Bacterial Heat-Stable Enterotoxins: Translation of Pathogenic Peptides into Novel Targeted Diagnostics and Therapeutics


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Abbreviations
| ABC: | Avidin Biotin Complex |
| CFTR: | cystic fibrosis transmembrane conductance regulator |
| cGMP: | Cyclic guanosine monophosphate |
| CMA: | cancer mucosa antigens |
| CNG: | cyclic nucleotide-gated channel |
| COX-2: | cyclooxygenase-2 |
| GC-C: | guanylyl cyclase C |
| IBD: | inflammatory bowel disease |
| IBS: | irritable bowel syndrome |
| MRP: | multidrug resistance proteins |
| PDE: | phosphodiesterase |
| PGE: | prostaglandin |
| PKA: | cAMP-dependent protein kinase |
| PKG: | cGMP-dependent protein kinase |
| PLA2: | phospholipase A2 |
| pRb: | phosphorylated retinoblastoma |
| SBM: | spontaneous bowel movement |
| ST: | heat-stable enterotoxin |
1. Introduction
1.1. Bacterial heat-stable enterotoxins
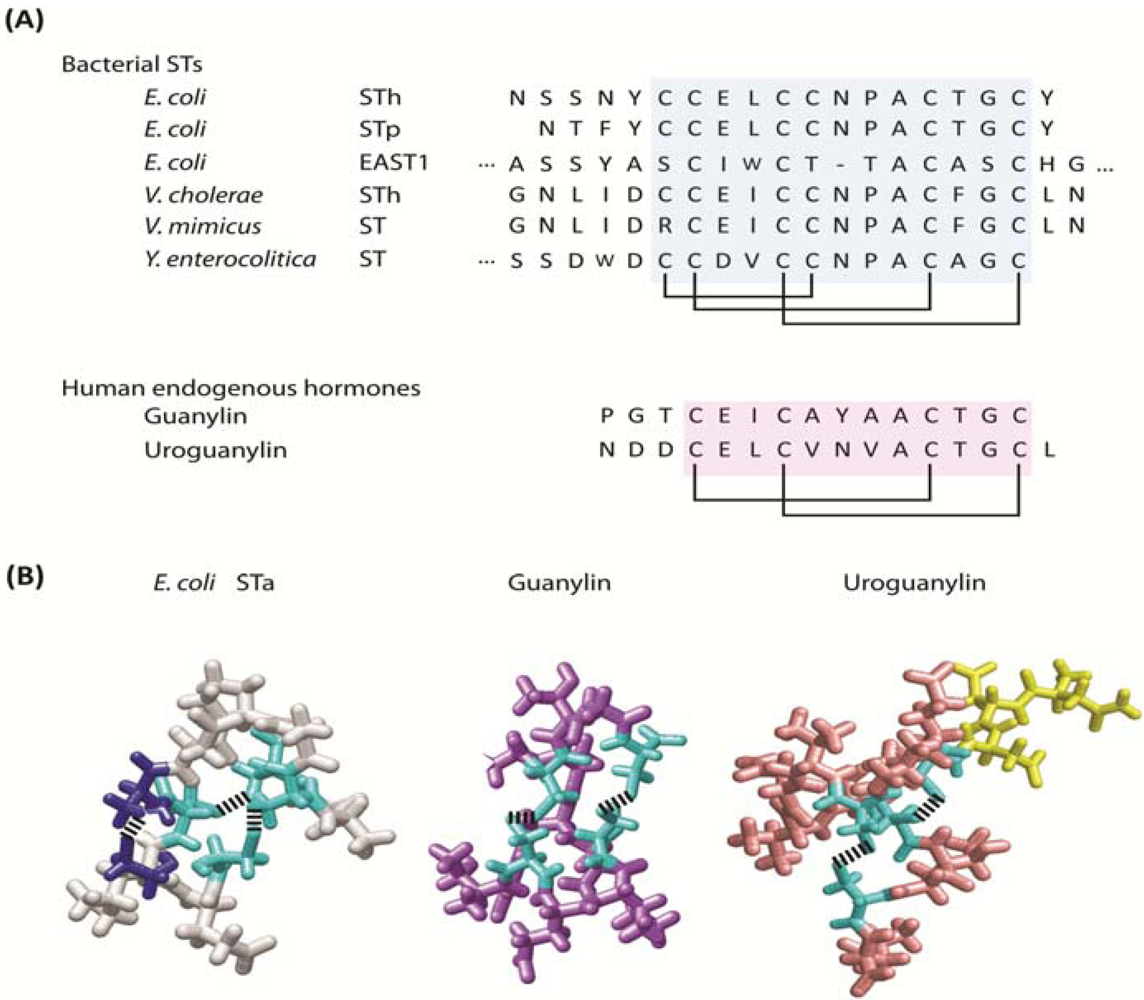
1.2. Molecular mimicry, convergent evolution and the guanylyl cyclase C paracrine hormone axis

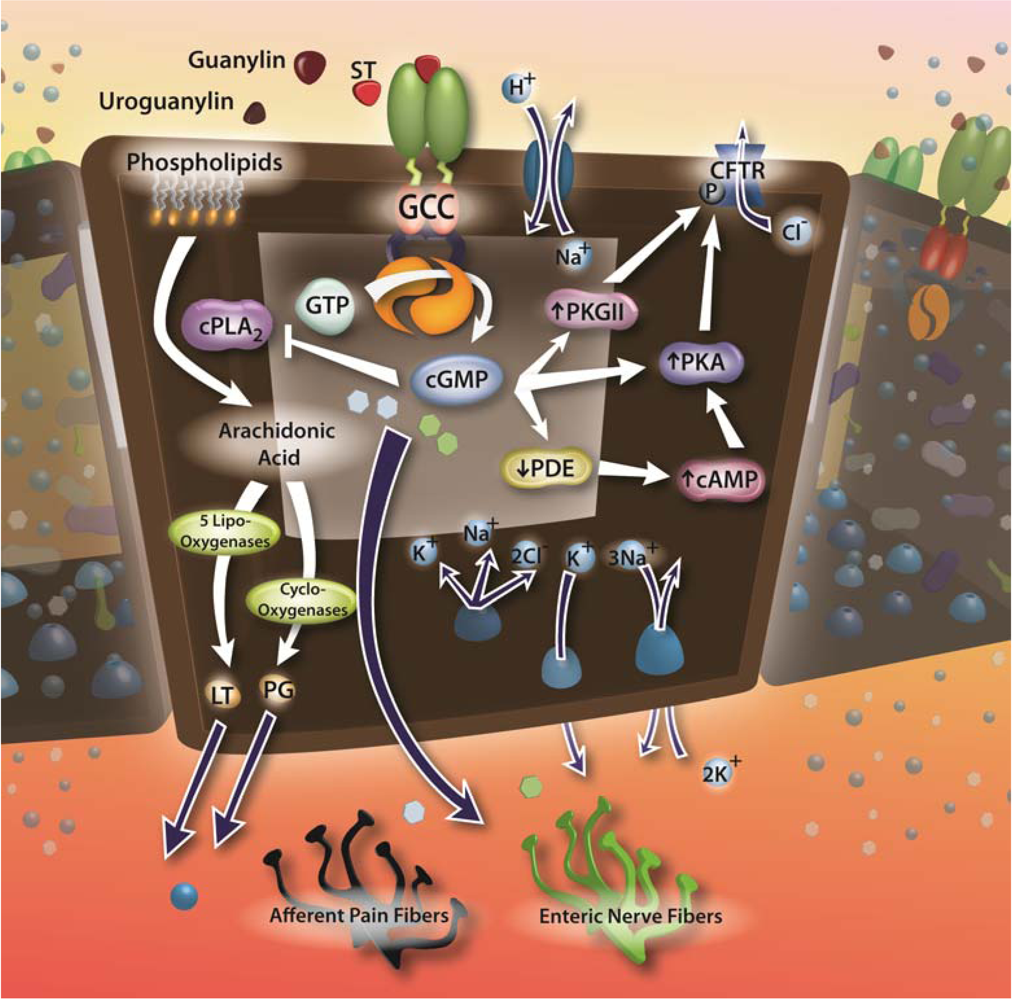
1.3. Guanylyl cyclase C and enterotoxin signaling circuits
2. Enterotoxin Signaling, Irritable Bowel Syndrome and Chronic Constipation
2.1. Enterotoxins, GC-C and fluid-electrolyte homeostasis

2.2. Enterotoxins and irritable bowel syndrome
2.3. Enterotoxin analogs for chronic constipation
2.4. Targeting GC-C in chronic inflammation
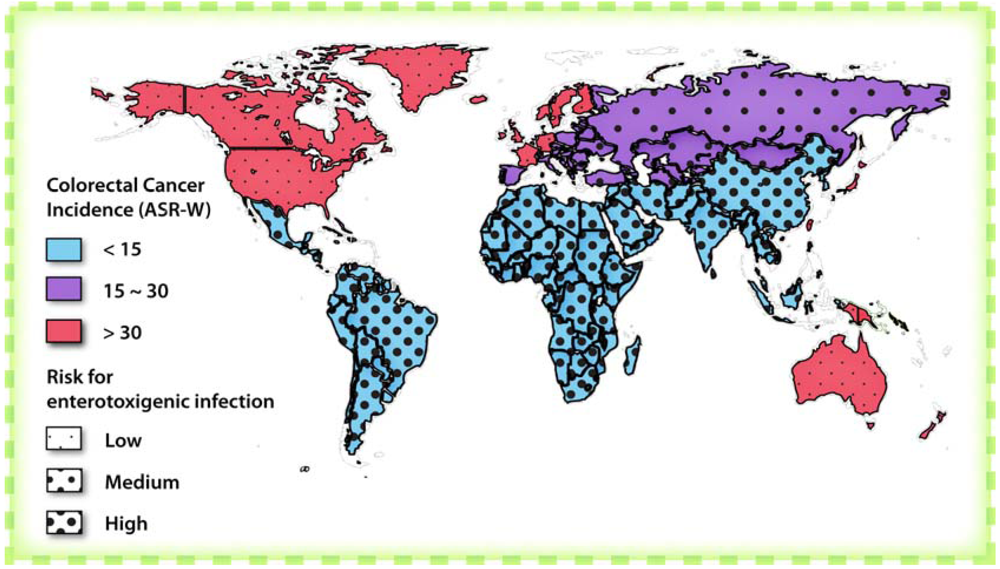
3. Targeting GC-C to Prevent Colorectal Cancer
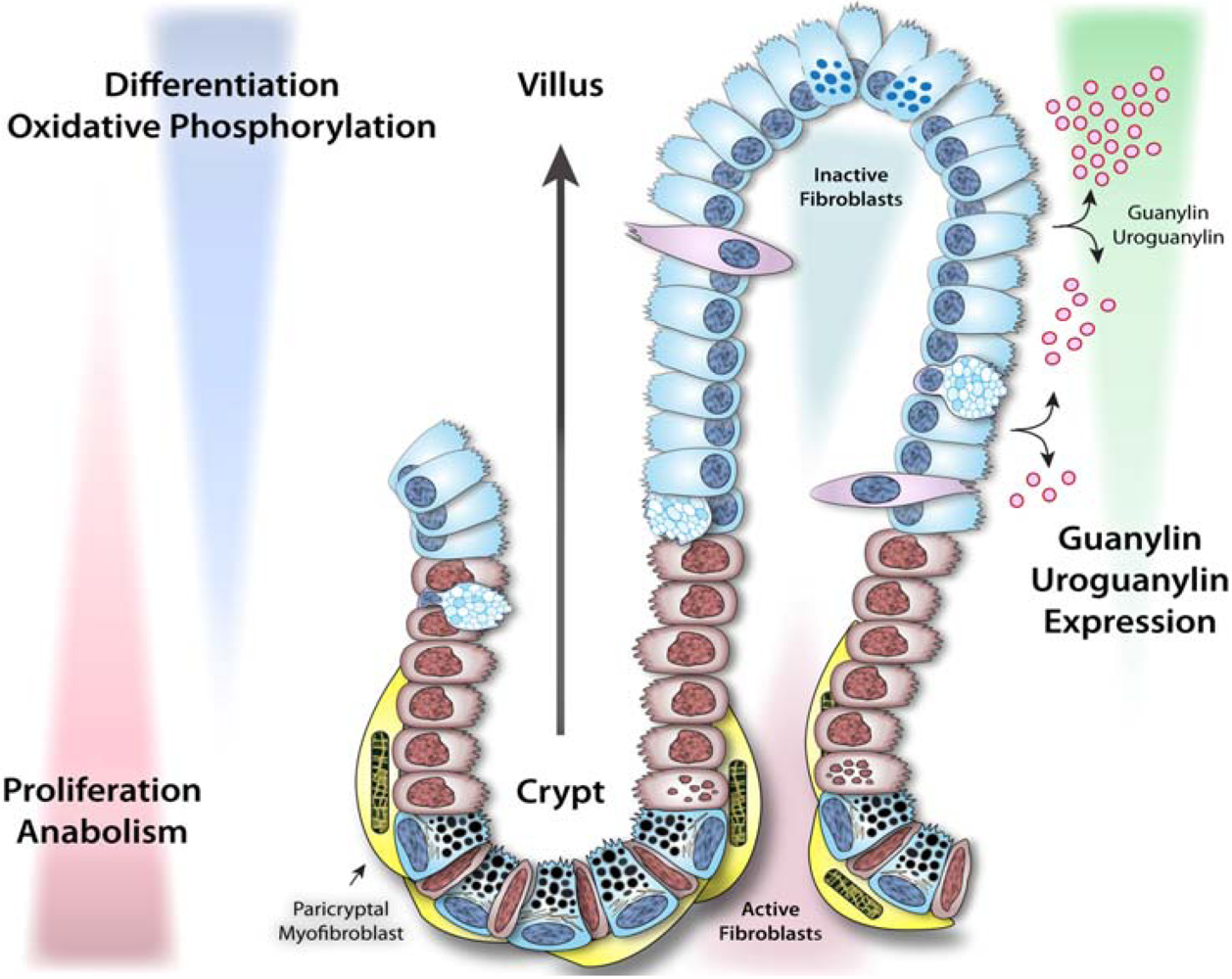
3.1. Dynamics of intestinal epithelial cells and crypt-villus homeostasis
3.2. Enterotoxigenic signaling pathways and intestinal tumorigenesis


3.3. GC-C paracrine hormone replacement to prevent colorectal cancer
4. Diagnostics and Therapeutics Targeted to GC-C for Metastatic Colorectal Cancer
4.1. GC-C as a marker of metastatic colorectal cancer
4.2. GC-C qRT-PCR as a molecular marker to stage patients with colorectal cancer
4.3. GC-C to deliver targeted diagnostics and therapeutics to metastatic colorectal cancer
4.4. GC-C-targeted colorectal cancer vaccines
5. Conclusions
References
- Burgess, M.N.; Bywater, R.J.; Cowley, C.M.; Mullan, N.A.; Newsome, P.M. Biological evaluation of a methanol-soluble, heat-stable Escherichia coli enterotoxin in infant mice, pigs, rabbits, and calves. Infect. Immun. 1978, 21, 526–531. [Google Scholar]
- Pai, C.H.; Mors, V. Production of enterotoxin by Yersinia enterocolitica. Infect. Immun. 1978, 19, 908–911. [Google Scholar]
- Olsson, E.; Soderlind, O. Comparison of different assays for definition of heat-stable enterotoxigenicity of Escherichia coli porcine strains. J. Clin. Microbiol. 1980, 11, 6–15. [Google Scholar]
- Lortie, L.A.; Dubreuil, J.D.; Harel, J. Characterization of Escherichia coli strains producing heat-stable enterotoxin b (STb) isolated from humans with diarrhea. J. Clin. Microbiol. 1991, 29, 656–659. [Google Scholar]
- Dreyfus, L.A.; Harville, B.; Howard, D.E.; Shaban, R.; Beatty, D.M.; Morris, S.J. Calcium influx mediated by the Escherichia coli heat-stable enterotoxin B (STB). Proc. Natl. Acad. Sci. USA 1993, 90, 3202–3206. [Google Scholar]
- Savarino, S.J.; Fasano, A.; Robertson, D.C.; Levine, M.M. Enteroaggregative Escherichia coli elaborate a heat-stable enterotoxin demonstrable in an in vitro rabbit intestinal model. J. Clin. Invest. 1991, 87, 1450–1455. [Google Scholar]
- Guglielmetti, P.; Bravo, L.; Zanchi, A.; Monte, R.; Lombardi, G.; Rossolini, G.M. Detection of the Vibrio cholerae heat-stable enterotoxin gene by polymerase chain reaction. Mol. Cell. Probes 1994, 8, 39–44. [Google Scholar]
- Arita, M.; Honda, T.; Miwatani, T.; Takeda, T.; Takao, T.; Shimonishi, Y. Purification and characterization of a heat-stable enterotoxin of Vibrio mimicus. FEMS Microbiol. Lett. 1991, 63, 105–110. [Google Scholar]
- Guarino, A.; Capano, G.; Malamisura, B.; Alessio, M.; Guandalini, S.; Rubino, A. Production of Escherichia coli STa-like heat-stable enterotoxin by Citrobacter freundii isolated from humans. J. Clin. Microbiol. 1987, 25, 110–114. [Google Scholar]
- Klipstein, F.A.; Engert, R.F.; Houghten, R.A. Immunological properties of purified Klebsiella pneumoniae heat-stable enterotoxin. Infect. Immun. 1983, 42, 838–841. [Google Scholar]
- Giannella, R.A. Escherichia coli heat-stable enterotoxins, guanylins, and their receptors: what are they and what do they do? J. Lab. Clin. Med. 1995, 125, 173–181. [Google Scholar] [PubMed]
- Alderete, J.F.; Robertson, D.C. Repression of heat-stable enterotoxin synthesis in enterotoxigenic Escherichia coli. Infect. Immun. 1977, 17, 629–633. [Google Scholar]
- Johnson, W.M.; Lior, H.; Johnson, K.G. Heat-stable enterotoxin from Escherichia coli: factors involved in growth and toxin production. Infect. Immun. 1978, 20, 352–359. [Google Scholar]
- Lucas, K.A.; Pitari, G.M.; Kazerounian, S.; Ruiz-Stewart, I.; Park, J.; Schulz, S.; Chepenik, K.P.; Waldman, S.A. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol. Rev. 2000, 52, 375–414. [Google Scholar]
- Birbe, R.; Palazzo, J.P.; Walters, R.; Weinberg, D.; Schulz, S.; Waldman, S.A. Guanylyl cyclase C is a marker of intestinal metaplasia, dysplasia, and adenocarcinoma of the gastrointestinal tract. Hum. Pathol. 2005, 36, 170–179. [Google Scholar]
- Schulz, S.; Green, C.K.; Yuen, P.S.; Garbers, D.L. Guanylyl cyclase is a heat-stable enterotoxin receptor. Cell 1990, 63, 941–948. [Google Scholar]
- Vaandrager, A.B.; van der Wiel, E.; Hom, M.L.; Luthjens, L.H.; de Jonge, H.R. Heat-stable enterotoxin receptor/guanylyl cyclase C is an oligomer consisting of functionally distinct subunits, which are non-covalently linked in the intestine. J. Biol. Chem. 1994, 269, 16409–16415. [Google Scholar]
- Lauber, T.; Nourse, A.; Schulz, A.; Marx, U.C. Native and recombinant proguanylin feature identical biophysical properties and are monomeric in solution. Biochemistry 2002, 41, 14602–14612. [Google Scholar]
- Kita, T.; Smith, C.E.; Fok, K.F.; Duffin, K.L.; Moore, W.M.; Karabatsos, P.J.; Kachur, J.F.; Hamra, F.K.; Pidhorodeckyj, N.V.; Forte, L.R.; et al. Characterization of human uroguanylin: a member of the guanylin peptide family. Am. J. Physiol. 1994, 266, F342–F348. [Google Scholar]
- Currie, M.G.; Fok, K.F.; Kato, J.; Moore, R.J.; Hamra, F.K.; Duffin, K.L.; Smith, C.E. Guanylin: an endogenous activator of intestinal guanylate cyclase. Proc. Natl. Acad. Sci. USA 1992, 89, 947–951. [Google Scholar]
- Hamra, F.K.; Forte, L.R.; Eber, S.L.; Pidhorodeckyj, N.V.; Krause, W.J.; Freeman, R.H.; Chin, D.T.; Tompkins, J.A.; Fok, K.F.; Smith, C.E.; et al. Uroguanylin: structure and activity of a second endogenous peptide that stimulates intestinal guanylate cyclase. Proc. Natl. Acad. Sci. USA 1993, 90, 10464–10468. [Google Scholar]
- Wiegand, R.C.; Kato, J.; Huang, M.D.; Fok, K.F.; Kachur, J.F.; Currie, M.G. Human guanylin: cDNA isolation, structure, and activity. FEBS Lett. 1992, 311, 150–154. [Google Scholar]
- Forte, L.R. Guanylin regulatory peptides: structures, biological activities mediated by cyclic GMP and pathobiology. Regul. Pept. 1999, 81, 25–39. [Google Scholar]
- Martin, S.; Adermann, K.; Forssmann, W.G.; Kuhn, M. Regulated, side-directed secretion of proguanylin from isolated rat colonic mucosa. Endocrinology 1999, 140, 5022–5029. [Google Scholar]
- Moss, N.G.; Fellner, R.C.; Qian, X.; Yu, S.J.; Li, Z.; Nakazato, M.; Goy, M.F. Uroguanylin, an intestinal natriuretic peptide, is delivered to the kidney as an unprocessed propeptide. Endocrinology 2008, 149, 4486–4498. [Google Scholar]
- Forte, L.R.; Thorne, P.K.; Eber, S.L.; Krause, W.J.; Freeman, R.H.; Francis, S.H.; Corbin, J.D. Stimulation of intestinal Cl- transport by heat-stable enterotoxin: activation of cAMP-dependent protein kinase by cGMP. Am. J. Physiol. 1992, 263, C607–C615. [Google Scholar]
- Sager, G. Cyclic GMP transporters. Neurochem. Int. 2004, 45, 865–873. [Google Scholar]
- Kuo, J.F.; Greengard, P. Stimulation of adenosine 3',5'-monophosphate-dependent and guanosine 3',5'-monophosphate-dependent protein kinases by some analogs of adenosine 3',5'-monophosphate. Biochem. Biophys. Res. Commun. 1970, 40, 1032–1038. [Google Scholar]
- Lohmann, S.M.; Vaandrager, A.B.; Smolenski, A.; Walter, U.; De Jonge, H.R. Distinct and specific functions of cGMP-dependent protein kinases. Trends Biochem. Sci. 1997, 22, 307–312. [Google Scholar]
- Corbin, J.D.; Lincoln, T.M. Comparison of cAMP and cGMP-dependent protein kinases. Adv. Cyclic Nucleotide Res. 1978, 9, 159–170. [Google Scholar]
- Walter, U. Distribution of cyclic-GMP-dependent protein kinase in various rat tissues and cell lines determined by a sensitive and specific radioimmunoassay. Eur. J. Biochem. 1981, 118, 339–346. [Google Scholar]
- Lincoln, T.M. cGMP-dependent protein kinase. Methods Enzymol. 1983, 99, 62–71. [Google Scholar]
- Francis, S.H.; Corbin, J.D. Purification of cGMP-binding protein phosphodiesterase from rat lung. Methods Enzymol. 1988, 159, 722–729. [Google Scholar]
- Lincoln, T.M.; Thompson, M.; Cornwell, T.L. Purification and characterization of two forms of cyclic GMP-dependent protein kinase from bovine aorta. J. Biol. Chem. 1988, 263, 17632–17637. [Google Scholar]
- Markert, T.; Vaandrager, A.B.; Gambaryan, S.; Pohler, D.; Hausler, C.; Walter, U.; De Jonge, H.R.; Jarchau, T.; Lohmann, S.M. Endogenous expression of type II cGMP-dependent protein kinase mRNA and protein in rat intestine. Implications for cystic fibrosis transmembrane conductance regulator. J. Clin. Invest. 1995, 96, 822–830. [Google Scholar] [PubMed]
- Sandberg, M.; Natarajan, V.; Ronander, I.; Kalderon, D.; Walter, U.; Lohmann, S.M.; Jahnsen, T. Molecular cloning and predicted full-length amino acid sequence of the type I beta isozyme of cGMP-dependent protein kinase from human placenta. Tissue distribution and developmental changes in rat. FEBS Lett. 1989, 255, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Tamura, N.; Ogawa, Y.; Yasoda, A.; Itoh, H.; Saito, Y.; Nakao, K. Two cardiac natriuretic peptide genes (atrial natriuretic peptide and brain natriuretic peptide) are organized in tandem in the mouse and human genomes. J. Mol. Cell. Cardiol. 1996, 28, 1811–1815. [Google Scholar]
- Wolfe, L.; Corbin, J.D.; Francis, S.H. Characterization of a novel isozyme of cGMP-dependent protein kinase from bovine aorta. J. Biol. Chem. 1989, 264, 7734–7741. [Google Scholar]
- Wernet, W.; Flockerzi, V.; Hofmann, F. The cDNA of the two isoforms of bovine cGMP-dependent protein kinase. FEBS Lett. 1989, 251, 191–196. [Google Scholar]
- Francis, S.H.; Corbin, J.D. Cyclic nucleotide-dependent protein kinases: intracellular receptors for cAMP and cGMP action. Crit. Rev. Clin. Lab. Sci. 1999, 36, 275–328. [Google Scholar]
- Guerrant, R.L.; Hughes, J.M.; Chang, B.; Robertson, D.C.; Murad, F. Activation of intestinal guanylate cyclase by heat-stable enterotoxin of Escherichia coli: studies of tissue specificity, potential receptors, and intermediates. J. Infect. Dis. 1980, 142, 220–228. [Google Scholar]
- Haberberger, R.L., Jr.; Mikhail, I.A.; Burans, J.P.; Hyams, K.C.; Glenn, J.C.; Diniega, B.M.; Sorgen, S.; Mansour, N.; Blacklow, N.R.; Woody, J.N. Travelers' diarrhea among United States military personnel during joint American-Egyptian armed forces exercises in Cairo, Egypt. Mil. Med. 1991, 156, 27–30. [Google Scholar]
- Carrithers, S.L.; Ott, C.E.; Hill, M.J.; Johnson, B.R.; Cai, W.; Chang, J.J.; Shah, R.G.; Sun, C.; Mann, E.A.; Fonteles, M.C.; Forte, L.R.; Jackson, B.A.; Giannella, R.A.; Greenberg, R.N. Guanylin and uroguanylin induce natriuresis in mice lacking guanylyl cyclase-C receptor. Kidney Int. 2004, 65, 40–53. [Google Scholar]
- Steinbrecher, K.A.; Mann, E.A.; Giannella, R.A.; Cohen, M.B. Increases in guanylin and uroguanylin in a mouse model of osmotic diarrhea are guanylate cyclase C-independent. Gastroenterology 2001, 121, 1191–1202. [Google Scholar]
- Schulz, S.; Lopez, M.J.; Kuhn, M.; Garbers, D.L. Disruption of the guanylyl cyclase-C gene leads to a paradoxical phenotype of viable but heat-stable enterotoxin-resistant mice. J. Clin. Invest. 1997, 100, 1590–1595. [Google Scholar]
- Mathias, J.R.; Carlson, G.M.; DiMarino, A.J. Intestinal myoelectric activity in response to live Vibrio cholerae and cholera enterotoxin. J. Clin. Invest. 1976, 58, 91–96. [Google Scholar]
- Burns, T.W.; Mathias, J.R.; Carlson, G.M. Effect of toxigenic Escherichia coli on myoelectric activity of small intestine. Am. J. Physiol. Endocrinol. Metab. 1978, 235, E311–E315. [Google Scholar]
- Sjogren, R.W.; Sherman, P.M.; Boedeker, E.C. Altered intestinal motility precedes diarrhea during Escherichia coli enteric infection. Am. J. Physiol. Gastrointest. Liver Physiol. 1989, 257, G725–G731. [Google Scholar]
- Mathias, J.R.; Nogueira, J.; Martin, J.L.; Carlson, G.M.; Giannella, R.A. Escherichia coli heat-stable toxin: its effect on motility of the small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 1982, 242, G360–G363. [Google Scholar]
- Roussel, A.J.; Woode, G.N.; Waldron, R.C.; Sriranganathan, N.; Jones, M.K. Myoelectric activity of the small intestine in enterotoxin-induced diarrhea of calves. Am. J. Vet. Res. 1992, 53, 1145–1148. [Google Scholar]
- Lundgren, O.; Svanvik, J.; Jivegard, L. Enteric nervous system. I. Physiology and pathophysiology of the intestinal tract. Dig. Dis. Sci. 1989, 34, 264–283. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, F.A.; Zhu, B. Effects of indomethacin, acetazolamide, ethacrynate sodium, and atropine on intestinal secretion mediated by Escherichia coli heat-stable enterotoxin in pig jejunum. Can. J. Physiol. Pharmacol. 1982, 60, 1281–1286. [Google Scholar]
- Beubler, E.; Badhri, P.; Schirgi, D. 5-HT receptor antagonists and heat-stable Escherichia coli enterotoxin-induced effects in the rat. Eur. J. Pharmacol. 1992, 219, 445–450. [Google Scholar]
- Beubler, E.; Schirgi-Degen, A.; Gamse, R. Inhibition of 5-hydroxytryptamine- and enterotoxin-induced fluid secretion by 5-HT receptor antagonists in the rat jejunum. Eur. J. Pharmacol. 1993, 248, 157–162. [Google Scholar]
- Rolfe, V.; Levin, R.J. Enterotoxin Escherichia coli STa activates a nitric oxide-dependent myenteric plexus secretory reflex in the rat ileum. J. Physiol. 1994, 475, 531–537. [Google Scholar]
- Hayden, U.L.; Greenberg, R.N.; Carey, H.V. Role of prostaglandins and enteric nerves in Escherichia coli heat-stable enterotoxin (STa)-induced intestinal secretion in pigs. Am. J. Vet. Res. 1996, 57, 211–215. [Google Scholar]
- Rolfe, V.E.; Levin, R.J. Vagotomy inhibits the jejunal fluid secretion activated by luminal ileal Escherichia coli STa in the rat in vivo. Gut 1999, 44, 615–619. [Google Scholar]
- Eklund, S.; Jodal, M.; Lundgren, O. The enteric nervous system participates in the secretory response to the heat stable enterotoxins of Escherichia coli in rats and cats. Neuroscience 1985, 14, 673–681. [Google Scholar]
- Forte, L.R., Jr. Uroguanylin and guanylin peptides: pharmacology and experimental therapeutics. Pharmacol. Ther. 2004, 104, 137–162. [Google Scholar]
- Grundmann, O.; Yoon, S.L. Irritable bowel syndrome: Epidemiology, diagnosis and treatment: An update for health-care practitioners. Clin. Gastroenterol. Hepatol. 2010, 25, 691–699. [Google Scholar]
- Cash, B.D.; Chey, W.D. Diagnosis of irritable bowel syndrome. Gastroenterol. Clin. North Am. 2005, 34, 205–220. [Google Scholar]
- Saito, Y.A.; Schoenfeld, P.; Locke, G.R., III. The epidemiology of irritable bowel syndrome in North America: A systematic review. Am. J. Gastroenterol. 2002, 97, 1910–1915. [Google Scholar]
- Saito, Y.A.; Talley, N.J.; Melton, L.J., III; Fett, S.; Zinsmeister, A.R.; Locke, G.R., III. The effect of new diagnostic criteria for irritable bowel syndrome on community prevalence estimates. Neurogastroenterol. Motil. 2003, 15, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Andrews, E.B.; Eaton, S.C.; Hollis, K.A.; Hopkins, J.S.; Ameen, V.; Hamm, L.R.; Cook, S.F.; Tennis, P.; Mangel, A.W. Prevalence and demographics of irritable bowel syndrome: Results from a large web-based survey. Aliment. Pharmacol. Ther. 2005, 22, 935–942. [Google Scholar]
- Hungin, A.P.S.; Chang, L.; Locke, G.R.; Dennis, E.H.; Barghout, V. Irritable bowel syndrome in the United States: Prevalence, symptom patterns and impact. Aliment. Pharmacol. Ther. 2005, 21, 1365–1375. [Google Scholar]
- Higgins, P.D.; Johanson, J.F. Epidemiology of constipation in North America: a systematic review. Am. J. Gastroenterol. 2004, 99, 750–759. [Google Scholar]
- Damon, H.; Dumas, P.; Mion, F. Impact of anal incontinence and chronic constipation on quality of life. Gastroenterol. Clin. Biol. 2004, 28, 16–20. [Google Scholar]
- Gralnek, I.M.; Hays, R.D.; Kilbourne, A.A.; Naliboff, B.; Mayer, E.A. The impact of irritable bowel syndrome on health-related quality of life. Gastroenterology 2000, 119, 654–660. [Google Scholar]
- Longstreth, G.F.; Wilson, A.; Knight, K.; Wong, J.; Chiou, C.F.; Barghout, V.; Frech, F.; Ofman, J.J. Irritable bowel syndrome, health care use, and costs: A U.S. managed care perspective. Am. J. Gastroenterol. 2003, 98, 600–607. [Google Scholar] [PubMed]
- Camilleri, M.; Williams, D.E. Economic burden of irritable bowel syndrome: Proposed strategies to control expenditures. Pharmacoeconomics 2000, 17, 331–338. [Google Scholar]
- Dennison, C.; Prasad, M.; Lloyd, A.; Bhattacharyya, S.K.; Dhawan, R.; Coyne, K. The health-related quality of life and economic burden of constipation. Pharmacoeconomics 2005, 23, 461–476. [Google Scholar]
- Leong, S.A.; Barghout, V.; Birnbaum, H.G.; Thibeault, C.E.; Ben-Hamadi, R.; Frech, F.; Ofman, J.J. The economic consequences of irritable bowel syndrome: A US employer perspective. Arch. Intern. Med. 2003, 163, 929–935. [Google Scholar]
- Yawn, B.P.; Lydick, E.; Locke, G.R.; Wollan, P.C.; Bertram, S.L.; Kurland, M.J. Do published guidelines for evaluation of Irritable Bowel Syndrome reflect practice? BMC Gastroenterol. 2001, 1, 11. [Google Scholar] [Green Version]
- Wahnschaffe, U.; Ullrich, R.; Riecken, E.O.; Schulzke, J.D. Celiac disease-like abnormalities in a subgroup of patients with irritable bowel syndrome. Gastroenterology 2001, 121, 1329–1338. [Google Scholar]
- Bryant, A.P.; Busby, R.W.; Cordero, E.A. MD-1100, a therapeutic agent in development for the treatment of IBS-C, enhances intestinal secretion and transit, decreases visceral pain and is minimally absorbed in rats. Gastroenterology 2005, 128, 464. [Google Scholar]
- Eutamene, H.; Bradesi, S.; Larauche, M.; Theodorou, V.; Beaufrand, C.; Ohning, G.; Fioramonti, J.; Cohen, M.; Bryant, A.P.; Kurtz, C.; Currie, M.G.; Mayer, E.A.; Bueno, L. Guanylate cyclase C-mediated antinociceptive effects of linaclotide in rodent models of visceral pain. Neurogastroenterol. Motil. 2010, 22, 312–e384. [Google Scholar]
- Kurtz, C.B.; Fitch, D.; Busby, R.W.; Fretzen, A.; Geis, S.; Currie, M.G. Effects of multidose administration of MD-1100 on safety, tolerability, exposure, and pharmacodynamics in healthy subjects. Gastroenterology 2006, 130, A26. [Google Scholar]
- Currie, M.G.; Kurtz, C.B.; Mahajan-Miklos, S.; Busby, R.; Fretzen, A.; Geis, S. Effects of single dose administration of MD-1100 on safety, tolerability, exposure, and stool consistency in healthy subjects. Am. J. Gastroenterol. 2005, 100, S328. [Google Scholar]
- Andresen, V.; Camilleri, M.; Busciglio, I.A.; Grudell, A.; Burton, D.; McKinzie, S.; Foxx-Orenstein, A.; Kurtz, C.B.; Sharma, V.; Johnston, J.M.; Currie, M.G.; Zinsmeister, A.R. Effect of 5 days linaclotide on transit and bowel function in females with constipation-predominant irritable bowel syndrome. Gastroenterology 2007, 133, 761–768. [Google Scholar]
- Johnston, J.M.; Kurtz, C.B.; Drossman, D.A.; Lembo, A.J.; Jeglinski, B.I.; MacDougall, J.E.; Antonelli, S.M.; Currie, M.G. Pilot study on the effect of linaclotide in patients with chronic constipation. Am. J. Gastroenterol. 2009, 104, 125–132. [Google Scholar]
- Lembo, A.J.; Kurtz, C.B.; Macdougall, J.E.; Lavins, B.J.; Currie, M.G.; Fitch, D.A.; Jeglinski, B.I.; Johnston, J.M. Efficacy of linaclotide for patients with chronic constipation. Gastroenterology 2010, 138, 886–895.e1. [Google Scholar] [PubMed]
- Synergy Pharmaceuticals pipeline: Basic science—GC-C agonists. Available online: http://www.synergybio.net/basic_science.htm (Accessed on 3 August 2010).
- Kubes, P.; Suzuki, M.; Granger, D.N. Nitric oxide: An endogenous modulator of leukocyte adhesion. Proc. Natl. Acad. Sci. USA 1991, 88, 4651–4655. [Google Scholar]
- Khoshakhlagh, P.; Bahrololoumi-Shapourabadi, M.; Mohammadirad, A.; Ashtaral-Nakhai, L.; Minaie, B.; Abdollahi, M. Beneficial Effect of Phosphodiesterase-5 Inhibitor in Experimental Inflammatory Bowel Disease; Molecular Evidence for Involvement of Oxidative Stress. Toxicol. Mech. Methods 2007, 17, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Mutlu, E.; Guzman, J.P.; Forsyth, C.; Banan, A. Phosphodiesterase 4 inhibitors and inflammatory bowel disease: Emerging therapies in inflammatory bowel disease. Expert Opin. Investig. Drugs 2007, 16, 1489–1506. [Google Scholar]
- Murthy, K.S.; Makhlouf, G.M. Differential Regulation of Phospholipase A2(PLA2)-dependent Ca2+ Signaling in Smooth Muscle by cAMP- and cGMP-dependent Protein Kinases. J. Biol. Chem. 1998, 273, 34519–34526. [Google Scholar]
- Pitari, G.M.; Li, P.; Lin, J.E.; Zuzga, D.; Gibbons, A.V.; Snook, A.E.; Schulz, S.; Waldman, S.A. The paracrine hormone hypothesis of colorectal cancer. Clin. Pharmacol. Ther. 2007, 82, 441–447. [Google Scholar]
- Gassler, N.; Newrzella, D.; Bohm, C.; Lyer, S.; Li, L.; Sorgenfrei, O.; van Laer, L.; Sido, B.; Mollenhauer, J.; Poustka, A.; Schirmacher, P.; Gretz, N. Molecular characterisation of non-absorptive and absorptive enterocytes in human small intestine. Gut 2006, 55, 1084–1089. [Google Scholar]
- Lin, J.E.; Li, P.; Snook, A.E.; Schulz, S.; Dasgupta, A.; Hyslop, T.M.; Gibbons, A.V.; Marszlowicz, G.; Pitari, G.M.; Waldman, S.A. The hormone receptor GUCY2C suppresses intestinal tumor formation by inhibiting AKT signaling. Gastroenterology 2010, 138, 241–254. [Google Scholar]
- Barker, N.; van de Wetering, M.; Clevers, H. The intestinal stem cell. Genes Dev. 2008, 22, 1856–1864. [Google Scholar]
- Humphries, A.; Wright, N.A. Colonic crypt organization and tumorigenesis. Nat. Rev. Cancer 2008, 8, 415–424. [Google Scholar]
- van den Brink, G.R.; Offerhaus, G.J. The morphogenetic code and colon cancer development. Cancer Cell 2007, 11, 109–117. [Google Scholar]
- Wang, P.Y.; Caspi, L.; Lam, C.K.; Chari, M.; Li, X.; Light, P.E.; Gutierrez-Juarez, R.; Ang, M.; Schwartz, G.J.; Lam, T.K. Upper intestinal lipids trigger a gut-brain-liver axis to regulate glucose production. Nature 2008, 452, 1012–1016. [Google Scholar]
- Murphy, K.G.; Bloom, S.R. Gut hormones and the regulation of energy homeostasis. Nature 2006, 444, 854–859. [Google Scholar]
- Rosen, C.J. Breaking into bone biology: serotonin's secrets. Nat. Med. 2009, 15, 145–146. [Google Scholar]
- Yadav, V.K.; Ryu, J.H.; Suda, N.; Tanaka, K.F.; Gingrich, J.A.; Schutz, G.; Glorieux, F.H.; Chiang, C.Y.; Zajac, J.D.; Insogna, K.L.; Mann, J.J.; Hen, R.; Ducy, P.; Karsenty, G. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell 2008, 135, 825–837. [Google Scholar]
- Koldovsky, O.; Dobiasova, M.; Hahn, P.; Kolinska, J.; Kraml, J.; Pacha, J. Development of gastrointestinal functions. Physiol. Res. 1995, 44, 341–348. [Google Scholar]
- Skipper, M.; Lewis, J. Getting to the guts of enteroendocrine differentiation. Nat. Genet. 2000, 24, 3–4. [Google Scholar]
- Bry, L.; Falk, P.; Huttner, K.; Ouellette, A.; Midtvedt, T.; Gordon, J.I. Paneth cell differentiation in the developing intestine of normal and transgenic mice. Proc. Natl. Acad. Sci. USA 1994, 91, 10335–10339. [Google Scholar]
- Pitari, G.M.; Zingman, L.V.; Hodgson, D.M.; Alekseev, A.E.; Kazerounian, S.; Bienengraeber, M.; Hajnoczky, G.; Terzic, A.; Waldman, S.A. Bacterial enterotoxins are associated with resistance to colon cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 2695–2699. [Google Scholar]
- Shailubhai, K.; Yu, H.H.; Karunanandaa, K.; Wang, J.Y.; Eber, S.L.; Wang, Y.; Joo, N.S.; Kim, H.D.; Miedema, B.W.; Abbas, S.Z.; Boddupalli, S.S.; Currie, M.G.; Forte, L.R. Uroguanylin treatment suppresses polyp formation in the Apc(Min/+) mouse and induces apoptosis in human colon adenocarcinoma cells via cyclic GMP. Cancer Res. 2000, 60, 5151–5157. [Google Scholar]
- Li, Z.; Taylor-Blake, B.; Light, A.R.; Goy, M.F. Guanylin, an endogenous ligand for C-type guanylate cyclase, is produced by goblet cells in the rat intestine. Gastroenterology 1995, 109, 1863–1875. [Google Scholar]
- Cohen, M.B.; Witte, D.P.; Hawkins, J.A.; Currie, M.G. Immunohistochemical localization of guanylin in the rat small intestine and colon. Biochem. Biophys. Res. Commun. 1995, 209, 803–808. [Google Scholar]
- Perkins, A.; Goy, M.F.; Li, Z. Uroguanylin is expressed by enterochromaffin cells in the rat gastrointestinal tract. Gastroenterology 1997, 113, 1007–1014. [Google Scholar]
- Nakazato, M.; Yamaguchi, H.; Date, Y.; Miyazato, M.; Kangawa, K.; Goy, M.F.; Chino, N.; Matsukura, S. Tissue distribution, cellular source, and structural analysis of rat immunoreactive uroguanylin. Endocrinology 1998, 139, 5247–5254. [Google Scholar]
- Birkenkamp-Demtroder, K.; Lotte Christensen, L.; Harder Olesen, S.; Frederiksen, C.M.; Laiho, P.; Aaltonen, L.A.; Laurberg, S.; Sorensen, F.B.; Hagemann, R.; Orntoft, T.F. Gene expression in colorectal cancer. Cancer Res. 2002, 62, 4352–4363. [Google Scholar]
- Cohen, M.B.; Hawkins, J.A.; Witte, D.P. Guanylin mRNA expression in human intestine and colorectal adenocarcinoma. Lab. Invest. 1998, 78, 101–108. [Google Scholar]
- Notterman, D.A.; Alon, U.; Sierk, A.J.; Levine, A.J. Transcriptional gene expression profiles of colorectal adenoma, adenocarcinoma, and normal tissue examined by oligonucleotide arrays. Cancer Res. 2001, 61, 3124–3130. [Google Scholar]
- Steinbrecher, K.A.; Wowk, S.A.; Rudolph, J.A.; Witte, D.P.; Cohen, M.B. Targeted inactivation of the mouse guanylin gene results in altered dynamics of colonic epithelial proliferation. Am. J. Pathol. 2002, 161, 2169–2178. [Google Scholar]
- Li, P.; Schulz, S.; Bombonati, A.; Palazzo, J.P.; Hyslop, T.M.; Xu, Y.; Barab, A.A.; Siracusa, L.D.; Pitari, G.M.; Waldman, S.A. Guanylyl cyclase C suppresses intestinal tumorigenesis by restricting proliferation and maintaining genomic integrity. Gastroenterology 2007, 133, 599–607. [Google Scholar]
- Li, P.; Lin, J.E.; Chervoneva, I.; Schulz, S.; Waldman, S.A.; Pitari, G.M. Homeostatic control of the crypt-villus axis by the bacterial enterotoxin receptor guanylyl cyclase C restricts the proliferating compartment in intestine. Am. J. Pathol. 2007, 171, 1847–1858. [Google Scholar]
- Yang, Q.; Bermingham, N.A.; Finegold, M.J.; Zoghbi, H.Y. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 2001, 294, 2155–2158. [Google Scholar]
- Aoki, K.; Tamai, Y.; Horiike, S.; Oshima, M.; Taketo, M.M. Colonic polyposis caused by mTOR-mediated chromosomal instability in Apc+/Delta716 Cdx2+/− compound mutant mice. Nat. Genet. 2003, 35, 323–330. [Google Scholar]
- Spruck, C.H.; Won, K.A.; Reed, S.I. Deregulated cyclin E induces chromosome instability. Nature 1999, 401, 297–300. [Google Scholar]
- Li, P.; Lin, J.E.; Snook, A.E.; Gibbons, A.; Zuzga, D.; Schulz, S.; Pitari, G.M.; Waldman, S.A. Colorectal cancer as a paracrine deficiency syndrome amenable to oral hormone replacement therapy. Clin. Transl. Sci. 2008, 1, 163–167. [Google Scholar]
- Pitari, G.M.; Baksh, R.I.; Harris, D.M.; Li, P.; Kazerounian, S.; Waldman, S.A. Interruption of homologous desensitization in cyclic guanosine 3',5'-monophosphate signaling restores colon cancer cytostasis by bacterial enterotoxins. Cancer Res. 2005, 65, 11129–11135. [Google Scholar]
- Pitari, G.M.; Di Guglielmo, M.D.; Park, J.; Schulz, S.; Waldman, S.A. Guanylyl cyclase C agonists regulate progression through the cell cycle of human colon carcinoma cells. Proc. Natl. Acad. Sci. USA 2001, 98, 7846–7851. [Google Scholar]
- Lin, J.E.; Li, P.; Pitari, G.M.; Schulz, S.; Waldman, S.A. Guanylyl cyclase C in colorectal cancer: susceptibility gene and potential therapeutic target. Future Oncol. 2009, 5, 509–522. [Google Scholar]
- Shen, W.; Hintze, T.H.; Wolin, M.S. Nitric oxide. An important signaling mechanism between vascular endothelium and parenchymal cells in the regulation of oxygen consumption. Circulation 1995, 92, 3505–3512. [Google Scholar] [PubMed]
- Nisoli, E.; Falcone, S.; Tonello, C.; Cozzi, V.; Palomba, L.; Fiorani, M.; Pisconti, A.; Brunelli, S.; Cardile, A.; Francolini, M.; Cantoni, O.; Carruba, M.O.; Moncada, S.; Clementi, E. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. USA 2004, 101, 16507–16512. [Google Scholar]
- Ndisang, J.F.; Jadhav, A. Upregulating the heme oxygenase system enhances insulin sensitivity and improves glucose metabolism in insulin-resistant diabetes in rats. Endocrinology 2009, 150, 2627–2636. [Google Scholar]
- Perk, H.; Armagan, A.; Naziroglu, M.; Soyupek, S.; Hoscan, M.B.; Sutcu, R.; Ozorak, A.; Delibas, N. Sildenafil citrate as a phosphodiesterase inhibitor has an antioxidant effect in the blood of men. J. Clin. Pharm. Ther. 2008, 33, 635–640. [Google Scholar]
- Garin-Laflam, M.P.; Steinbrecher, K.A.; Rudolph, J.A.; Mao, J.; Cohen, M.B. Activation of guanylate cyclase C signaling pathway protects intestinal epithelial cells from acute radiation-induced apoptosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G740–G749. [Google Scholar]
- Carrithers, S.L.; Barber, M.T.; Biswas, S.; Parkinson, S.J.; Park, P.K.; Goldstein, S.D.; Waldman, S.A. Guanylyl cyclase C is a selective marker for metastatic colorectal tumors in human extraintestinal tissues. Proc. Natl. Acad. Sci. USA 1996, 93, 14827–14832. [Google Scholar]
- Buc, E.; Vartanian, M.D.; Darcha, C.; Dechelotte, P.; Pezet, D. Guanylyl cyclase C as a reliable immunohistochemical marker and its ligand Escherichia coli heat-stable enterotoxin as a potential protein-delivering vehicle for colorectal cancer cells. Eur. J. Cancer. 2005, 41, 1618–1627. [Google Scholar]
- Gold, P.; Freedman, S.O. Demonstration of Tumor-Specific Antigens in Human Colonic Carcinomata by Immunological Tolerance and Absorption Techniques. J. Exp. Med. 1965, 121, 439–462. [Google Scholar]
- Arakawa, F.; Shibaguchi, H.; Xu, Z.; Kuroki, M. Targeting of T cells to CEA-expressing tumor cells by chimeric immune receptors with a highly specific single-chain anti-CEA activity. Anticancer Res. 2002, 22, 4285–4289. [Google Scholar]
- Goldenberg, D.M.; Neville, A.M.; Carter, A.C.; Go, V.L.; Holyoke, E.D.; Isselbacher, K.J.; Schein, P.S.; Schwartz, M. CEA (carcinoembryonic antigen): its role as a marker in the management of cancer. J. Cancer Res. Clin. Oncol. 1981, 101, 239–242. [Google Scholar]
- Iddings, D.; Ahmad, A.; Elashoff, D.; Bilchik, A. The prognostic effect of micrometastases in previously staged lymph node negative (N0) colorectal carcinoma: a meta-analysis. Ann. Surg. Oncol. 2006, 13, 1386–1392. [Google Scholar]
- Nicastri, D.G.; Doucette, J.T.; Godfrey, T.E.; Hughes, S.J. Is occult lymph node disease in colorectal cancer patients clinically significant? A review of the relevant literature. J. Mol. Diagn. 2007, 9, 563–571. [Google Scholar]
- Compton, C.C.; Greene, F.L. The staging of colorectal cancer: 2004 and beyond. CA Cancer J. Clin. 2004, 54, 295–308. [Google Scholar]
- Andre, T.; Boni, C.; Mounedji-Boudiaf, L.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C.; Zaninelli, M.; Clingan, P.; Bridgewater, J.; Tabah-Fisch, I.; de Gramont, A. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N. Engl. J. Med. 2004, 350, 2343–2351. [Google Scholar]
- Mamounas, E.; Wieand, S.; Wolmark, N.; Bear, H.D.; Atkins, J.N.; Song, K.; Jones, J.; Rockette, H. Comparative efficacy of adjuvant chemotherapy in patients with Dukes' B versus Dukes' C colon cancer: results from four National Surgical Adjuvant Breast and Bowel Project adjuvant studies (C-01, C-02, C-03, and C-04). J. Clin. Oncol. 1999, 17, 1349–1355. [Google Scholar] [PubMed]
- Meyerhardt, J.A.; Mayer, R.J. Systemic therapy for colorectal cancer. N. Engl. J. Med. 2005, 352, 476–487. [Google Scholar]
- Quasar Collaborative, G.; Gray, R.; Barnwell, J.; McConkey, C.; Hills, R.K.; Williams, N.S.; Kerr, D.J. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007, 370, 2020–2029. [Google Scholar]
- Wolpin, B.M.; Meyerhardt, J.A.; Mamon, H.J.; Mayer, R.J. Adjuvant treatment of colorectal cancer. CA Cancer J. Clin. 2007, 57, 168–185. [Google Scholar]
- Benson, A.B., III; Schrag, D.; Somerfield, M.R.; Cohen, A.M.; Figueredo, A.T.; Flynn, P.J.; Krzyzanowska, M.K.; Maroun, J.; McAllister, P.; Van Cutsem, E.; Brouwers, M.; Charette, M.; Haller, D.G. American Society of Clinical Oncology recommendations on adjuvant chemotherapy for stage II colon cancer. J. Clin. Oncol. 2004, 22, 3408–3419. [Google Scholar] [PubMed]
- Gill, S.; Loprinzi, C.L.; Sargent, D.J.; Thome, S.D.; Alberts, S.R.; Haller, D.G.; Benedetti, J.; Francini, G.; Shepherd, L.E.; Francois Seitz, J.; Labianca, R.; Chen, W.; Cha, S.S.; Heldebrant, M.P.; Goldberg, R.M. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J. Clin. Oncol. 2004, 22, 1797–1806. [Google Scholar] [PubMed]
- Greene, F. References. In AJCC Cancer Staging Handbook: From the AJCC Cancer Staging Manual, 7th; Edge, S.B., Byrd, D.R., Compton, C.C., Fritz, A.G., Greene, F.L., Trotti, A., Eds.; Springer: New York, NY, USA, 2002; Volume 6, pp. 27–38, 153–218. [Google Scholar]
- Schulz, S.; Hyslop, T.; Haaf, J.; Bonaccorso, C.; Nielsen, K.; Witek, M.E.; Birbe, R.; Palazzo, J.; Weinberg, D.; Waldman, S.A. A validated quantitative assay to detect occult micrometastases by reverse transcriptase-polymerase chain reaction of guanylyl cyclase C in patients with colorectal cancer. Clin. Cancer Res. 2006, 12, 4545–4552. [Google Scholar]
- Witek, M.E.; Nielsen, K.; Walters, R.; Hyslop, T.; Palazzo, J.; Schulz, S.; Waldman, S.A. The putative tumor suppressor Cdx2 is overexpressed by human colorectal adenocarcinomas. Clin. Cancer Res. 2005, 11, 8549–8556. [Google Scholar]
- Waldman, S.A.; Hyslop, T.; Schulz, S.; Barkun, A.; Nielsen, K.; Haaf, J.; Bonaccorso, C.; Li, Y.; Weinberg, D.S. Association of GUCY2C expression in lymph nodes with time to recurrence and disease-free survival in pN0 colorectal cancer. JAMA 2009, 301, 745–752. [Google Scholar]
- SEER Stat Fact Sheets: Colon and Rectum. Available online: http://seer.cancer.gov/statfacts/html/colorect.html (Accessed on 3 August 2010).
- Gambhir, S.S. Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer 2002, 2, 683–693. [Google Scholar]
- Weissleder, R. Molecular imaging in cancer. Science 2006, 312, 1168–1171. [Google Scholar]
- Wolfe, H.R.; Mendizabal, M.; Lleong, E.; Cuthbertson, A.; Desai, V.; Pullan, S.; Fujii, D.K.; Morrison, M.; Pither, R.; Waldman, S.A. In vivo imaging of human colon cancer xenografts in immunodeficient mice using a guanylyl cyclase C—specific ligand. J. Nucl. Med. 2002, 43, 392–399. [Google Scholar]
- Gali, H.; Sieckman, G.L.; Hoffman, T.J.; Kiefer, G.E.; Chin, D.T.; Forte, L.R.; Volkert, W.A. Synthesis and in vitro evaluation of an 111In-labeled ST-peptide enterotoxin (ST) analogue for specific targeting of guanylin receptors on human colonic cancers. Anticancer Res. 2001, 21, 2785–2792. [Google Scholar]
- Gali, H.; Sieckman, G.L.; Hoffman, T.J.; Owen, N.K.; Mazuru, D.G.; Forte, L.R.; Volkert, W.A. Chemical synthesis of Escherichia coli ST(h) analogues by regioselective disulfide bond formation: biological evaluation of an (111)In-DOTA-Phe(19)-ST(h) analogue for specific targeting of human colon cancers. Bioconjug. Chem. 2002, 13, 224–231. [Google Scholar]
- Giblin, M.F.; Gali, H.; Sieckman, G.L.; Owen, N.K.; Hoffman, T.J.; Forte, L.R.; Volkert, W.A. In vitro and in vivo comparison of human Escherichia coli heat-stable peptide analogues incorporating the 111In-DOTA group and distinct linker moieties. Bioconjug. Chem. 2004, 15, 872–880. [Google Scholar]
- Giblin, M.F.; Sieckman, G.L.; Shelton, T.D.; Hoffman, T.J.; Forte, L.R.; Volkert, W.A. In vitro and in vivo evaluation of 177Lu- and 90Y-labeled E. coli heat-stable enterotoxin for specific targeting of uroguanylin receptors on human colon cancers. Nucl. Med. Biol. 2006, 33, 481–488. [Google Scholar] [PubMed]
- Giblin, M.F.; Sieckman, G.L.; Watkinson, L.D.; Daibes-Figueroa, S.; Hoffman, T.J.; Forte, L.R.; Volkert, W.A. Selective targeting of E. coli heat-stable enterotoxin analogs to human colon cancer cells. Anticancer Res. 2006, 26, 3243–3251. [Google Scholar] [PubMed]
- Liu, D.; Overbey, D.; Watkinson, L.D.; Daibes-Figueroa, S.; Hoffman, T.J.; Forte, L.R.; Volkert, W.A.; Giblin, M.F. In vivo imaging of human colorectal cancer using radiolabeled analogs of the uroguanylin peptide hormone. Anticancer Res. 2009, 29, 3777–3783. [Google Scholar]
- Urbanski, R.; Carrithers, S.L.; Waldman, S.A. Internalization of E. coli ST mediated by guanylyl cyclase C in T84 human colon carcinoma cells. Biochim. Biophys. Acta 1995, 1245, 29–36. [Google Scholar] [PubMed]
- Mejia, A.; Schulz, S.; Hyslop, T.; Weinberg, D.S.; Waldman, S.A. GUCY2C reverse transcriptase PCR to stage pN0 colorectal cancer patients. Expert Rev. Mol. Diagn. 2009, 9, 777–785. [Google Scholar]
- Walker, L.S.; Abbas, A.K. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol. 2002, 2, 11–19. [Google Scholar]
- Speetjens, F.M.; Kuppen, P.J.; Welters, M.J.; Essahsah, F.; Voet van den Brink, A.M.; Lantrua, M.G.; Valentijn, A.R.; Oostendorp, J.; Fathers, L.M.; Nijman, H.W.; Drijfhout, J.W.; van de Velde, C.J.; Melief, C.J.; van der Burg, S.H. Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clin. Cancer Res. 2009, 15, 1086–1095. [Google Scholar]
- Elkord, E.; Dangoor, A.; Burt, D.J.; Southgate, T.D.; Daayana, S.; Harrop, R.; Drijfhout, J.W.; Sherlock, D.; Hawkins, R.E.; Stern, P.L. Immune evasion mechanisms in colorectal cancer liver metastasis patients vaccinated with TroVax (MVA-5T4). Cancer Immunol. Immunother. 2009, 58, 1657–1667. [Google Scholar]
- Gulley, J.L.; Arlen, P.M.; Tsang, K.Y.; Yokokawa, J.; Palena, C.; Poole, D.J.; Remondo, C.; Cereda, V.; Jones, J.L.; Pazdur, M.P.; Higgins, J.P.; Hodge, J.W.; Steinberg, S.M.; Kotz, H.; Dahut, W.L.; Schlom, J. Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clin. Cancer Res. 2008, 14, 3060–3069. [Google Scholar]
- Hartman, Z.C.; Wei, J.; Osada, T.; Glass, O.; Lei, G.; Yang, X.Y.; Peplinski, S.; Kim, D.W.; Xia, W.; Spector, N.; Marks, J.; Barry, W.; Hobeika, A.; Devi, G.; Amalfitano, A.; Morse, M.A.; Lyerly, H.K.; Clay, T.M. An adenoviral vaccine encoding full-length inactivated human Her2 exhibits potent immunogenicty and enhanced therapeutic efficacy without oncogenicity. Clin. Cancer Res. 2010, 16, 1466–1477. [Google Scholar]
- Ullenhag, G.J.; Frodin, J.E.; Mosolits, S.; Kiaii, S.; Hassan, M.; Bonnet, M.C.; Moingeon, P.; Mellstedt, H.; Rabbani, H. Immunization of colorectal carcinoma patients with a recombinant canarypox virus expressing the tumor antigen Ep-CAM/KSA (ALVAC-KSA) and granulocyte macrophage colony- stimulating factor induced a tumor-specific cellular immune response. Clin. Cancer Res. 2003, 9, 2447–2456. [Google Scholar]
- Hodge, J.W.; Higgins, J.; Schlom, J. Harnessing the unique local immunostimulatory properties of modified vaccinia Ankara (MVA) virus to generate superior tumor-specific immune responses and antitumor activity in a diversified prime and boost vaccine regimen. Vaccine 2009, 27, 4475–4482. [Google Scholar]
- Slingluff, C.L., Jr.; Petroni, G.R.; Olson, W.; Czarkowski, A.; Grosh, W.W.; Smolkin, M.; Chianese-Bullock, K.A.; Neese, P.Y.; Deacon, D.H.; Nail, C.; Merrill, P.; Fink, R.; Patterson, J.W.; Rehm, P.K. Helper T-cell responses and clinical activity of a melanoma vaccine with multiple peptides from MAGE and melanocytic differentiation antigens. J. Clin. Oncol. 2008, 26, 4973–4980. [Google Scholar]
- Belyakov, I.M.; Berzofsky, J.A. Immunobiology of mucosal HIV infection and the basis for development of a new generation of mucosal AIDS vaccines. Immunity 2004, 20, 247–253. [Google Scholar]
- Mowat, A.M.; Viney, J.L. The anatomical basis of intestinal immunity. Immunol. Rev. 1997, 156, 145–166. [Google Scholar]
- Mora, J.R.; Cheng, G.; Picarella, D.; Briskin, M.; Buchanan, N.; von Andrian, U.H. Reciprocal and dynamic control of CD8 T cell homing by dendritic cells from skin- and gut-associated lymphoid tissues. J. Exp. Med. 2005, 201, 303–316. [Google Scholar]
- Snook, A.E.; Stafford, B.J.; Eisenlohr, L.C.; Rothstein, J.L.; Waldman, S.A. Mucosally Restricted Antigens as Novel Immunological Targets for Antitumor Therapy. Biomark. Med. 2007, 1, 187–202. [Google Scholar]
- Snook, A.E.; Eisenlohr, L.C.; Rothstein, J.L.; Waldman, S.A. Cancer mucosa antigens as a novel immunotherapeutic class of tumor-associated antigen. Clin. Pharmacol. Ther. 2007, 82, 734–739. [Google Scholar]
- Snook, A.E.; Li, P.; Stafford, B.J.; Faul, E.J.; Huang, L.; Birbe, R.C.; Bombonati, A.; Schulz, S.; Schnell, M.J.; Eisenlohr, L.C.; Waldman, S.A. Lineage-specific T-cell responses to cancer mucosa antigen oppose systemic metastases without mucosal inflammatory disease. Cancer Res. 2009, 69, 3537–3544. [Google Scholar]
- Snook, A.E.; Stafford, B.J.; Li, P.; Tan, G.; Huang, L.; Birbe, R.; Schulz, S.; Schnell, M.J.; Thakur, M.; Rothstein, J.L.; Eisenlohr, L.C.; Waldman, S.A. Guanylyl cyclase C-induced immunotherapeutic responses opposing tumor metastases without autoimmunity. J. Natl. Cancer Inst. 2008, 100, 950–961. [Google Scholar]
- Belyakov, I.M.; Ahlers, J.D. What role does the route of immunization play in the generation of protective immunity against mucosal pathogens? J. Immunol. 2009, 183, 6883–6892. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lin, J.E.; Valentino, M.; Marszalowicz, G.; Magee, M.S.; Li, P.; Snook, A.E.; Stoecker, B.A.; Chang, C.; Waldman, S.A. Bacterial Heat-Stable Enterotoxins: Translation of Pathogenic Peptides into Novel Targeted Diagnostics and Therapeutics. Toxins 2010, 2, 2028-2054. https://doi.org/10.3390/toxins2082028
Lin JE, Valentino M, Marszalowicz G, Magee MS, Li P, Snook AE, Stoecker BA, Chang C, Waldman SA. Bacterial Heat-Stable Enterotoxins: Translation of Pathogenic Peptides into Novel Targeted Diagnostics and Therapeutics. Toxins. 2010; 2(8):2028-2054. https://doi.org/10.3390/toxins2082028
Chicago/Turabian StyleLin, Jieru E., Michael Valentino, Glen Marszalowicz, Michael S. Magee, Peng Li, Adam E. Snook, Brian A. Stoecker, Chang Chang, and Scott A. Waldman. 2010. "Bacterial Heat-Stable Enterotoxins: Translation of Pathogenic Peptides into Novel Targeted Diagnostics and Therapeutics" Toxins 2, no. 8: 2028-2054. https://doi.org/10.3390/toxins2082028