Bioavailability of a Lipidic Formulation of Curcumin in Healthy Human Volunteers


Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Methods
2.2.1. Characterization of CRM-LF
2.2.1.1. Analysis of CRM in CRM-LF
2.2.1.2. Globule Size Analysis of CRM-LF upon Dilution
2.2.2. Pharmacokinetic Study
2.2.2.1. Demographic Data of Healthy Volunteers
2.2.2.2. Study Design
2.2.2.3. Dosing and Blood Sampling
2.2.2.4. Sample Processing
2.2.2.5. Analysis of CRM in Plasma by the LC-MS/MS Method
2.2.2.6. Data Analysis
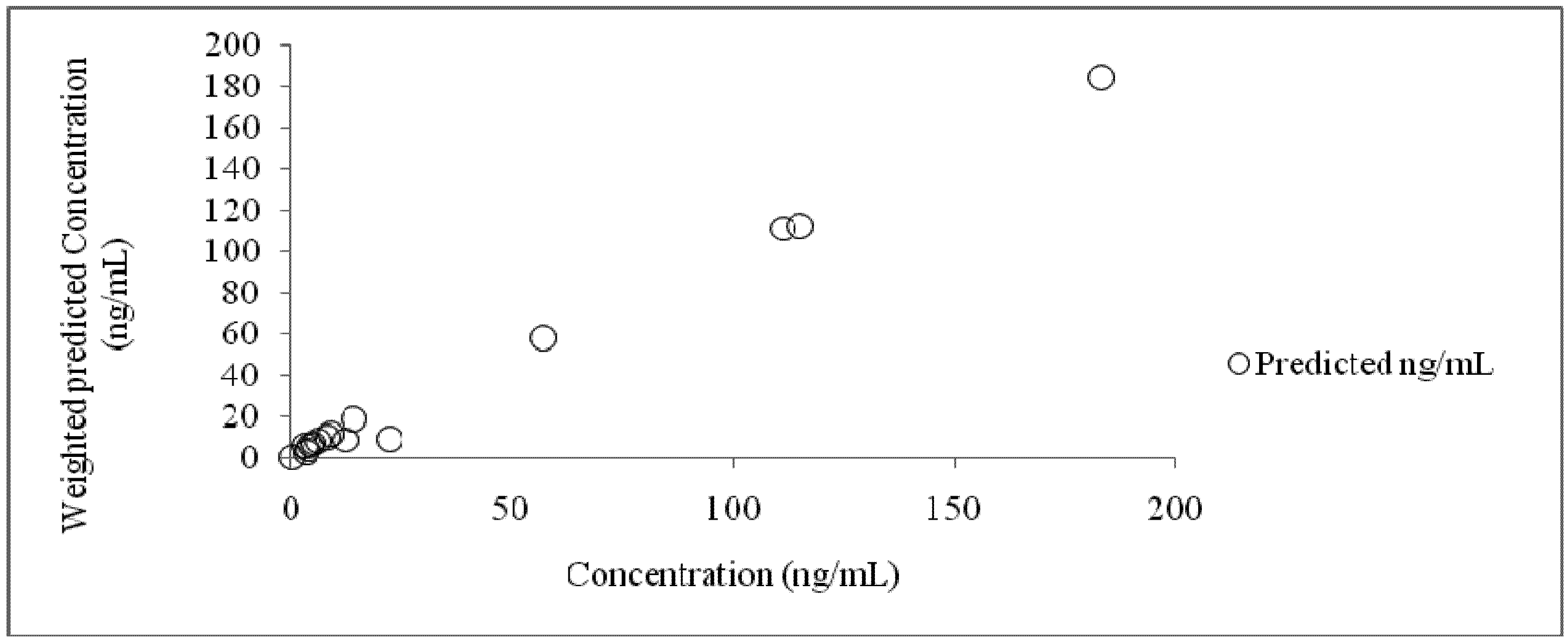
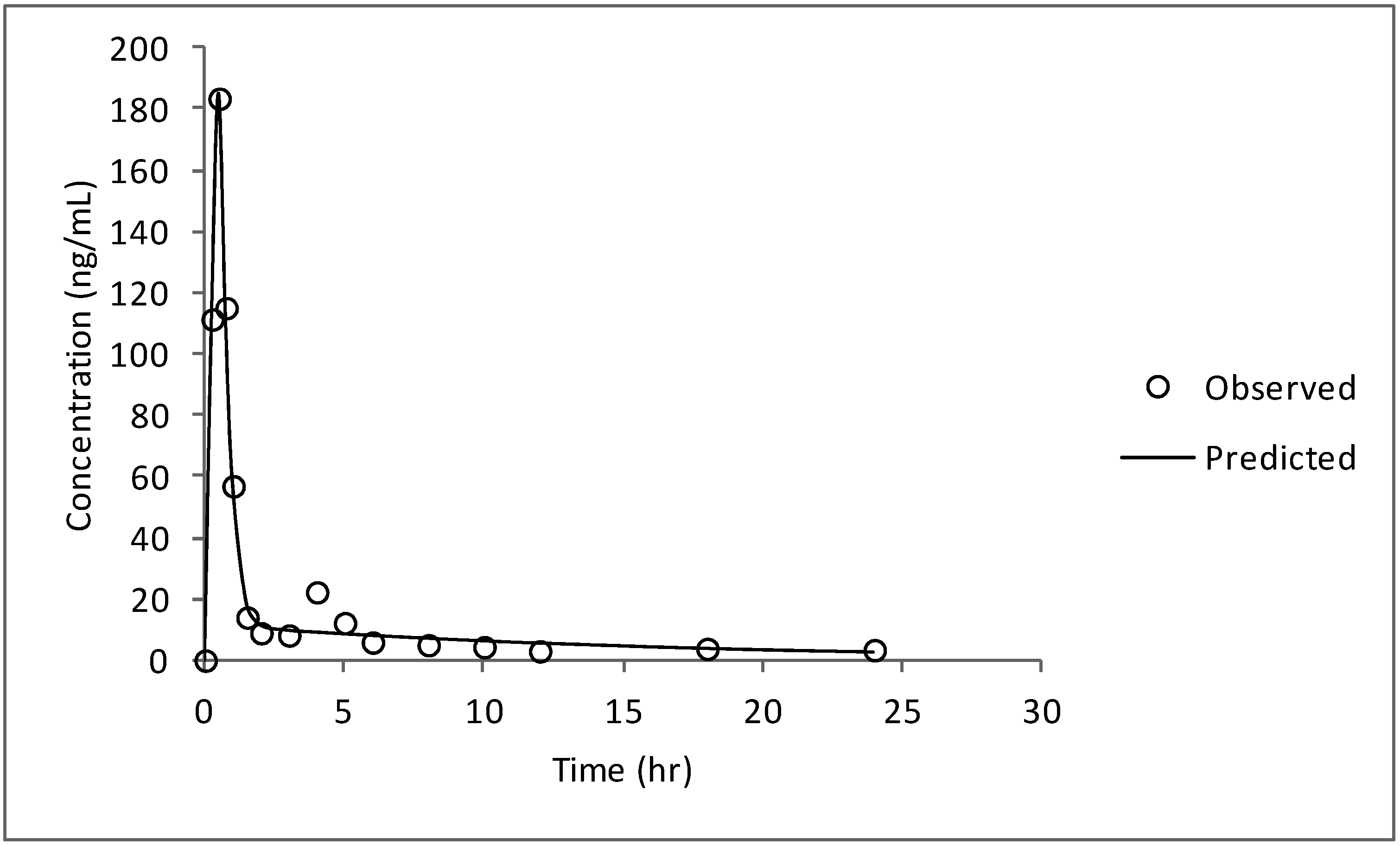
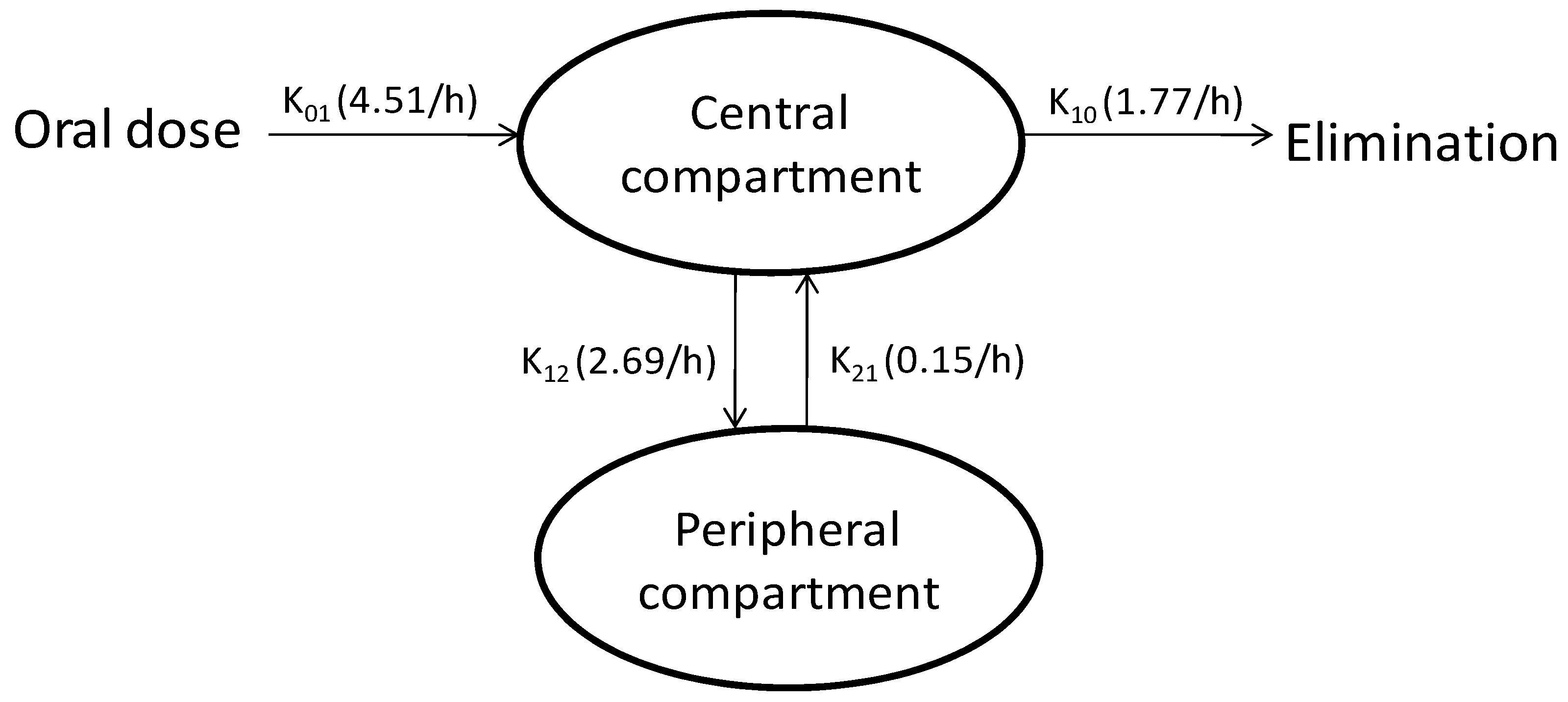
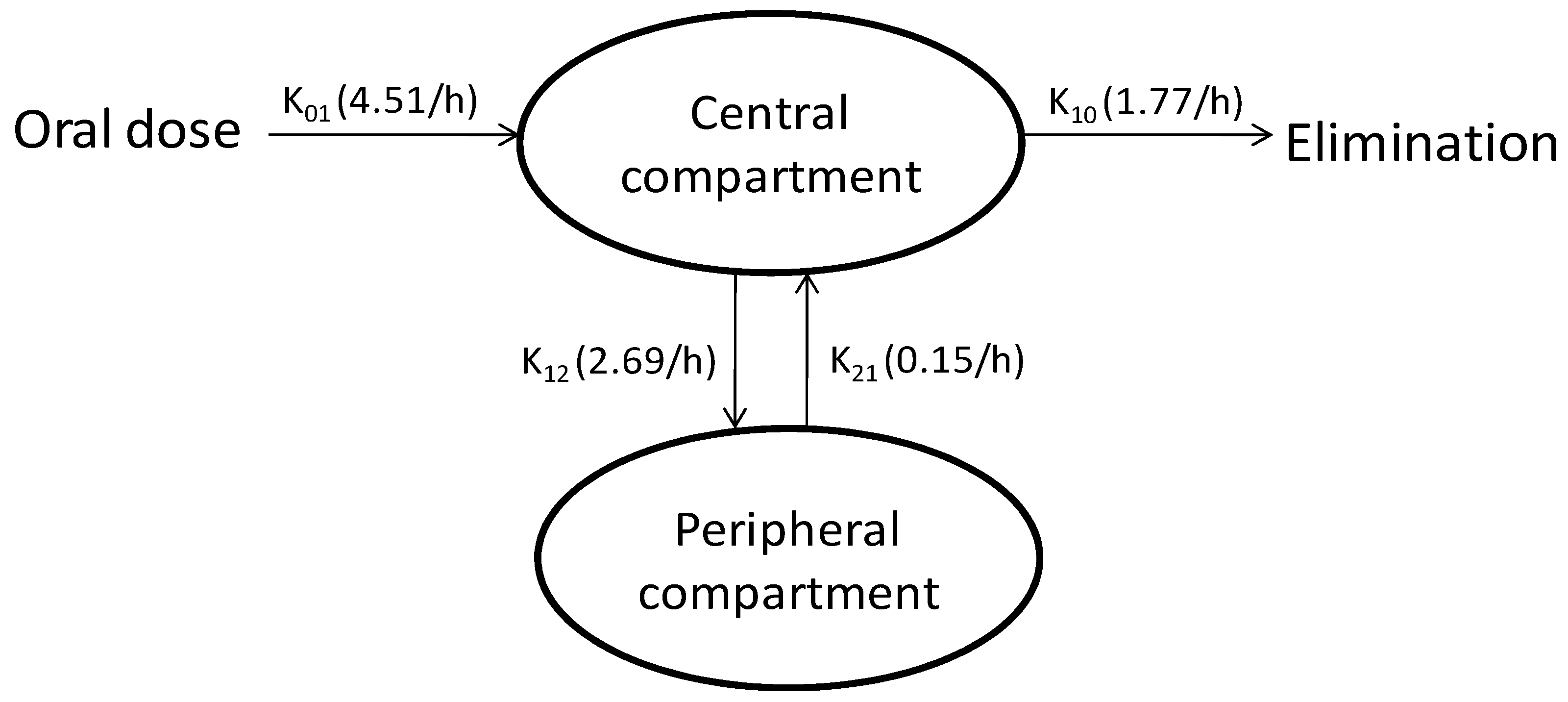
2.2.2.7. Compartmental Modeling
3. Results and Discussion
3.1. Characterization of CRM-LF
3.1.1. Analysis of CRM in CRM-LF
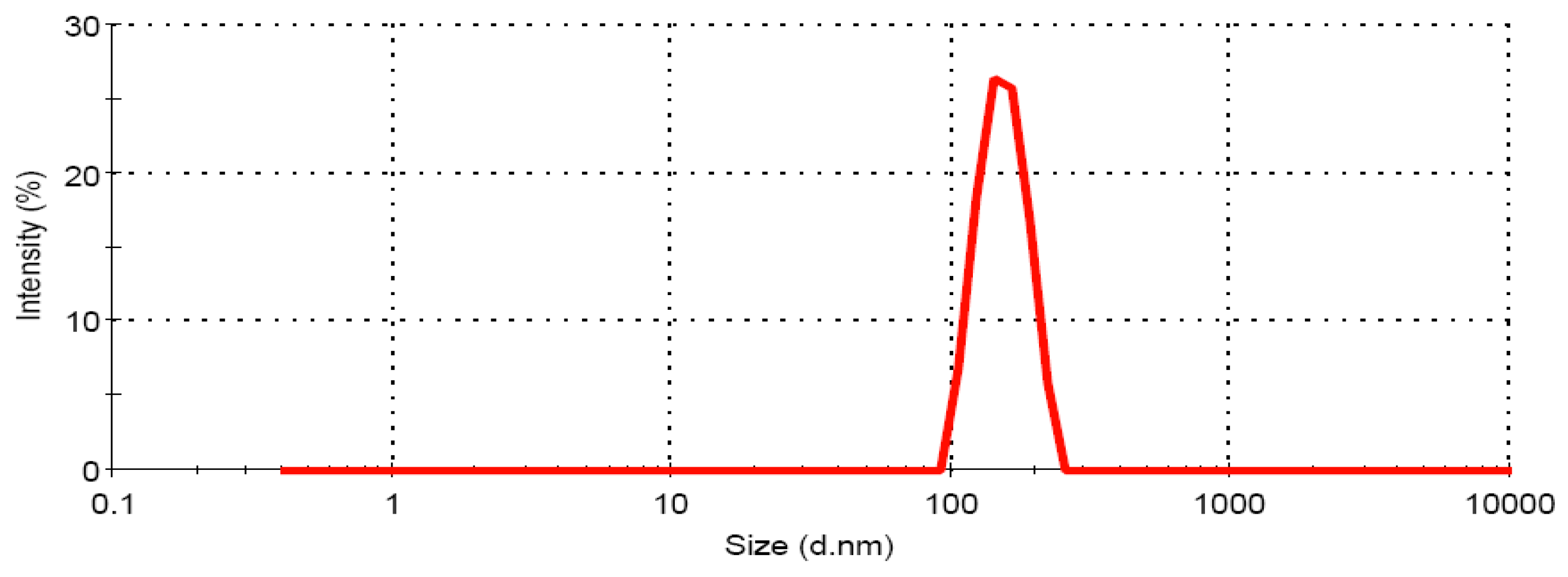
3.1.2. Globule size Analysis of CRM-LF upon Dilution
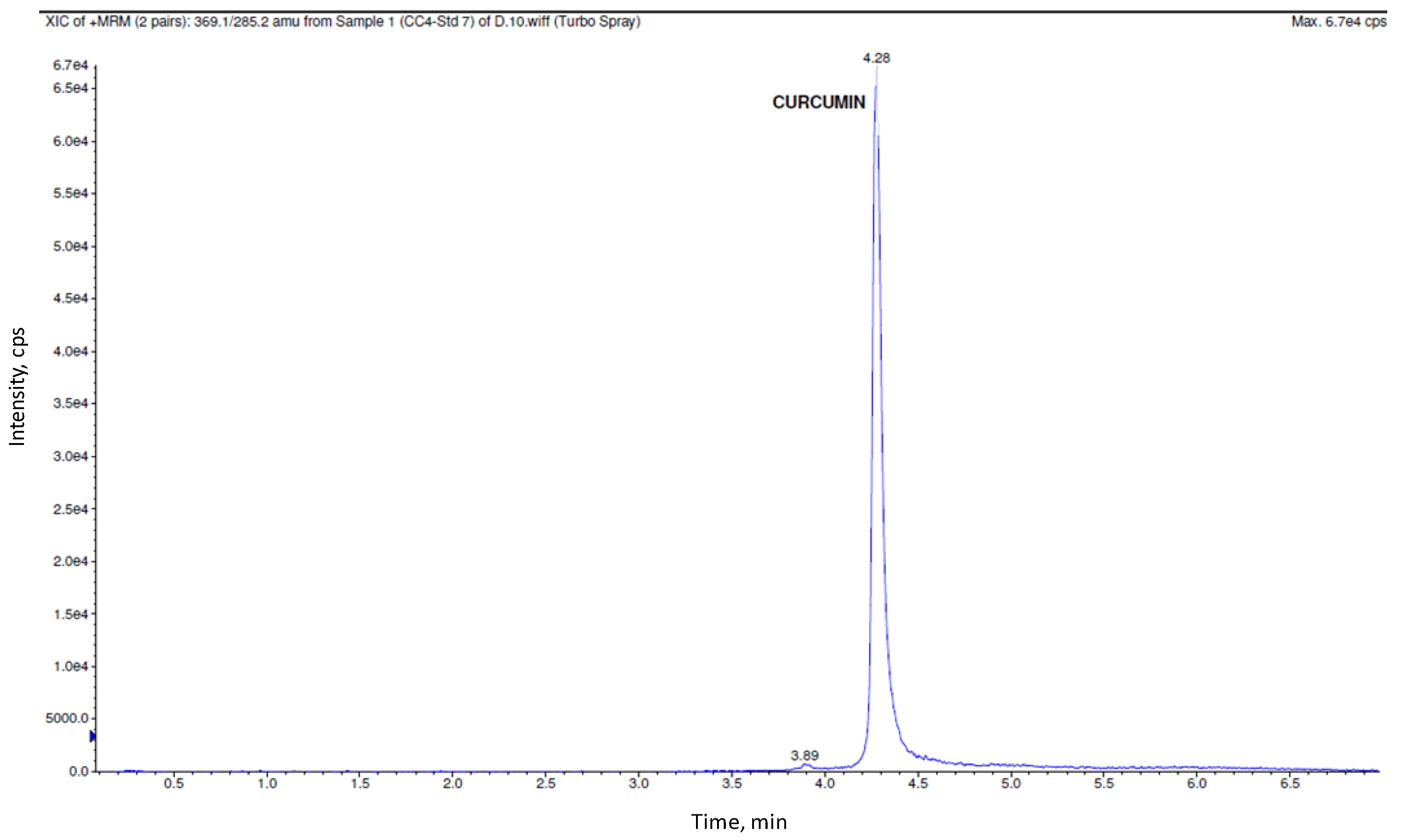
3.2. Bioanalytical Method Validation
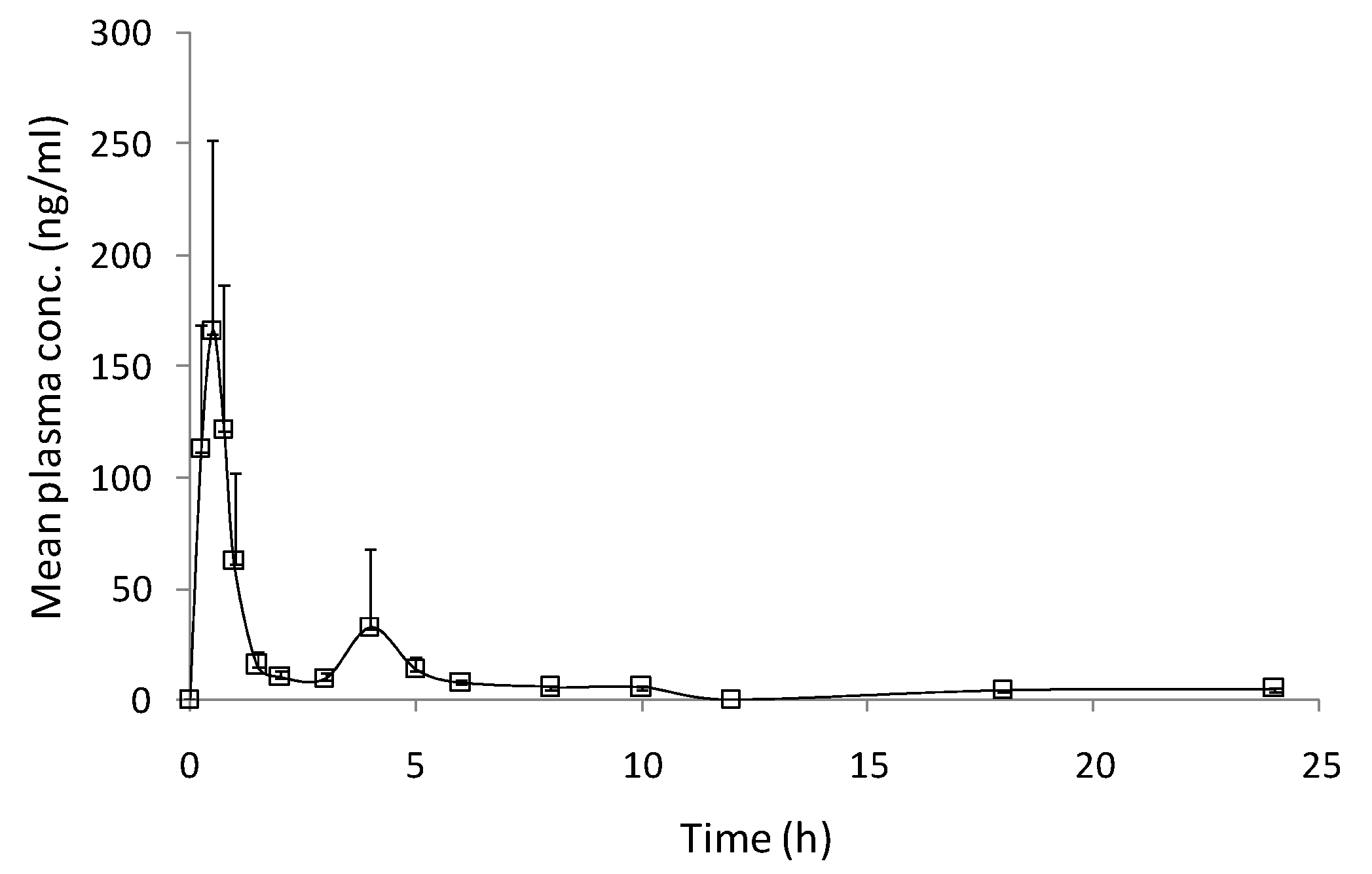
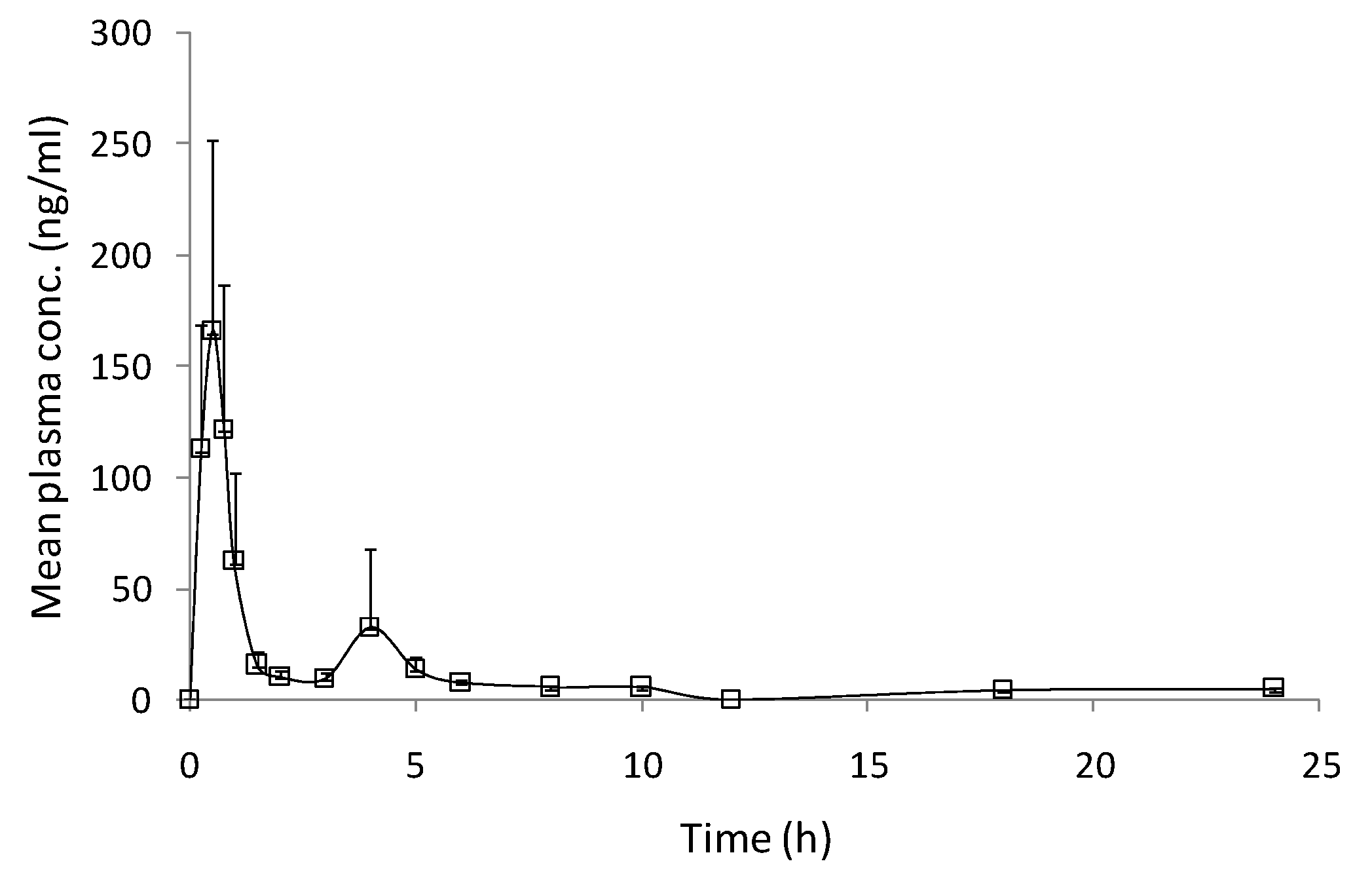
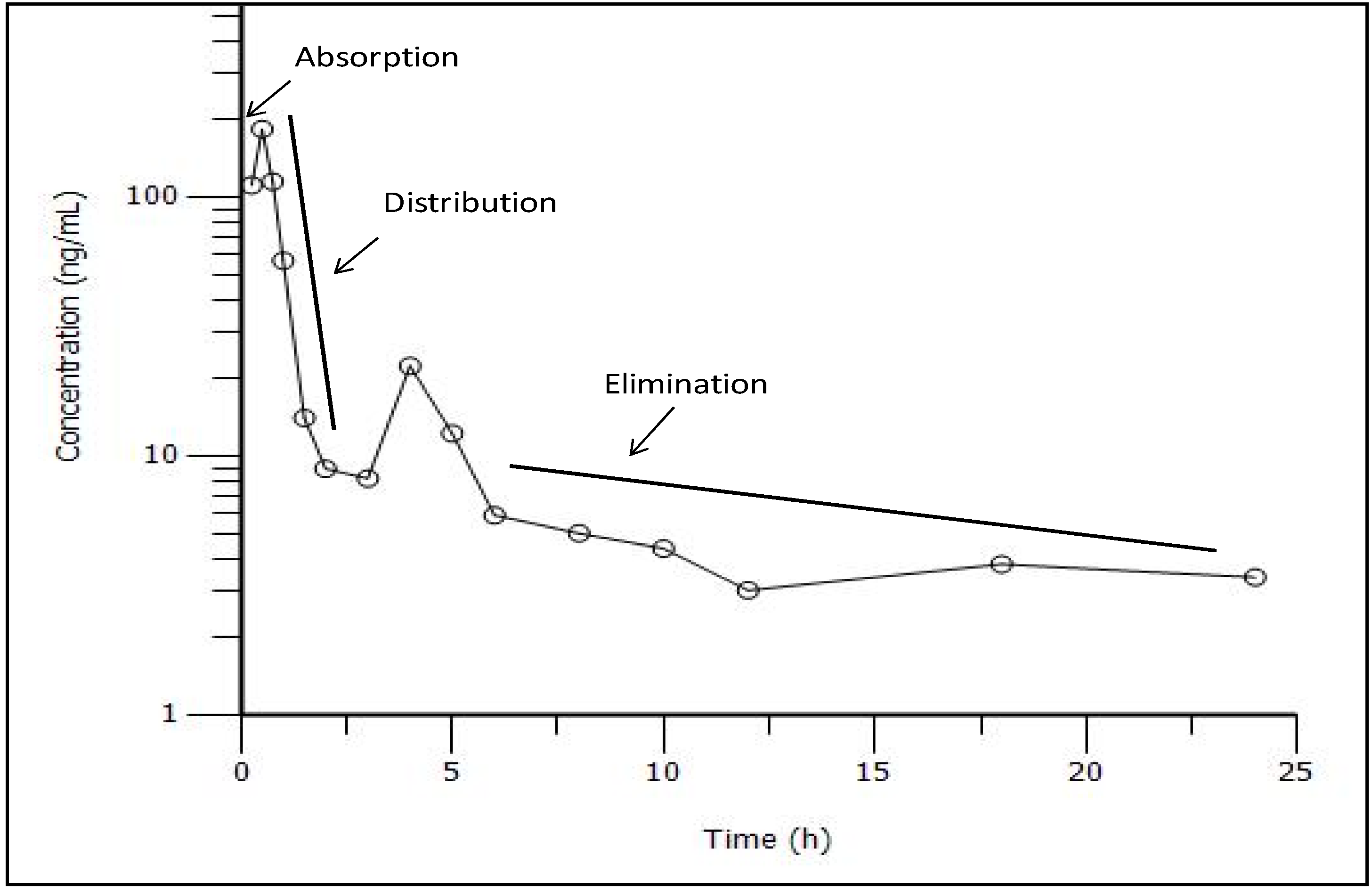
3.3. Pharmacokinetic Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacokinetic parameters | Value (mean ± SEM) |
|---|---|
| Cmax (ng/mL) | 183.35 ± 37.54 |
| Tmax (h) | 0.60 ± 0.05 |
| t1/2 (h) | 12.85 ± 4.57 |
| AUC0–t (ng.h/mL) | 231.31 ± 24.45 |
| AUC0–∞ (ng.h/mL) | 321.12 ± 25.55 |

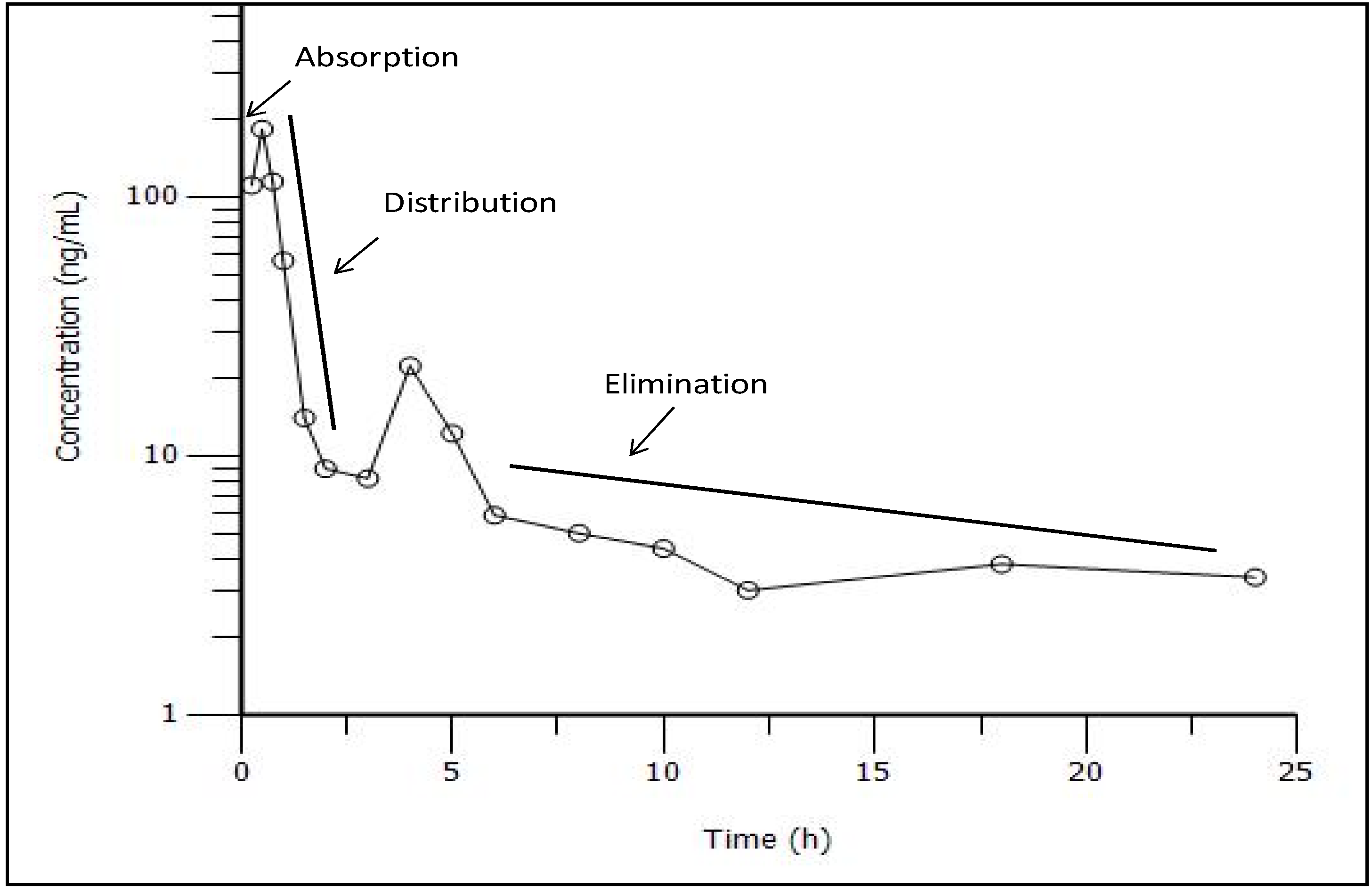
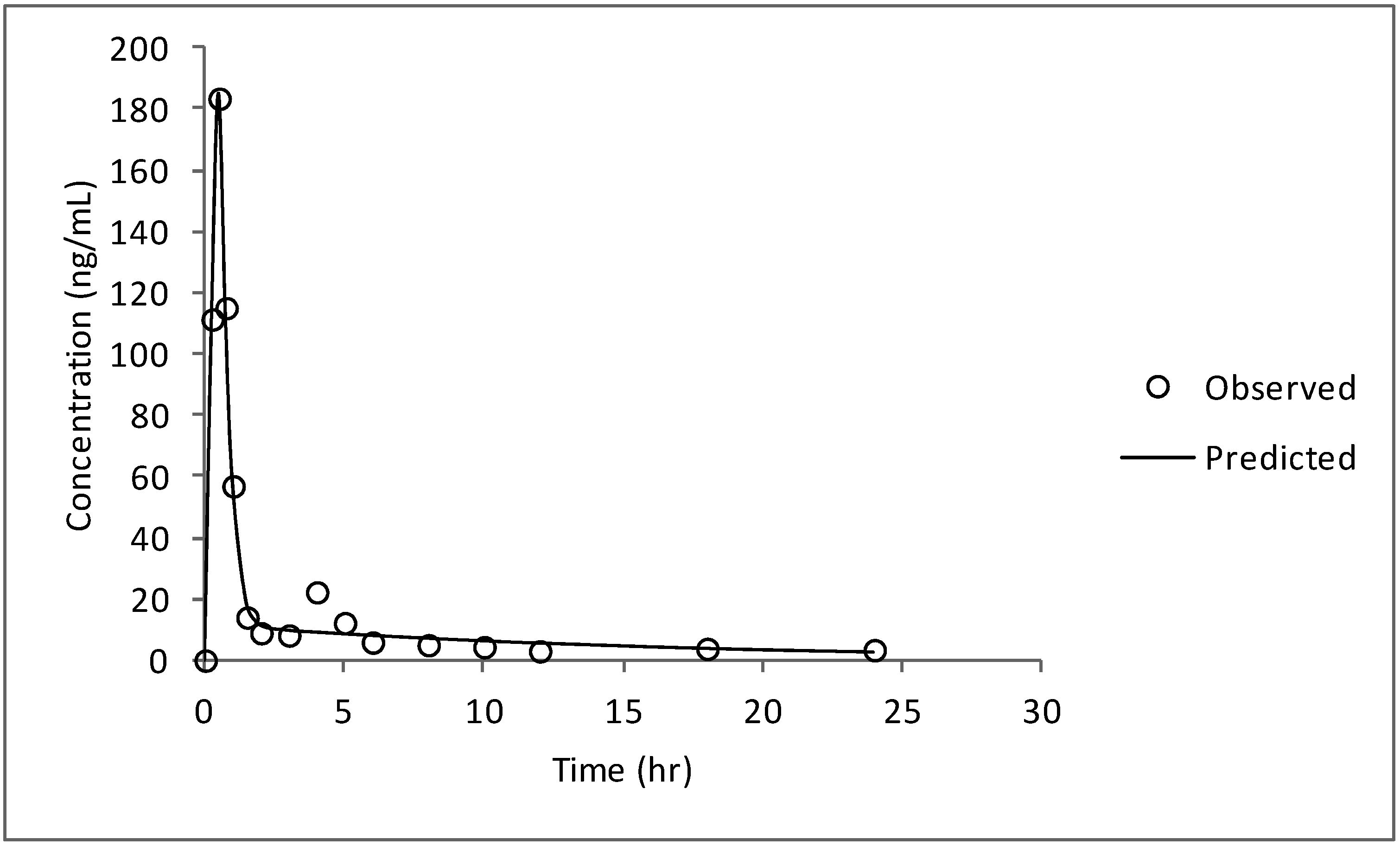
3.4. Compartmental Modeling
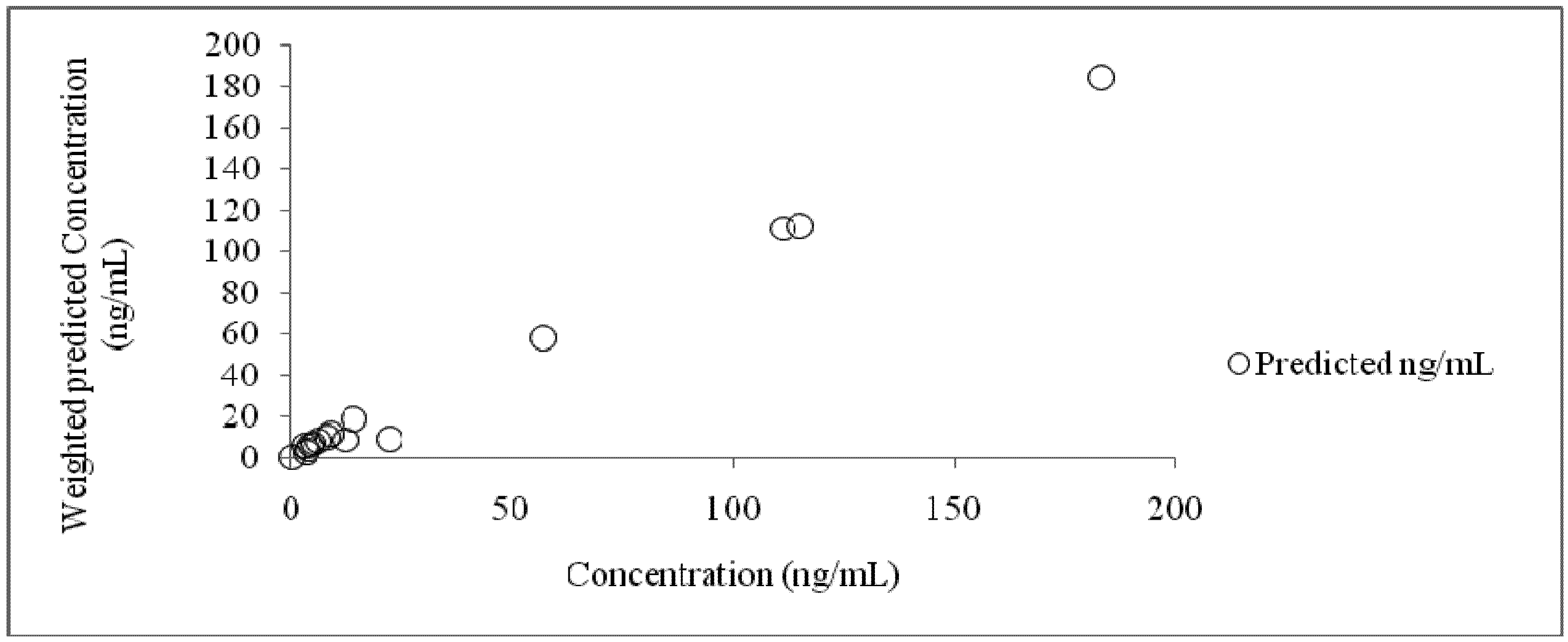
| Time (h) | Concentration (ng/mL) | Predicted (ng/mL) | Residual (ng/mL) |
|---|---|---|---|
| 0 | 0 | 0 | 0 |
| 0.25 | 111.42 | 111.34 | 0.07 |
| 0.5 | 183.51 | 184.81 | −1.30 |
| 0.75 | 115.13 | 112.37 | 2.75 |
| 1 | 56.92 | 58.14 | −1.22 |
| 1.5 | 14.04 | 18.58 | −4.54 |
| 2 | 8.94 | 11.50 | −2.56 |
| 3 | 8.19 | 9.77 | −1.58 |
| 4 | 22.29 | 9.16 | 13.12 |
| 5 | 12.23 | 8.61 | 3.61 |
| 6 | 5.9 | 8.09 | −2.19 |
| 8 | 5.03 | 7.15 | −2.12 |
| 10 | 4.39 | 6.31 | −1.92 |
| 12 | 3.03 | 5.57 | −2.54 |
| 18 | 3.82 | 3.84 | −0.02 |
| 24 | 3.4 | 2.64 | 0.75 |
| Parameter | Units | Estimate |
|---|---|---|
| Absorption phase | ||
| Cmax | ng/mL | 195.87 |
| Tmax | h | 0.41 |
| AUC | h.ng/mL | 297.42 |
| K01 | 1/h | 4.51 |
| Tlag | h | 0.18 |
| Distribution phase | ||
| K12 | 1/h | 2.69 |
| K21 | 1/h | 0.15 |
| Elimination phase | ||
| K10 | 1/h | 1.77 |
4. Conclusions
Conflict of Interest
Acknowledgments
References
- Ratnam, D.V.; Ankola, D.D.; Bhardwaj, V.; Sahana, D.K.; Ravi Kumar, M.N.V. Role of antioxidants in prophylaxis and therapy: A pharmaceutical perspective. J. Control. Rel. 2006, 113, 189–207. [Google Scholar] [CrossRef]
- De Toma, A.S.; Salamekh, S.; Ramamoorthy, A.; Lim, M.H. Misfolded proteins in Alzheimer’s disease and type II diabetes. Chem. Soc. Rev. 2012, 41, 608–621. [Google Scholar]
- Popovych, N.; Brender, J.R.; Soong, R.; Vivekanandan, S.; Hartman, K.; Basrur, V.; Macdonald, P.M.; Ramamoorthy, A. Site specific interaction of the polyphenol EGCG with the SEVI amyloid precursor peptide PAP(248−286). J. Phys. Chem. 2012, 116, 3650–3658. [Google Scholar]
- Huang, R.; Vivekanandan, S.; Brender, J.R.; Abe, Y.; Naito, A.; Ramamoorthy, A. NMR characterization of monomeric and oligomeric conformations of human calcitonin and its interaction with EGCG. J. Mol. Biol. 2012, 416, 108–120. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Sundaram, C.; Malani, N.; Ichikawa, H. Curcumin: The Indian solid gold. Adv. Exp. Med. Biol. 2007, 595, 1–75. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Harikumar, K.B. Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int. J. Biochem. Cell Biol. 2009, 41, 40–59. [Google Scholar] [CrossRef]
- Wang, Y.J.; Pan, M.H.; Cheng, A.L.; Lin, L.I.; Ho, Y.S.; Hsieh, C.Y.; Lin, J.K. Stability of curcumin in buffer solutions and characterization of its degradation products. J. Pharm. Biomed. Anal. 1997, 15, 1867–1876. [Google Scholar] [CrossRef]
- Pan, M.H.; Huang, T.M.; Lin, J.K. Biotransformation of curcumin through reduction and glucuronidation in mice. Drug Metab. Dispos. 1999, 27, 486–494. [Google Scholar]
- Wahlang, B.; Pawar, Y.B.; Bansal, A.K. Identification of permeability-related hurdles in oral delivery of curcumin using the Caco-2 cell model. Eur. J. Pharm. Biopharm. 2011, 77, 275–282. [Google Scholar] [CrossRef]
- Sharma, R.A.; Euden, S.A.; Platton, S.L.; Cooke, D.N.; Shafayat, A.; Hewitt, H.R.; Marczylo, T.H.; Morgan, B.; Hemingway, D.; Plummer, S.M.; et al. Phase I clinical trial of oral curcumin: biomarkers of systemic activity and compliance. Clin. Cancer Res. 2004, 10, 6847–6854. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 94, 79–83. [Google Scholar]
- Chattopadhyay, I.; Biswas, K.; Bandyopadhyay, U.; Banerjee, R.K. Turmeric and curcumin: Biological actions and medicinal applications. Curr. Sci. 2004, 87, 44–53. [Google Scholar]
- Munjal, B.; Pawar, Y.B.; Patel, S.B.; Bansal, A.K. Comparative oral bioavailability advantage from curcumin formulations. Drug Deliv. Transl. Res. 2011, 1, 322–331. [Google Scholar] [CrossRef]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. 2008, 14, 4491–4499. [Google Scholar]
- Sharma, R.A.; McLelland, H.R.; Hill, K.A.; Ireson, C.R.; Euden, S.A.; Manson, M.M.; Pirmohamed, M.; Marnett, L.J.; Gescher, A.J.; Steward, W.P. Pharmacodynamic and pharmacokinetic study of oral Curcuma extract in patients with colorectal cancer. Clin. Cancer Res. 2001, 7, 1894–1900. [Google Scholar]
- Lao, C.D.; Ruffin, M.T.; Normolle, D.; Heath, D.D.; Murray, S.I.; Bailey, J.M.; Boggs, M.E.; Crowell, J.; Rock, C.L.; Brenner, D.E. Dose escalation of a curcuminoid formulation. BMC Complement. Altern. Med. 2006, 6, 10. [Google Scholar] [CrossRef]
- Antony, B.; Merina, B.; Iyer, V.S.; Judy, N.; Lennertz, K.; Joyal, S. A pilot cross-over study to evaluate human oral bioavailability of BCM-95® CG (BiocurcumaxTM), a novel bioenhanced preparation of curcumin. Ind. J. Pharm. Sci. 2008, 70, 445–449. [Google Scholar] [CrossRef]
- Gota, V.S.; Maru, G.B.; Soni, T.G.; Gandhi, T.R.; Kochar, N.; Agarwal, M.G. Safety and pharmacokinetics of a solid lipid curcumin particle formulation in osteosarcoma patients and healthy volunteers. J. Agric. Food Chem. 2010, 58, 2095–2099. [Google Scholar]
- Wahlang, B.; Kabra, D.; Pawar, Y.B.; Tikoo, K.; Bansal, A.K. Contribution of formulation and excipients towards enhanced permeation of curcumin. Arzneimittelforschung 2012, 62, 88–93. [Google Scholar] [CrossRef]
- Pawar, Y.B. Department of Pharmaceutics, NIPER, SAS Nagar, Punjab, India. 2011; Unpublished research data. [Google Scholar]
- Lopez-Lazaro, M. Anticancer and carcinogenic properties of curcumin: Considerations for its clinical development as a cancer chemopreventive and chemotherapeutic agent. Mol. Nutr. Food Res. 2008, 52, S103–S127. [Google Scholar]
- Martin-Cordero, C.; Lopez-Lazaro, M.; Galvez, M.; Ayuso, M.J. Curcumin as a DNA topoisomerase II poison. J. Enz. Inhib. Med. Chem. 2003, 18, 505–509. [Google Scholar] [CrossRef]
- Wang, H.; Mao, Y.; Chen, A.Y.; Zhou, N.; LaVoie, E.J.; Liu, L.F. Stimulation of topoisomerase II-mediated DNA damage via a mechanism involving protein thiolation. Biochem. 2001, 40, 3316–3323. [Google Scholar] [CrossRef]
- Barry, J.; Fritz, M.; Brender, J.R.; Smith, P.E.S.; Lee, D.K.; Ramamoorthy, A. Determining the Effects of Lipophilic Drugs on Membrane Structure by Solid-State NMR Spectroscopy: The Case of the Antioxidant Curcumin. J. Am. Chem. Soc. 2009, 131, 4490–4498. [Google Scholar]
- Paolo, A.D.; Barbara, C.; Chella, A.; Angeletti, C.A.; Tacca, M.D. Pharmacokinetics of azithromycin in lung tissue, bronchial washing, and plasma in patients given multiple oral doses of 500 and 1000 mg daily. Pharmacol. Res. 2002, 46, 545–550. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pawar, Y.B.; Munjal, B.; Arora, S.; Karwa, M.; Kohli, G.; Paliwal, J.K.; Bansal, A.K. Bioavailability of a Lipidic Formulation of Curcumin in Healthy Human Volunteers. Pharmaceutics 2012, 4, 517-530. https://doi.org/10.3390/pharmaceutics4040517
Pawar YB, Munjal B, Arora S, Karwa M, Kohli G, Paliwal JK, Bansal AK. Bioavailability of a Lipidic Formulation of Curcumin in Healthy Human Volunteers. Pharmaceutics. 2012; 4(4):517-530. https://doi.org/10.3390/pharmaceutics4040517
Chicago/Turabian StylePawar, Yogesh B., Bhushan Munjal, Saurabh Arora, Manoj Karwa, Gunjan Kohli, Jyoti K. Paliwal, and Arvind K. Bansal. 2012. "Bioavailability of a Lipidic Formulation of Curcumin in Healthy Human Volunteers" Pharmaceutics 4, no. 4: 517-530. https://doi.org/10.3390/pharmaceutics4040517
APA StylePawar, Y. B., Munjal, B., Arora, S., Karwa, M., Kohli, G., Paliwal, J. K., & Bansal, A. K. (2012). Bioavailability of a Lipidic Formulation of Curcumin in Healthy Human Volunteers. Pharmaceutics, 4(4), 517-530. https://doi.org/10.3390/pharmaceutics4040517