pharmacol. Ther. 1997, 61, 544–553. [Google Scholar]
- Borin, M.T.; Chambers, J.H.; Carel, B.J.; Freimuth, W.W.; Aksentijevich, S.; Piergies, A.A. Pharmacokinetic study of the interaction between rifabutin and delavirdine mesylate in HIV-1 infected patients. Antiviral Res 1997, 35, 53–63. [Google Scholar]
- Crauwels, H.M.; van Heeswijk, R.P.G.; Kestens, D.; Stevens, M.; Buelens, A.; Boven, K.; hoetelmans, R.M.W. The pharmacokinetic interaction between rifabutin and TMC278, an investigational NNRTI. Presented at the XVIIth International AIDS Conference, Mexico City, Mexico, 3–8 August 2008. Abstract TUPE0080.
- Marzolini, C.; Telenti, A.; Decosterd, L.A.; Greub, G.; Biollaz, J.; Buclin, T. Efavirenz plasma levels can predict treatment failure and central nervous system side effects in HIV-1-infected patients. AIDS 2001, 15, 71–75. [Google Scholar]
- Fletcher, C.V.; Anderson, P.L.; Kakuda, T.N.; Schacker, T.W.; Henry, K.; Gross, C.R.; Brundage, R.C. Concentration-controlled compared with conventional antiretroviral therapy for HIV infection. AIDS 2002, 16, 551–560. [Google Scholar]
- Burger, D.; Hugen, P.; Reiss, P.; Gyssens, I.; Schneider, M.; Kroon, F.; Schreij, G.; Brinkman, K.; Richter, C.; Prins, J.; Aarnoutse, R.; Lange, J. Therapeutic drug monitoring of nelfinavir and indinavir in treatment-naive HIV-1-infected individuals. AIDS 2003, 17, 1157–1165. [Google Scholar]
- Acosta, E.P.; Gerber, J.G. Position paper on therapeutic drug monitoring of antiretroviral agents. AIDS Res. Hum. Retrovirus. 2002, 18, 825–834. [Google Scholar]
- LaPorte, C.J.L.; Back, B.J.; Blaschke, T.; Boucher, C.A.B.; Fletcher, C.V.; Flexner, C.; Gerber, J.G.; Kashuba, A.D.M.; Schapiro, J.; Burger, D.M. Updated guideline to perform therapeutic drug monitoring for antiretroviral agents. Rev. Antivir. Ther. 2006, 3, 4–14. [Google Scholar]
- Acosta, E.P.; Henry, K.; Baken, L.; Page, L.M.; Fletcher, C.V. Indinavir concentrations and antiviral effect. Pharmacotherapy 1999, 19, 708–712. [Google Scholar]
- Marzolini, C.; Buclin, T.; Decosterd, L.A.; Biollaz, J.; Telenti, A. Nelfinavir plasma levels under twice-daily and three-times-daily regimens: high interpatient and low intrapatient variability. Ther. Drug Monit. 2001, 23, 394–398. [Google Scholar]
- Gardner, E.M.; Burman, W.J.; Steiner, J.F.; Anderson, P.L.; Bangsberg, D.R. Antiretroviral medication adherence and the development of class-specific antiretroviral resistance. AIDS 2009, 23, 1035–1046. [Google Scholar]
- Pretorius, E.; Klinker, H.; Rosenkranz, B. The role of therapeutic drug monitoring in the management of patients with human immunodeficiency virus infection. Ther. Drug Monit. 2011, 33, 265–274. [Google Scholar]
- Veldkamp, A.I.; Weverling, G.J.; Lange, J.M.; Montaner, J.S.; Reiss, P.; Cooper, D.A.; Vella, S.; Hall, D.; Beijnen, J.H.; Hoetelmans, R.M. High exposure to nevirapine in plasma is associated with an improved virological response in HIV-1-infected individuals. AIDS 2001, 15, 1089–1095. [Google Scholar]
- McFayden, L.; Jacqmin, P.; Wade, J.; Weatherley, B. Maraviroc exposure response analysis: phase 3 antiviral efficacy in treatment-experienced HIV+ patients. Presented at the 16th Population Approach Group in Europe Meeting, Kobenhavn, Denmark, 13–15 June 2007. Abstract 1172.
- Boffito, M.; Acosta, E.; Burger, D.; Fletcher, C.V.; Flexner, C.; Garaffo, R.; Gatti, G.; Kurowski, M.; Perno, C.F.; Peytavin, G.; Regazzi, M.; Back, D. Therapeutic drug monitoring and drug-drug interactions involving antiretroviral drugs. Antivir. Ther. 2005, 10, 469–477. [Google Scholar]
- Boffito, M.; Acosta, E.; Burger, D.; Fletcher, C.V.; Flexner, C.; Garaffo, R.; Gatti, G.; Kurowski, M.; Perno, C.F.; Peytavin, G.; Regazzi, M.; Back, D. Current status and future prospects of therapeutic drug monitoring and applied clinical pharmacology in antiretroviral therapy. Antivir. Ther. 2005, 10, 375–392. [Google Scholar]
- Kappelhoff, B.S.; Crommentuyn, K.M.; de Maat, M.M.; Mulder, J.W.; Huitema, A.D.; Beijnen, J.H. Practical guidelines to interpret plasma concentrations of antiretroviral drugs. Clin. Pharmacokinet. 2004, 43, 845–853. [Google Scholar]
- Ivanovic, J.; Nicastri, E.; Ascenzi, P.; Bellagamba, R.; De Marinis, E.; Notari, S.; Pucillo, L.P.; Tozzi, V.; Ippolito, G.; Narciso, P. Therapeutic drug monitoring in the management of HIV-infected patients. Curr. Med. Chem. 2008, 15, 1925–1939. [Google Scholar]
- Thompson, M.A.; Aberg, J.A.; Cahn, P.; Montaner, J.S.; Rizzardini, G.; Telenti, A.; Gatell, J.M.; Gunthard, H.F.; Hammer, S.M.; Hirsch, M.S.; et al. Antiretroviral treatment of adult HIV infection: 2010 recommendations of the International AIDS Society-USA panel. JAMA 2010, 304, 321–333. [Google Scholar]
- European AIDS Clinical Society Treatment Guidelines. Available online: http://www.europeanaidsclinicalsociety.org/images/stories/EACSPdf/1_treatment_of_hiv_infected_adults.pdf (accessed on 20 June 2011).
- Antinori, A.; Marcotullio, S.; Ammassari, A.; Andreoni, M.; Angarano, G.; Carosi, G.; Cinque, P.; d'Arminio Monforte, A.; Di Perri, G.; Ensoli, B.; et al. Italian guidelines for the use of antiretroviral agents and the diagnostic-clinical management of HIV-1 infected persons. New Microbiol. 2011, 34, 109–146. [Google Scholar]
- Miller, C.D.; El-Kholi, R.; Faragon, J.J.; Lodise, T.P. Prevalence and risk factors for clinically significant drug interactions with antiretroviral therapy. Pharmacotherapy 2007, 27, 1379–1386. [Google Scholar]
- Patel, N.; Abdelsayed, S.; Veve, M.; Miller, C.D. Predictors of clinically significant drug-drug interactions among patients treated with nonnucleoside reverse transcriptase inhibitor-, protease inhibitor-, and raltegravir-based antiretroviral regimens. Ann. Pharmacother. 2011, 45, 317–324. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rathbun, R.C.; Liedtke, M.D. Antiretroviral Drug Interactions: Overview of Interactions Involving New and Investigational Agents and the Role of Therapeutic Drug Monitoring for Management. Pharmaceutics 2011, 3, 745-781. https://doi.org/10.3390/pharmaceutics3040745
Rathbun RC, Liedtke MD. Antiretroviral Drug Interactions: Overview of Interactions Involving New and Investigational Agents and the Role of Therapeutic Drug Monitoring for Management. Pharmaceutics. 2011; 3(4):745-781. https://doi.org/10.3390/pharmaceutics3040745
Chicago/Turabian StyleRathbun, R. Chris, and Michelle D. Liedtke. 2011. "Antiretroviral Drug Interactions: Overview of Interactions Involving New and Investigational Agents and the Role of Therapeutic Drug Monitoring for Management" Pharmaceutics 3, no. 4: 745-781. https://doi.org/10.3390/pharmaceutics3040745
APA StyleRathbun, R. C., & Liedtke, M. D. (2011). Antiretroviral Drug Interactions: Overview of Interactions Involving New and Investigational Agents and the Role of Therapeutic Drug Monitoring for Management. Pharmaceutics, 3(4), 745-781. https://doi.org/10.3390/pharmaceutics3040745