Effects of 1,25(OH)2D3 on Cancer Cells and Potential Applications in Combination with Established and Putative Anti-Cancer Agents



Abstract
:1. Introduction
1.1. 1,25(OH)2D3 in Combination with Anti-Metabolites
1.2. 1,25(OH)2D3 in Combination with Platinum Compounds
1.3. 1,25(OH)2D3 in Combination with Taxanes
1.4. 1,25(OH)2D3 in Combination with Tyrosine Kinase Inhibitors
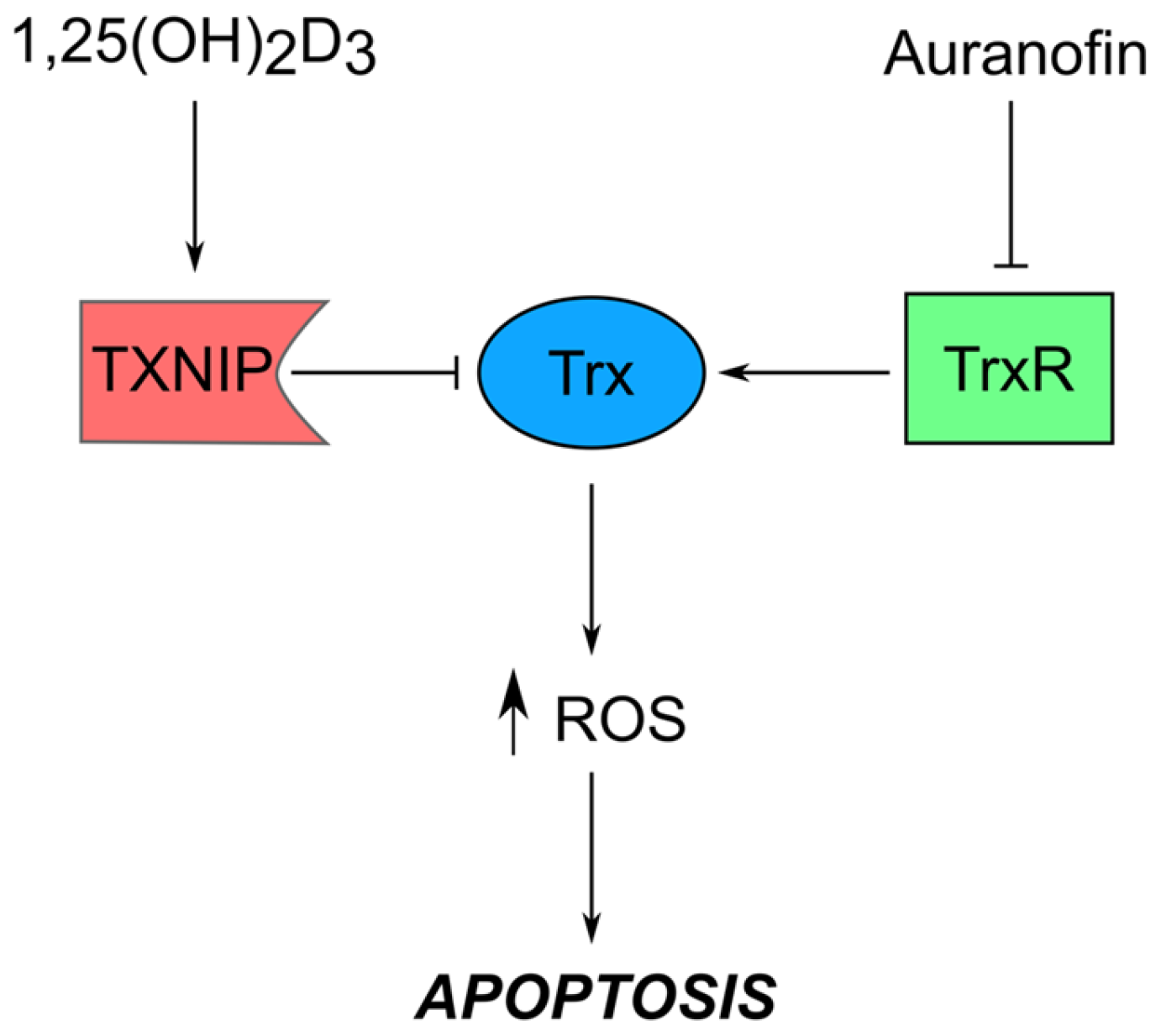
1.5. 1,25(OH)2D3 in Combination with Non-Classical Anti-Cancer Drugs
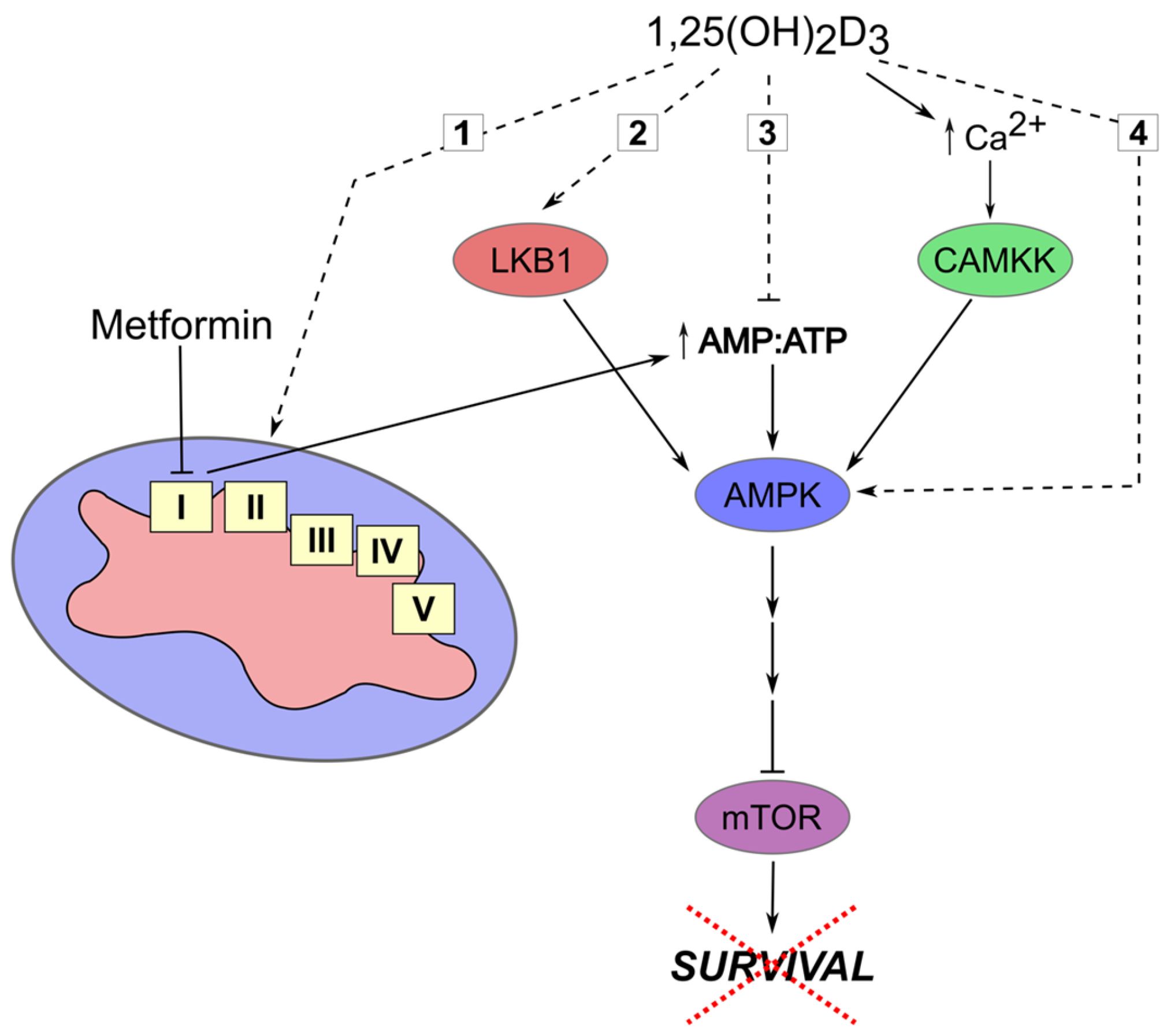
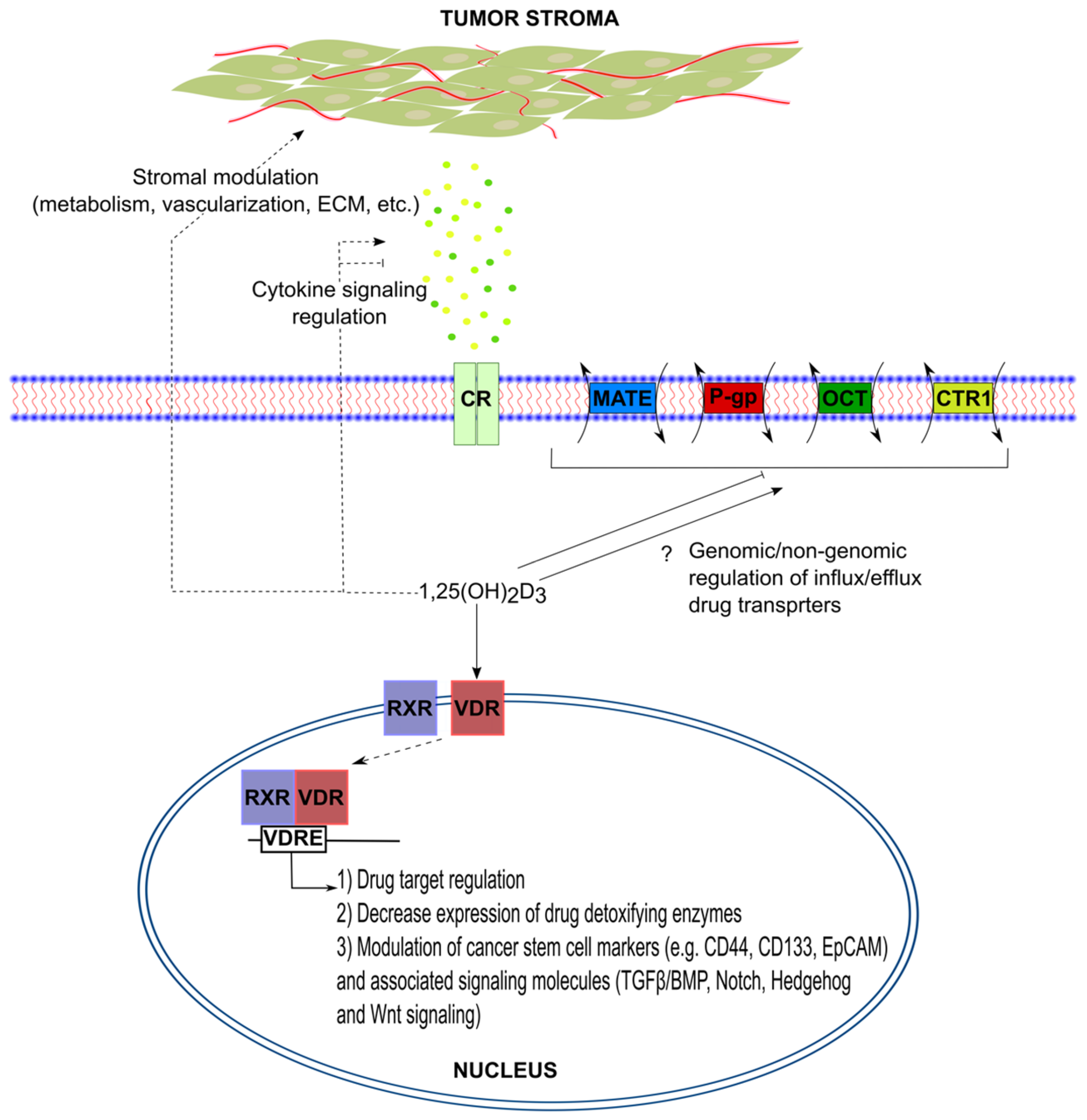
1.6. Overcoming Chemoresistance with 1,25(OH)2D3
2. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 1,25(OH)2D3 | 1,25-dihydroxyvitamin D3 |
| 5-FU | 5-Fluorouracil |
| ABC | ATP binding cassette |
| AMPK | AMP-activated protein kinase |
| BMP | Bone morphogenetic protein |
| CAMKK | Calcium/calmodulin-dependent protein kinase kinase |
| CMF | Cyclophosphamide—Methotrexate—5-Flurouracil |
| CR | Cytokine receptor |
| CSCs | Cancer stem cells |
| CTR1 | Copper transporter 1 |
| FDA | Food and Drug Administration |
| FOLFOX | 5-Flurouracil—Folinic acid—Oxaliplatin |
| HIF1α | Hypoxia-Inducible factor 1-α |
| LKB1 | Liver-kinase B1 |
| MATE | Multidrug and toxic compound extrusion |
| mTOR | Mammalian target of rapamycin |
| OCT | Organic cation transporter |
| P-gp | P-glycoprotein |
| PCa | Pancreatic cancer |
| ROS | Reactive oxygen species |
| RXR | Retinoid X receptor |
| TGF-β | Transforming growth factor-β |
| TKI | Tyrosine kinase inhibitor |
| Trx | Thioredoxin |
| TrxR | Thioredoxin reductase |
| TXNIP/VDUP1 | Thioredoxin-interacting protein/vitamin D3-upregulated protein 1 |
| VDR | Vitamin D receptor |
| VDRE | Vitamin D response elements |
References
- Corrie, P.G. Cytotoxic chemotherapy: Clinical aspects. Medicine 2008, 36, 24–28. [Google Scholar] [CrossRef]
- Chabner, B.A.; Roberts, T.G., Jr. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.; Krishnan, A.V.; Swami, S.; Giovannucci, E.; Feldman, B.J. The role of vitamin D in reducing cancer risk and progression. Nat. Rev. Cancer 2014, 14, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Hossein-nezhad, A.; Holick, M.F. Vitamin d for health: A global perspective. Mayo Clin. Proc. 2013, 88, 720–755. [Google Scholar] [CrossRef] [PubMed]
- El Abdaimi, K.; Papavasiliou, V.; Rabbani, S.A.; Rhim, J.S.; Goltzman, D.; Kremer, R. Reversal of hypercalcemia with the vitamin D analogue eb1089 in a human model of squamous cancer. Cancer Res. 1999, 59, 3325–3328. [Google Scholar] [PubMed]
- Brown, A.J.; Ritter, C.R.; Finch, J.L.; Morrissey, J.; Martin, K.J.; Murayama, E.; Nishii, Y.; Slatopolsky, E. The noncalcemic analogue of vitamin D, 22-oxacalcitriol, suppresses parathyroid hormone synthesis and secretion. J. Clin. Investig. 1989, 84, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Leyssens, C.; Verlinden, L.; Verstuyf, A. The future of vitamin D analogs. Front. Physiol. 2014, 5, 122. [Google Scholar] [CrossRef] [PubMed]
- Ramagopalan, S.V.; Heger, A.; Berlanga, A.J.; Maugeri, N.J.; Lincoln, M.R.; Burrell, A.; Handunnetthi, L.; Handel, A.E.; Disanto, G.; Orton, S.M.; et al. A chip-seq defined genome-wide map of vitamin D receptor binding: Associations with disease and evolution. Genome Res. 2010, 20, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Danilenko, M.; Wang, Q.; Wang, X.; Levy, J.; Sharoni, Y.; Studzinski, G.P. Carnosic acid potentiates the antioxidant and prodifferentiation effects of 1alpha,25-dihydroxyvitamin D3 in leukemia cells but does not promote elevation of basal levels of intracellular calcium. Cancer Res. 2003, 63, 1325–1332. [Google Scholar] [PubMed]
- Wang, Q.; Salman, H.; Danilenko, M.; Studzinski, G.P. Cooperation between antioxidants and 1,25-dihydroxyvitamin D3 in induction of leukemia hl60 cell differentiation through the JNK/AP-1/Egr-1 pathway. J. Cell. Physiol. 2005, 204, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Swami, S.; Krishnan, A.V.; Peehl, D.M.; Feldman, D. Genistein potentiates the growth inhibitory effects of 1,25-dihydroxyvitamin D3 in du145 human prostate cancer cells: Role of the direct inhibition of cyp24 enzyme activity. Mol. Cell. Endocrinol. 2005, 241, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Lind, M.J. Principles of cytotoxic chemotherapy. Medicine 2008, 36, 19–23. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006, 17 (Suppl. S5), v7–v12. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Gallick, G.E. Gemcitabine resistance in pancreatic cancer: Picking the key players. Clin. Cancer Res. 2008, 14, 1284–1285. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.D.; Ma, Y.; Flynn, G.; Muindi, J.R.; Kong, R.X.; Trump, D.L.; Johnson, C.S. Calcitriol enhances gemcitabine anti-tumor activity in vitro and in vivo by promoting apoptosis in a human pancreatic carcinoma model system. Cell Cycle 2010, 9, 3022–3029. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, V.; Zhou, Y.; Yen, T.J. A synthetic lethal screen identifies the vitamin D receptor as a novel gemcitabine sensitizer in pancreatic cancer cells. Cell Cycle 2014, 13, 3839–3856. [Google Scholar] [CrossRef] [PubMed]
- El-Shemi, A.G.; Refaat, B.; Kensara, O.A.; Mohamed, A.M.; Idris, S.; Ahmad, J. Paricalcitol enhances the chemopreventive efficacy of 5-fluorouracil on an intermediate-term model of azoxymethane-induced colorectal tumors in rats. Cancer Prev. Res. 2016, 9, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Refaat, B.; El-Shemi, A.G.; Kensara, O.A.; Mohamed, A.M.; Idris, S.; Ahmad, J.; Khojah, A. Vitamin D3 enhances the tumouricidal effects of 5-fluorouracil through multipathway mechanisms in azoxymethane rat model of colon cancer. J. Exp. Clin. Cancer Res. 2015, 34, 71. [Google Scholar] [CrossRef] [PubMed]
- Milczarek, M.; Filip-Psurska, B.; Swietnicki, W.; Kutner, A.; Wietrzyk, J. Vitamin D analogs combined with 5-fluorouracil in human HT-29 colon cancer treatment. Oncol. Rep. 2014, 32, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.; vanCamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yu, W.D.; Hershberger, P.A.; Flynn, G.; Kong, R.X.; Trump, D.L.; Johnson, C.S. 1alpha,25-dihydroxyvitamin D3 potentiates cisplatin antitumor activity by p73 induction in a squamous cell carcinoma model. Mol. Cancer Ther. 2008, 7, 3047–3055. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, P.A.; McGuire, T.F.; Yu, W.D.; Zuhowski, E.G.; Schellens, J.H.; Egorin, M.J.; Trump, D.L.; Johnson, C.S. Cisplatin potentiates 1,25-dihydroxyvitamin D3-induced apoptosis in association with increased mitogen-activated protein kinase kinase kinase 1 (MEKK-1) expression. Mol. Cancer Ther. 2002, 1, 821–829. [Google Scholar] [PubMed]
- Jorgensen, A.; Blomberg Jensen, M.; Nielsen, J.E.; Juul, A.; Rajpert-De Meyts, E. Influence of vitamin D on cisplatin sensitivity in testicular germ cell cancer-derived cell lines and in a NTera2 xenograft model. J. Steroid Biochem. Mol. Biol. 2013, 136, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.D.; van Ginkel, P.R.; Darjatmoko, S.R.; Lindstrom, M.J.; Albert, D.M. Use of combination therapy with cisplatin and calcitriol in the treatment of y-79 human retinoblastoma xenograft model. Br. J. Ophthalmol. 2009, 93, 1105–1108. [Google Scholar] [CrossRef] [PubMed]
- Pelczynska, M.; Switalska, M.; Maciejewska, M.; Jaroszewicz, I.; Kutner, A.; Opolski, A. Antiproliferative activity of vitamin D compounds in combination with cytostatics. Anticancer Res. 2006, 26, 2701–2705. [Google Scholar] [PubMed]
- Milczarek, M.; Rosinska, S.; Psurski, M.; Maciejewska, M.; Kutner, A.; Wietrzyk, J. Combined colonic cancer treatment with vitamin D analogs and irinotecan or oxaliplatin. Anticancer Res. 2013, 33, 433–444. [Google Scholar] [PubMed]
- Zhang, Z.; Zhang, H.; Hu, Z.; Wang, P.; Wan, J.; Li, B. Synergy of 1,25-dihydroxyvitamin D3 and carboplatin in growth suppression of skov-3 cells. Oncol. Lett. 2014, 8, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Wani, M.C.; Horwitz, S.B. Nature as a remarkable chemist: A personal story of the discovery and development of taxol. Anticancer Drugs 2014, 25, 482–487. [Google Scholar] [CrossRef]
- Rowinsky, E.K. The development and clinical utility of the taxane class of antimicrotubule chemotherapy agents. Annu. Rev. Med. 1997, 48, 353–374. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, W.; Uytingco, M.S.; Christakos, S.; Wieder, R. 1,25-dihydroxyvitamin D3 and all-trans-retinoic acid sensitize breast cancer cells to chemotherapy-induced cell death. Cancer Res. 2000, 60, 2040–2048. [Google Scholar] [PubMed]
- Hershberger, P.A.; Yu, W.D.; Modzelewski, R.A.; Rueger, R.M.; Johnson, C.S.; Trump, D.L. Calcitriol (1,25-dihydroxycholecalciferol) enhances paclitaxel antitumor activity in vitro and in vivo and accelerates paclitaxel-induced apoptosis. Clin. Cancer Res. 2001, 7, 1043–1051. [Google Scholar] [PubMed]
- Ting, H.J.; Hsu, J.; Bao, B.Y.; Lee, Y.F. Docetaxel-induced growth inhibition and apoptosis in androgen independent prostate cancer cells are enhanced by 1alpha,25-dihydroxyvitamin D3. Cancer Lett. 2007, 247, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Scholar, E.M. Role of tyrosine kinase inhibitors in cancer therapy. J. Pharmacol. Exp. Ther. 2005, 315, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Yokoyama, A. Sunitinib, an orally available receptor tyrosine kinase inhibitor, induces monocytic differentiation of acute myelogenous leukemia cells that is enhanced by 1,25-dihydroxyvitamin D3. Leukemia 2009, 23, 2171–2173. [Google Scholar] [CrossRef] [PubMed]
- Lainey, E.; Wolfromm, A.; Sukkurwala, A.Q.; Micol, J.B.; Fenaux, P.; Galluzzi, L.; Kepp, O.; Kroemer, G. Egfr inhibitors exacerbate differentiation and cell cycle arrest induced by retinoic acid and vitamin D3 in acute myeloid leukemia cells. Cell Cycle 2013, 12, 2978–2991. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, K.D.; Shaurova, T.; Merzianu, M.; Suresh, A.; Kuriakose, M.A.; Johnson, C.S.; Hershberger, P.A.; Seshadri, M. Impact of short-term 1,25-dihydroxyvitamin D3 on the chemopreventive efficacy of erlotinib against oral cancer. Cancer Prev. Res. 2015, 8, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.R.; Morris, A.D. Metformin in cancer treatment and prevention. Annu. Rev. Med. 2015, 66, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, S.; Hawley, S.A.; Green, K.A.; Saner, N.; Mustard, K.J.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta activates ampk without forming a stable complex: Synergistic effects of Ca2+ and amp. Biochem. J. 2010, 426, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.S.; Li, H.X.; Li, C.Y.; Zhang, S.Y.; Chen, J.; Wang, Q.L.; Gao, J.M.; Liang, J.Q.; Gao, M.T.; Wu, Y.J. Synergistic antitumor activity of vitamin D3 combined with metformin in human breast carcinoma MDA-MB-231 cells involves m-TOR related signaling pathways. Pharmazie 2015, 70, 117–122. [Google Scholar] [PubMed]
- Li, H.X.; Gao, J.M.; Liang, J.Q.; Xi, J.M.; Fu, M.; Wu, Y.J. Vitamin D3 potentiates the growth inhibitory effects of metformin in DU145 human prostate cancer cells mediated by AMPK/mTOR signalling pathway. Clin. Exp. Pharmacol. Physiol. 2015, 42, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.S.; Li, H.X.; Li, C.Y.; Zhang, S.Y.; Chen, J.; Wang, Q.L.; Gao, J.M.; Liang, J.Q.; Gao, M.T.; Wu, Y.J. Vitamin D3 enhances antitumor activity of metformin in human bladder carcinoma SW-780 cells. Pharmazie 2015, 70, 123–128. [Google Scholar] [PubMed]
- Li, W.; Wang, Q.L.; Liu, X.; Dong, S.H.; Li, H.X.; Li, C.Y.; Guo, L.S.; Gao, J.M.; Berger, N.A.; Li, L.; et al. Combined use of vitamin D3 and metformin exhibits synergistic chemopreventive effects on colorectal neoplasia in rats and mice. Cancer Prev. Res. 2015, 8, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an old drug for a golden new age. Drugs R D 2015, 15, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Ott, I. On the medicinal chemistry of gold complexes as anticancer drugs. Coordin. Chem. Rev. 2009, 253, 1670–1681. [Google Scholar] [CrossRef]
- Arner, E.S.; Holmgren, A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006, 16, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chng, W.J. Roles of thioredoxin binding protein (TXNIP) in oxidative stress, apoptosis and cancer. Mitochondrion 2013, 13, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.S.; DeLuca, H.F. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochim. Biophys. Acta 1994, 1219, 26–32. [Google Scholar] [CrossRef]
- Stoltzman, C.A.; Peterson, C.W.; Breen, K.T.; Muoio, D.M.; Billin, A.N.; Ayer, D.E. Glucose sensing by mondoa:Mlx complexes: A role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6912–6917. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; O’Shea, J.M.; Kaadige, M.R.; Cunha, S.; Wilde, B.R.; Cohen, A.L.; Welm, A.L.; Ayer, D.E. Metabolic reprogramming in triple-negative breast cancer through myc suppression of txnip. Proc. Natl. Acad. Sci. USA 2015, 112, 5425–5430. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.F.; Koh, M.Y.; Williams, R.R.; James, B.; Wang, H.; Tate, W.R.; Gallegos, A.; von Hoff, D.D.; Han, H.; Powis, G. Identification of thioredoxin-interacting protein 1 as a hypoxia-inducible factor 1alpha-induced gene in pancreatic cancer. Pancreas 2008, 36, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kim, M.; Kim, N.H.; Oh, M.K.; Cho, J.K.; Jin, J.Y.; Kim, I.S. Auranofin promotes retinoic acid- or dihydroxyvitamin D3-mediated cell differentiation of promyelocytic leukaemia cells by increasing histone acetylation. Br. J. Pharmacol. 2008, 154, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.; Borden, K.L.B. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. Met amplification occurs with or without t790m mutations in egfr mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of met amplification in egfr mutant nsclc. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Wang, L.H.; Clark, R.E.; Pirmohamed, M. Active transport of imatinib into and out of cells: Implications for drug resistance. Blood 2004, 104, 3739–3745. [Google Scholar] [CrossRef] [PubMed]
- So, J.Y.; Suh, N. Targeting cancer stem cells in solid tumors by vitamin D. J. Steroid Biochem. Mol. Biol. 2015, 148, 79–85. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Drug | Resistance Mechanism | References |
|---|---|---|
| Anti-metabolites | ||
| 5-FU | Aberrant expression of: | [13,15] |
| Gemcitabine | Thymidylate synthase Thymidine phosphorylase Dihydropyrimide dehydrogenase Human equilibrative nucleoside transporter 1 | |
| Platins | ||
| Cisplatin | Aberrant expression of: | [23] |
| Carboplatin | Copper transporter (CTR1) ATPase copper transporting alpha (ATP7A) ATPase copper transporting beta (ATP7B) ATP binding cassette subfamily C member 2 (ABCC2) Excision repair cross-complementing-1 (ERCC1) mutL homolog 1(MLH1) | |
| Taxanes | ||
| Paclitaxel | Increased P-glycoprotein (P-gp) expression | [58] |
| Docetaxel | Altered microtubule dynamics and binding of drug to target | |
| TKIs | ||
| Gefitinib | Mutations in target | [37,59,60,61] |
| Erlotinib | Induced expression of MET and/or HGF | |
| Sunitinib | Aberrant drug influx/efflux (OCT1 and/or ABCB1) | |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abu el Maaty, M.A.; Wölfl, S. Effects of 1,25(OH)2D3 on Cancer Cells and Potential Applications in Combination with Established and Putative Anti-Cancer Agents. Nutrients 2017, 9, 87. https://doi.org/10.3390/nu9010087
Abu el Maaty MA, Wölfl S. Effects of 1,25(OH)2D3 on Cancer Cells and Potential Applications in Combination with Established and Putative Anti-Cancer Agents. Nutrients. 2017; 9(1):87. https://doi.org/10.3390/nu9010087
Chicago/Turabian StyleAbu el Maaty, Mohamed A., and Stefan Wölfl. 2017. "Effects of 1,25(OH)2D3 on Cancer Cells and Potential Applications in Combination with Established and Putative Anti-Cancer Agents" Nutrients 9, no. 1: 87. https://doi.org/10.3390/nu9010087