
Augmenting the Efficacy of Immunotoxins and Other Targeted Protein Toxins by Endosomal Escape Enhancers


Abstract
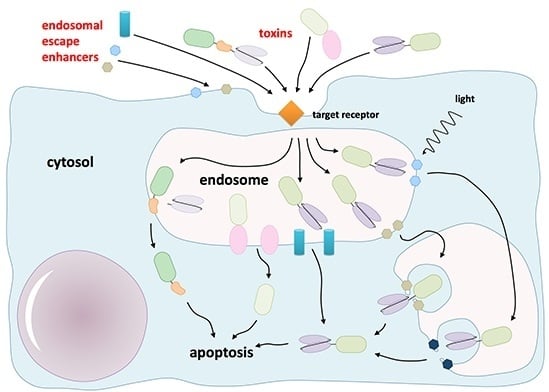
:
1. Introduction
2. Chemical Enhancers
2.1. Lysosomotropic Amines
2.2. Carboxylic Ionophores
2.3. Calcium Channel Antagonists
2.4. Other Organic Compounds
3. Enhancers of Viral and Bacterial Origin
4. Enhancers of Eukaryotic Origin
4.1. Proteins and Peptides
4.2. Secondary Metabolites
5. Synthetic Peptide Enhancers
6. Physicochemical Techniques
7. Discussion
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fuchs, H.; Bachran, C. Design of targeted protein toxins. In Drug Delivery in Oncology–From Basic Research to Cancer Therapy; Kratz, F., Senter, P., Steinhagen, H., Eds.; Wiley-VCH: Weinheim, Germany, 2011; Volume 3, pp. 1443–1487. [Google Scholar]
- Alewine, C.; Hassan, R.; Pastan, I. Advances in anticancer immunotoxin therapy. Oncologist 2015, 20, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Holliger, P.; Prospero, T.; Winter, G. “Diabodies”: Small bivalent and bispecific antibody fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar] [CrossRef] [PubMed]
- May, C.; Sapra, P.; Gerber, H.P. Advances in bispecific biotherapeutics for the treatment of cancer. Biochem. Pharmacol. 2012, 84, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, C.K.; Sollazzo, M. Of minibody, camel and bacteriophage. Comb. Chem. High Throughput Screen. 2001, 4, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Olsnes, S.; Sandvig, K.; Petersen, O.W.; van Deurs, B. Immunotoxins—Entry into cells and mechanisms of action. Immunol. Today 1989, 10, 291–295. [Google Scholar] [PubMed]
- Pirker, R.; FitzGerald, D.J.; Hamilton, T.C.; Ozols, R.F.; Laird, W.; Frankel, A.E.; Willingham, M.C.; Pastan, I. Characterization of immunotoxins active against ovarian cancer cell lines. J. Clin. Investig. 1985, 76, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Ravel, S.; Colombatti, M.; Casellas, P. Internalization and intracellular fate of anti-CD5 monoclonal antibody and anti-CD5 ricin A-chain immunotoxin in human leukemic T cells. Blood 1992, 79, 1511–1517. [Google Scholar] [PubMed]
- Fuchs, H.; Bachran, C.; Flavell, D.J. Diving through membranes: molecular cunning to enforce the endosomal escape of antibody-targeted anti-tumor toxins. Antibodies 2013, 2, 209–235. [Google Scholar] [CrossRef]
- Selbo, P.K.; Bostad, M.; Olsen, C.E.; Edwards, V.T.; Hogset, A.; Weyergang, A.; Berg, K. Photochemical internalisation, a minimally invasive strategy for light-controlled endosomal escape of cancer stem cell-targeting therapeutics. Photochem. Photobiol. Sci. 2015, 14, 1433–1450. [Google Scholar] [CrossRef] [PubMed]
- Wales, R.; Roberts, L.M.; Lord, J.M. Addition of an endoplasmic reticulum retrieval sequence to ricin A chain significantly increases its cytotoxicity to mammalian cells. J. Biol. Chem. 1993, 268, 23986–23990. [Google Scholar] [PubMed]
- Zhang, D.; Wang, J.; Xu, D. Cell-penetrating peptides as noninvasive transmembrane vectors for the development of novel multifunctional drug-delivery systems. J. Control. Release 2016, 229, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Wu, M. Enhancement of immunotoxin activity using chemical and biological reagents. Br. J. Cancer 1997, 75, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Seglen, P.O.; Grinde, B.; Solheim, A.E. Inhibition of the lysosomal pathway of protein degradation in isolated rat hepatocytes by ammonia, methylamine, chloroquine and leupeptin. Eur. J. Biochem. 1979, 95, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Casellas, P.; Brown, J.P.; Gros, O.; Gros, P.; Hellstrom, I.; Jansen, F.K.; Poncelet, P.; Roncucci, R.; Vidal, H.; Hellstrom, K.E. Human melanoma cells can be killed in vitro by an immunotoxin specific for melanoma-associated antigen p97. Int. J. Cancer 1982, 30, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Casellas, P.; Bourrie, B.J.; Gros, P.; Jansen, F.K. Kinetics of cytotoxicity induced by immunotoxins. Enhancement by lysosomotropic amines and carboxylic ionophores. J. Biol. Chem. 1984, 259, 9359–9364. [Google Scholar] [PubMed]
- Zhang, Y.; Schulte, W.; Pink, D.; Phipps, K.; Zijlstra, A.; Lewis, J.D.; Waisman, D.M. Sensitivity of cancer cells to truncated diphtheria toxin. PLoS ONE 2010, 5, e10498. [Google Scholar] [CrossRef] [PubMed]
- Umata, T.; Moriyama, Y.; Futai, M.; Mekada, E. The cytotoxic action of diphtheria toxin and its degradation in intact Vero cells are inhibited by bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase. J. Biol. Chem. 1990, 265, 21940–21945. [Google Scholar] [PubMed]
- Marcil, J.; Ravindranath, N.; Sairam, M.R. Cytotoxic activity of lutropin-gelonin conjugate in mouse Leydig tumor cells: potentiation of the hormonotoxin activity by different drugs. Mol. Cell. Endocrinol. 1993, 92, 83–90. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Houston, L.L. Inhibition of human acute lymphoblastic leukemia cells by immunotoxins: Potentiation by chloroquine. Science 1984, 223, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Lizzi, A.R.; D’Alessandro, A.M.; Zeolla, N.; Brisdelli, F.; D’Andrea, G.; Pitari, G.; Oratore, A.; Bozzi, A.; Ippoliti, R. The effect of AZT and chloroquine on the activities of ricin and a saporin-transferrin chimeric toxin. Biochem. Pharmacol. 2005, 70, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Siena, S.; Villa, S.; Bregni, M.; Bonnadonna, G.; Gianni, A.M. Amantadine potentiates T lymphocyte killing by an anti-pan-T cell (CD5) ricin A-chain immunotoxin. Blood 1987, 69, 345–348. [Google Scholar] [PubMed]
- Geden, S.E.; Gardner, R.A.; Fabbrini, M.S.; Ohashi, M.; Phanstiel Iv, O.; Teter, K. Lipopolyamine treatment increases the efficacy of intoxication with saporin and an anticancer saporin conjugate. FEBS J. 2007, 274, 4825–4836. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, S.; Seth, P.; Pirker, R.; FitzGerald, D.; Gottesman, M.M.; Pastan, I. Potentiation of cytotoxic activity of immunotoxins on cultured human cells. Cancer Res. 1985, 45, 1005–1007. [Google Scholar] [PubMed]
- Siena, S.; Lappi, D.A.; Bregni, M.; Formosa, A.; Villa, S.; Soria, M.; Bonadonna, G.; Gianni, A.M. Synthesis and characterization of an antihuman T-lymphocyte saporin immunotoxin (OKT1-SAP) with in vivo stability into nonhuman primates. Blood 1988, 72, 756–765. [Google Scholar] [PubMed]
- Siena, S.; Bregni, M.; Formosa, A.; Martineau, D.; Lappi, D.A.; Bonadonna, G.; Gianni, A.M. Evaluation of antihuman T lymphocyte saporin immunotoxins potentially useful in human transplantation. Transplantation 1988, 46, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Myers, C.D.; Thorpe, P.E.; Ross, W.C.; Cumber, A.J.; Katz, F.E.; Tax, W.; Greaves, M.F. An immunotoxin with therapeutic potential in T cell leukemia: WT1-ricin A. Blood 1984, 63, 1178–1185. [Google Scholar] [PubMed]
- Kronke, M.; Schlick, E.; Waldmann, T.A.; Vitetta, E.S.; Greene, W.C. Selective killing of human T-lymphotropic virus-I infected leukemic T-cells by monoclonal anti-interleukin 2 receptor antibody-ricin A chain conjugates: Potentiation by ammonium chloride and monensin. Cancer Res. 1986, 46, 3295–3298. [Google Scholar] [PubMed]
- Faguet, G.B.; Agee, J.F. Four ricin chain A-based immunotoxins directed against the common chronic lymphocytic leukemia antigen: In vitro characterization. Blood 1993, 82, 536–543. [Google Scholar] [PubMed]
- Vollmar, A.M.; Banker, D.E.; Mendelsohn, J.; Herschman, H.R. Toxicity of ligand and antibody-directed ricin A-chain conjugates recognizing the epidermal growth factor receptor. J. Cell. Physiol. 1987, 131, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterhout, Y.V.; Preijers, F.W.; Wessels, H.M.; de Witte, T. Cytotoxicity of CD3-ricin A chain immunotoxins in relation to cellular uptake and degradation kinetics. Cancer Res. 1992, 52, 5921–5925. [Google Scholar] [PubMed]
- Mujoo, K.; Reisfeld, R.A.; Cheung, L.; Rosenblum, M.G. A potent and specific immunotoxin for tumor cells expressing disialoganglioside GD2. Cancer Immunol. Immunother. 1991, 34, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Ramakrishnan, S.; Houston, L.L. Immunotoxin-mediated elimination of clonogenic tumor cells in the presence of human bone marrow. J. Immunol. 1985, 134, 2010–2016. [Google Scholar] [PubMed]
- Niesen, J.; Hehmann-Titt, G.; Woitok, M.; Fendel, R.; Barth, S.; Fischer, R.; Stein, C. A novel fully-human cytolytic fusion protein based on granzyme B shows in vitro cytotoxicity and ex vivo binding to solid tumors overexpressing the epidermal growth factor receptor. Cancer Lett. 2016, 374, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.; Firman, P.; Schneider, H. Sodium ion-proton exchange reactions of the carboxylic acid ionophore monensin. J. Am. Chem. Soc. 1985, 107, 4297–4300. [Google Scholar] [CrossRef]
- Grinde, B. Effect of carboxylic ionophores on lysosomal protein degradation in rat hepatocytes. Exp. Cell Res. 1983, 149, 27–35. [Google Scholar] [CrossRef]
- Derbyshire, E.J.; Stahel, R.A.; Wawrzynczak, E.J. Potentiation of a weakly active ricin A chain immunotoxin recognizing the neural cell adhesion molecule. Clin. Exp. Immunol. 1992, 89, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Bjorn, M.J.; Houston, L.L. Recombinant ricin A chain conjugated to monoclonal antibodies: Improved tumor cell inhibition in the presence of lysosomotropic compounds. Cancer Res. 1989, 49, 613–617. [Google Scholar] [PubMed]
- Colombatti, M.; Dell’Arciprete, L.; Chignola, R.; Tridente, G. Carrier protein-monensin conjugates: Enhancement of immunotoxin cytotoxicity and potential in tumor treatment. Cancer Res. 1990, 50, 1385–1391. [Google Scholar] [PubMed]
- Casellas, P.; Jansen, F.K. Immunotoxin enhancers. Cancer Treat. Res. 1988, 37, 351–369. [Google Scholar] [PubMed]
- Hertler, A.A.; Schlossman, D.M.; Borowitz, M.J.; Blythman, H.E.; Casellas, P.; Frankel, A.E. An anti-CD5 immunotoxin for chronic lymphocytic leukemia: Enhancement of cytotoxicity with human serum albumin-monensin. Int. J. Cancer 1989, 43, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Griffin, T.; Raso, V. Monensin in lipid emulsion for the potentiation of ricin A chain immunotoxins. Cancer Res. 1991, 51, 4316–4322. [Google Scholar] [PubMed]
- Griffin, T.; Rybak, M.E.; Recht, L.; Singh, M.; Salimi, A.; Raso, V. Potentiation of antitumor immunotoxins by liposomal monensin. J. Natl. Cancer Inst. 1993, 85, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Ferdous, A.J.; Stembridge, N.Y.; Singh, M. Role of monensin PLGA polymer nanoparticles and liposomes as potentiator of ricin A immunotoxins in vitro. J. Control. Release 1998, 50, 71–78. [Google Scholar] [CrossRef]
- Raso, V.; Lawrence, J. Carboxylic ionophores enhance the cytotoxic potency of ligand- and antibody-delivered ricin A chain. J. Exp. Med. 1984, 160, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Griffin, T.W.; Childs, L.R.; FitzGerald, D.J.; Levin, L.V. Enhancement of the cytotoxic effect of anti-carcinoembryonic antigen immunotoxins by adenovirus and carboxylic ionophores. J. Natl. Cancer Inst. 1987, 79, 679–685. [Google Scholar] [PubMed]
- Griffin, T.W.; Pagnini, P.G.; Houston, L.L. Enhancement of the specific cytotoxicity of a breast cancer-associated antigen immunotoxin by the carboxylic ionophore monensin. J. Biol. Response Mod. 1987, 6, 537–545. [Google Scholar] [PubMed]
- Roth, J.A.; Ames, R.S.; Fry, K.; Lee, H.M.; Scannon, P.J. Mediation of reduction of spontaneous and experimental pulmonary metastases by ricin A-chain immunotoxin 45-2D9-RTA with potentiation by systemic monensin in mice. Cancer Res. 1988, 48, 3496–3501. [Google Scholar] [PubMed]
- Derbyshire, E.J.; Wawrzynczak, E.J. An anti-mucin immunotoxin BrE-3-ricin A-chain is potently and selectively toxic to human small-cell lung cancer. Int. J. Cancer 1992, 52, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Handa, J.T.; Houston, L.L.; Jaffe, G.J. Monensin enhances the cytotoxic effect of antitransferrin receptor immunotoxin on cultured RPE cells. Curr. Eye Res. 1993, 12, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, S.; Gottesman, M.M.; Hanover, J.A.; Fitzgerald, D.J.; Willingham, M.C.; Pastan, I. Verapamil enhances the toxicity of conjugates of epidermal growth factor with Pseudomonas exotoxin and antitransferrin receptor with Pseudomonas exotoxin. J. Cell. Physiol. 1984, 120, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Pirker, R.; FitzGerald, D.J.; Willingham, M.C.; Pastan, I. Enhancement of the activity of immunotoxins made with either ricin A chain or Pseudomonas exotoxin in human ovarian and epidermoid carcinoma cell lines. Cancer Res. 1988, 48, 3919–3923. [Google Scholar] [PubMed]
- Pirker, R.; FitzGerald, D.J.; Raschack, M.; Frank, Z.; Willingham, M.C.; Pastan, I. Enhancement of the activity of immunotoxins by analogues of verapamil. Cancer Res. 1989, 49, 4791–4795. [Google Scholar] [PubMed]
- Jaffrezou, J.P.; Levade, T.; Thurneyssen, O.; Chiron, M.; Bordier, C.; Attal, M.; Chatelain, P.; Laurent, G. In vitro and in vivo enhancement of ricin-A chain immunotoxin activity by novel indolizine calcium channel blockers: delayed intracellular degradation linked to lipidosis induction. Cancer Res. 1992, 52, 1352–1359. [Google Scholar] [PubMed]
- Jaffrezou, J.P.; Levade, T.; Kuhlein, E.; Thurneyssen, O.; Chiron, M.; Grandjean, H.; Carriere, D.; Laurent, G. Enhancement of ricin A chain immunotoxin activity by perhexiline on established and fresh leukemic cells. Cancer Res. 1990, 50, 5558–5566. [Google Scholar] [PubMed]
- Wu, Y.N.; Gadina, M.; Tao-Cheng, J.H.; Youle, R.J. Retinoic acid disrupts the Golgi apparatus and increases the cytosolic routing of specific protein toxins. J. Cell Biol. 1994, 125, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Hudson, T.H.; Grillo, F.G. Brefeldin-A enhancement of ricin A-chain immunotoxins and blockade of intact ricin, modeccin, and abrin. J. Biol. Chem. 1991, 266, 18586–18592. [Google Scholar] [PubMed]
- Andersson, Y.; Engebraaten, O.; Fodstad, O. Synergistic anti-cancer effects of immunotoxin and cyclosporin in vitro and in vivo. Br. J. Cancer 2009, 101, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Davol, P.A.; Bizuneh, A.; Frackelton, A.R., Jr. Wortmannin, a phosphoinositide 3-kinase inhibitor, selectively enhances cytotoxicity of receptor-directed-toxin chimeras in vitro and in vivo. Anticancer Res. 1999, 19, 1705–1713. [Google Scholar] [PubMed]
- Park, T.G.; Jeong, J.H.; Kim, S.W. Current status of polymeric gene delivery systems. Adv. Drug Deliv. Rev. 2006, 58, 467–486. [Google Scholar] [CrossRef] [PubMed]
- Lai, P.S.; Pai, C.L.; Peng, C.L.; Shieh, M.J.; Berg, K.; Lou, P.J. Enhanced cytotoxicity of saporin by polyamidoamine dendrimer conjugation and photochemical internalization. J. Biomed. Mater. Res. Part A 2008, 87, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Pattrick, N.G.; Richardson, S.C.; Casolaro, M.; Ferruti, P.; Duncan, R. Poly(amidoamine)-mediated intracytoplasmic delivery of ricin A-chain and gelonin. J. Control. Release 2001, 77, 225–232. [Google Scholar] [CrossRef]
- Yefenof, E.; Abboud, G.; Epszteyn, S.; Vitetta, E.S. Treatment of premalignancy: Prevention of lymphoma in radiation leukemia virus-inoculated mice by cyclosporin A and immunotoxin. Proc. Natl. Acad. Sci. USA 1992, 89, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Jaffrezou, J.P.; Sikic, B.I.; Laurent, G. Cyclosporin A and cyclosporin SDZ PSC 833 enhance anti-CD5 ricin A-chain immunotoxins in human leukemic T cells. Blood 1994, 83, 482–489. [Google Scholar] [PubMed]
- Greber, U.F. Virus and host mechanics support membrane penetration and cell entry. J. Virol. 2016, 90, 3802–3805. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Whittaker, G.R. Fusion of Enveloped Viruses in Endosomes. Traffic 2016, 17, 593–614. [Google Scholar] [CrossRef] [PubMed]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, U. Cell-penetrating peptides: Design, synthesis, and applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef] [PubMed]
- Vives, E.; Schmidt, J.; Pelegrin, A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim. Biophys. Acta 2008, 1786, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Reissmann, S. Cell penetration: Scope and limitations by the application of cell-penetrating peptides. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef] [PubMed]
- Foged, C.; Nielsen, H.M. Cell-penetrating peptides for drug delivery across membrane barriers. Expert Opin. Drug Deliv. 2008, 5, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, M.; Birch, D.; Nielsen, H.M. Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef] [PubMed]
- Salerno, J.C.; Ngwa, V.M.; Nowak, S.J.; Chrestensen, C.A.; Healey, A.N.; McMurry, J.L. Novel cell-penetrating peptide-adaptors effect intracellular delivery and endosomal escape of protein cargos. J. Cell Sci. 2016, 129, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.; Bachran, C.; Li, T.; Heisler, I.; Durkop, H.; Sutherland, M. A cleavable molecular adapter reduces side effects and concomitantly enhances efficacy in tumor treatment by targeted toxins in mice. J. Control. Release 2007, 117, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.C.; Zhang, J.; Min, K.A.; He, H.; David, A.E.; Huang, Y.; Yang, V.C. PTD-Modified ATTEMPTS for Enhanced Toxin-based Cancer Therapy: An in vivo Proof-of-Concept Study. Pharm. Res. 2015, 32, 2690–2703. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.C.; Zhang, J.; Min, K.A.; Lee, K.; Moon, C.; Balthasar, J.P.; Yang, V.C. Combination of antibody targeting and PTD-mediated intracellular toxin delivery for colorectal cancer therapy. J. Control. Release 2014, 194, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.C.; Zhao, J.; Zhang, J.; Huang, Y.; He, H.; Wang, M.; Min, K.A.; Yang, V.C. Recombinant TAT-gelonin fusion toxin: Synthesis and characterization of heparin/protamine-regulated cell transduction. J. Biomed. Mater. Res. Part A 2015, 103, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, D.J.P.; Padmanabhan, R.; Pastan, I.; Willingham, M.C. Adenovirus-induced release of epidermal growth factor and pseudomonas toxin into the cytosol of KB cells during receptor-mediated endocytosis. Cell 1983, 32, 607–617. [Google Scholar] [CrossRef]
- Seth, P.; Fitzgerald, D.; Ginsberg, H.; Willingham, M.; Pastan, I. Evidence that the penton base of adenovirus is involved in potentiation of toxicity of Pseudomonas exotoxin conjugated to epidermal growth factor. Mol. Cell. Biol. 1984, 4, 1528–1533. [Google Scholar] [CrossRef] [PubMed]
- Lorenzetti, I.; Meneguzzi, A.; Fracasso, G.; Potrich, C.; Costantini, L.; Chiesa, E.; Legname, G.; Menestrina, G.; Tridente, G.; Colombatti, M. Genetic grafting of membrane-acting peptides to the cytotoxin dianthin augments its ability to de-stabilize lipid bilayers and enhances its cytotoxic potential as the component of transferrin-toxin conjugates. Int. J. Cancer 2000, 86, 582–589. [Google Scholar] [CrossRef]
- Chignola, R.; Anselmi, C.; Serra, M.D.; Franceschi, A.; Fracasso, G.; Pasti, M.; Chiesa, E.; Lord, J.M.; Tridente, G.; Colombatti, M. Self-potentiation of Ligand-Toxin Conjugates Containing Ricin A Chain Fused with Viral Structures. J. Biol. Chem. 1995, 270, 23345–23351. [Google Scholar] [CrossRef] [PubMed]
- Tolstikov, V.V.; Cole, R.; Fang, H.; Pincus, S.H. Influence of endosome-destabilizing peptides on efficacy of anti-HIV immunotoxins. Bioconjug. Chem. 1997, 8, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Heisler, I.; Keller, J.; Tauber, R.; Sutherland, M.; Fuchs, H. A cleavable adapter to reduce nonspecific cytotoxicity of recombinant immunotoxins. Int. J. Cancer 2003, 103, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Hetzel, C.; Bachran, C.; Fischer, R.; Fuchs, H.; Barth, S.; Stöcker, M. Small cleavable adapters enhance the specific cytotoxicity of a humanized immunotoxin directed against CD64-positive cells. J. Immunother. 2008, 31, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Weldon, J.E.; Skarzynski, M.; Therres, J.A.; Ostovitz, J.R.; Zhou, H.; Kreitman, R.J.; Pastan, I. Designing the furin-cleavable linker in recombinant immunotoxins based on Pseudomonas exotoxin A. Bioconjug. Chem. 2015, 26, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Fitzgerald, D.J.; Adhya, S.; Pastan, I. Functional domains of Pseudomonas exotoxin identified by deletion analysis of the gene expressed in E. coli. Cell 1987, 48, 129–136. [Google Scholar] [CrossRef]
- Michalska, M.; Wolf, P. Pseudomonas Exotoxin A: Optimized by evolution for effective killing. Front. Microbiol. 2015, 6, 963. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.; Elsasser-Beile, U. Pseudomonas exotoxin A: From virulence factor to anti-cancer agent. Int. J. Med. Microbiol. IJMM 2009, 299, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Bachran, C.; Heisler, I.; Fuchs, H.; Sutherland, M. Influence of protein transduction domains on target-specific chimeric proteins. Biochem. Biophys. Res. Commun. 2005, 337, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Bachanova, V.; Frankel, A.E.; Cao, Q.; Lewis, D.; Grzywacz, B.; Verneris, M.R.; Ustun, C.; Lazaryan, A.; McClune, B.; Warlick, E.D.; et al. Phase I study of a bispecific ligand-directed toxin targeting CD22 and CD19 (DT2219) for refractory B-cell malignancies. Clin. Cancer Res. 2015, 21, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Bachran, C.; Abdelazim, S.; Fattah, R.J.; Liu, S.; Leppla, S.H. Recombinant expression and purification of a tumor-targeted toxin in Bacillus anthracis. Biochem. Biophys. Res. Commun. 2013, 430, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Arons, E.; Stetler-Stevenson, M.; Fitzgerald, D.J.; Wilson, W.H.; Pastan, I. Recombinant immunotoxins and other therapies for relapsed/refractory hairy cell leukemia. Leuk. Lymphoma 2011, 52 (Suppl. 2), 82–86. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Pastan, I. Immunoconjugates in the management of hairy cell leukemia. Best Pract. Res. Clin. Haematol. 2015, 28, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Vallera, D.A.; Hall, W.A. Diphtheria toxin-based targeted toxin therapy for brain tumors. J. Neuro Oncol. 2013, 114, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Madhumathi, J.; Devilakshmi, S.; Sridevi, S.; Verma, R.S. Immunotoxin therapy for hematologic malignancies: Where are we heading? Drug Discov. Today 2016, 21, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; Onda, M.; Pastan, I. Immunogenicity of therapeutic recombinant immunotoxins. Immunol. Rev. 2016, 270, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Liu, Y.; Gao, S.; Lin, S.; Gu, X.; Pomper, M.G.; Wang, P.C.; Shan, L. A bivalent recombinant immunotoxin with high potency against tumors with EGFR and EGFRvIII expression. Cancer Biol. Ther. 2015, 16, 1764–1774. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pratts, S.G.; Zhang, H.; Spencer, P.J.; Yu, R.; Tonsho, M.; Shah, J.A.; Tanabe, T.; Powell, H.R.; Huang, C.A.; et al. Treg depletion in non-human primates using a novel diphtheria toxin-based anti-human CCR4 immunotoxin. Mol. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wei, M.; Zhang, H.; Chen, H.; Germana, S.; Huang, C.A.; Madsen, J.C.; Sachs, D.H.; Wang, Z. Diphtheria-toxin based anti-human CCR4 immunotoxin for targeting human CCR4+ cells in vivo. Mol. Oncol. 2015, 9, 1458–1470. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Tiefenthaler, G.; Schiller, C.; Weiss, E.H.; Georges, G.; Brinkmann, U. Prospects of bacterial and plant protein-based immunotoxins for treatment of cancer. Cancer Genom. Proteom. 2014, 11, 25–38. [Google Scholar]
- Weng, A.; Thakur, M.; Beceren-Braun, F.; Bachran, D.; Bachran, C.; Riese, S.B.; Jenett-Siems, K.; Gilabert-Oriol, R.; Melzig, M.F.; Fuchs, H. The toxin component of targeted anti-tumor toxins determines their efficacy increase by saponins. Mol. Oncol. 2012, 6, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Piascik, P. FDA approves fusion protein for treatment of lymphoma. J. Am. Pharm. Assoc. 1999, 39, 571–572. [Google Scholar] [CrossRef]
- Ratts, R.; Trujillo, C.; Bharti, A.; vanderSpek, J.; Harrison, R.; Murphy, J.R. A conserved motif in transmembrane helix 1 of diphtheria toxin mediates catalytic domain delivery to the cytosol. Proc. Natl. Acad. Sci. USA 2005, 102, 15635–15640. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Jacquez, P. Roles of Anthrax Toxin Receptor 2 in Anthrax Toxin Membrane Insertion and Pore Formation. Toxins 2016, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Aaronson, H.; Mitola, D.J.; Leppla, S.H.; Bugge, T.H. Potent antitumor activity of a urokinase-activated engineered anthrax toxin. Proc. Natl. Acad. Sci. USA 2003, 100, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Netzel-Arnett, S.; Birkedal-Hansen, H.; Leppla, S.H. Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res. 2000, 60, 6061–6067. [Google Scholar] [PubMed]
- Wising, C.; Molne, L.; Jonsson, I.M.; Ahlman, K.; Lagergard, T. The cytolethal distending toxin of Haemophilus ducreyi aggravates dermal lesions in a rabbit model of chancroid. Microbes Infect. Inst. Pasteur 2005, 7, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Bachran, C.; Hasikova, R.; Leysath, C.E.; Sastalla, I.; Zhang, Y.; Fattah, R.J.; Liu, S.; Leppla, S.H. Cytolethal distending toxin B as a cell-killing component of tumor-targeted anthrax toxin fusion proteins. Cell Death Dis. 2014, 5, e1003. [Google Scholar] [CrossRef] [PubMed]
- Mozola, C.C.; Caparon, M.G. Dual modes of membrane binding direct pore formation by Streptolysin O. Mol. Microbiol. 2015, 97, 1036–1050. [Google Scholar] [CrossRef] [PubMed]
- Verherstraeten, S.; Goossens, E.; Valgaeren, B.; Pardon, B.; Timbermont, L.; Haesebrouck, F.; Ducatelle, R.; Deprez, P.; Wade, K.R.; Tweten, R.; et al. Perfringolysin O: The Underrated Clostridium perfringens Toxin? Toxins 2015, 7, 1702–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seveau, S. Multifaceted activity of listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Sub Cell. Biochem. 2014, 80, 161–195. [Google Scholar]
- Pirie, C.M.; Liu, D.V.; Wittrup, K.D. Targeted cytolysins synergistically potentiate cytoplasmic delivery of gelonin immunotoxin. Mol. Cancer Ther. 2013, 12, 1774–1782. [Google Scholar] [CrossRef] [PubMed]
- Provoda, C.J.; Stier, E.M.; Lee, K.D. Tumor cell killing enabled by listeriolysin O-liposome-mediated delivery of the protein toxin gelonin. J. Biol. Chem. 2003, 278, 35102–35108. [Google Scholar] [CrossRef] [PubMed]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell Internalization of the Third Helix of the Antennapedia Homeodomain Is Receptor-independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar] [CrossRef] [PubMed]
- Abes, S.; Turner, J.J.; Ivanova, G.D.; Owen, D.; Williams, D.; Arzumanov, A.; Clair, P.; Gait, M.J.; Lebleu, B. Efficient splicing correction by PNA conjugation to an R6-Penetratin delivery peptide. Nucleic Acids Res. 2007, 35, 4495–4502. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, P.; El-Andaloussi, S.; Sütlü, T.; Johansson, H.; Langel, Ü. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007, 21, 2664–2671. [Google Scholar] [CrossRef] [PubMed]
- Dupont, E.; Prochiantz, A.; Joliot, A. Penetratin Story: An Overview. Methods Mol. Biol. 2015, 1324, 29–37. [Google Scholar] [PubMed]
- Rosado, C.J.; Buckle, A.M.; Law, R.H.; Butcher, R.E.; Kan, W.T.; Bird, C.H.; Ung, K.; Browne, K.A.; Baran, K.; Bashtannyk-Puhalovich, T.A.; et al. A common fold mediates vertebrate defense and bacterial attack. Science 2007, 317, 1548–1551. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Wan, L.; Chen, L.; Li, S.; Lu, Y.; Huang, Q.; Wang, L.; Li, Y.; Cheng, J.; Lu, X. Selective depletion of activated T cells by recombinant immunotoxin containing anti-CTLA-4 single-chain fragment of variable antibody and N-terminal fragment of perforin. Transplant. Proc. 2006, 38, 2151–2153. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Jiang, S.; Han, B.; Sun, T.; Li, Z.; Zhao, L.; Gao, Q.; Sun, J. Cytotoxic T lymphocyte-dependent tumor growth inhibition by a vascular endothelial growth factor-superantigen conjugate. Biochem. Biophys. Res. Commun. 2012, 427, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Kurschus, F.C.; Kleinschmidt, M.; Fellows, E.; Dornmair, K.; Rudolph, R.; Lilie, H.; Jenne, D.E. Killing of target cells by redirected granzyme B in the absence of perforin. FEBS Lett. 2004, 562, 87–92. [Google Scholar] [CrossRef]
- Stahnke, B.; Thepen, T.; Stocker, M.; Rosinke, R.; Jost, E.; Fischer, R.; Tur, M.K.; Barth, S. Granzyme B-H22(scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol. Cancer Ther. 2008, 7, 2924–2932. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, S.; Letzian, S.; Jost, E.; Mladenov, R.; Hristodorov, D.; Huhn, M.; Fischer, R.; Barth, S.; Thepen, T. Granzyme M as a novel effector molecule for human cytolytic fusion proteins: CD64-specific cytotoxicity of Gm-H22(scFv) against leukemic cells. Cancer Lett. 2013, 341, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, S.; Rosinke, R.; Jost, E.; Hehmann-Titt, G.; Huhn, M.; Melmer, G.; Barth, S.; Thepen, T. Targeted ex vivo reduction of CD64-positive monocytes in chronic myelomonocytic leukemia and acute myelomonocytic leukemia using human granzyme B-based cytolytic fusion proteins. Int. J. Cancer 2014, 135, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Mohamedali, K.A.; Marks, J.W.; Cheung, L.H.; Hittelman, W.N.; Rosenblum, M.G. Construction and characterization of novel, completely human serine protease therapeutics targeting Her2/neu. Mol. Cancer Ther. 2013, 12, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.A.; Lord, J.M. Ricin trafficking in cells. Toxins 2015, 7, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Vitetta, E.S.; Cushley, W.; Uhr, J.W. Synergy of ricin A chain-containing immunotoxins and ricin B chain-containing immunotoxins in in vitro killing of neoplastic human B cells. Proc. Natl. Acad. Sci. USA 1983, 80, 6332–6335. [Google Scholar] [CrossRef]
- Vitetta, E.S.; Fulton, R.J.; Uhr, J.W. Cytotoxicity of a cell-reactive immunotoxin containing ricin A chain is potentiated by an anti-immunotoxin containing ricin B chain. J. Exp. Med. 1984, 160, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Embleton, M.J.; Charleston, A.; Robins, R.A.; Pimm, M.V.; Baldwin, R.W. Recombinant ricin toxin A chain cytotoxicity against carcinoembryonic antigen expressing tumour cells mediated by a bispecific monoclonal antibody and its potentiation by ricin toxin B chain. Br. J. Cancer 1991, 63, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, S.; Hansen, H.P.; Hehmann-Titt, G.; Huhn, M.; Fischer, R.; Barth, S.; Thepen, T. Efficacy of an adapted granzyme B-based anti-CD30 cytolytic fusion protein against PI-9-positive classical Hodgkin lymphoma cells in a murine model. Blood Cancer J. 2013, 3, e106. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.; Bachran, D.; Panjideh, H.; Schellmann, N.; Weng, A.; Melzig, M.F.; Sutherland, M.; Bachran, C. Saponins as tool for improved targeted tumor therapies. Curr. Drug Targets 2009, 10, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Hebestreit, P.; Melzig, M.F. Cytotoxic activity of the seeds from Agrostemma githago var. githago. Planta Medica 2003, 69, 921–925. [Google Scholar] [PubMed]
- Melzig, M.F.; Hebestreit, P.; Gaidi, G.; Lacaille-Dubois, M.A. Structure-activity-relationship of saponins to enhance toxic effects of agrostin. Planta Medica 2005, 71, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.; Bachran, C.; Fuchs, H.; Krause, E.; Stephanowitz, H.; Melzig, M.F. Enhancement of saporin cytotoxicity by Gypsophila saponins--more than stimulation of endocytosis. Chem. Biol. Interact. 2009, 181, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.; Jenett-Siems, K.; Gorick, C.; Melzig, M.F. Enhancement of cytotoxicity of ribosome-inactivating-protein type I by saponinum album is not based on stimulation of phagocytosis. J. Pharm. Pharmacol. 2008, 60, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.; Thakur, M.; von Mallinckrodt, B.; Beceren-Braun, F.; Gilabert-Oriol, R.; Wiesner, B.; Eichhorst, J.; Bottger, S.; Melzig, M.F.; Fuchs, H. Saponins modulate the intracellular trafficking of protein toxins. J. Control. Release 2012, 164, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Bachran, D.; Schneider, S.; Bachran, C.; Weng, A.; Melzig, M.F.; Fuchs, H. The endocytic uptake pathways of targeted toxins are influenced by synergistically acting Gypsophila saponins. Mol. Pharm. 2011, 8, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.E.; Bachran, C.; Fuchs, H.; Weng, A.; Melzig, M.F.; Flavell, S.U.; Flavell, D.J. Triterpenoid saponin augmention of saporin-based immunotoxin cytotoxicity for human leukaemia and lymphoma cells is partially immunospecific and target molecule dependent. Immunopharmacol. Immunotoxicol. 2015, 37, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Bachran, D.; Schneider, S.; Bachran, C.; Urban, R.; Weng, A.; Melzig, M.F.; Hoffmann, C.; Kaufmann, A.M.; Fuchs, H. Epidermal growth factor receptor expression affects the efficacy of the combined application of saponin and a targeted toxin on human cervical carcinoma cells. Int. J. Cancer 2010, 127, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Gilabert-Oriol, R.; Thakur, M.; von Mallinckrodt, B.; Hug, T.; Wiesner, B.; Eichhorst, J.; Melzig, M.F.; Fuchs, H.; Weng, A. Modified trastuzumab and cetuximab mediate efficient toxin delivery while retaining antibody-dependent cell-mediated cytotoxicity in target cells. Mol. Pharm. 2013, 10, 4347–4357. [Google Scholar] [CrossRef] [PubMed]
- Heisler, I.; Sutherland, M.; Bachran, C.; Hebestreit, P.; Schnitger, A.; Melzig, M.F.; Fuchs, H. Combined application of saponin and chimeric toxins drastically enhances the targeted cytotoxicity on tumor cells. J. Control. Release 2005, 106, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Bachran, C.; Durkop, H.; Sutherland, M.; Bachran, D.; Muller, C.; Weng, A.; Melzig, M.F.; Fuchs, H. Inhibition of tumor growth by targeted toxins in mice is dramatically improved by saponinum album in a synergistic way. J. Immunother. 2009, 32, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Mergel, K.; Weng, A.; von Mallinckrodt, B.; Gilabert-Oriol, R.; Durkop, H.; Melzig, M.F.; Fuchs, H. Targeted tumor therapy by epidermal growth factor appended toxin and purified saponin: An evaluation of toxicity and therapeutic potential in syngeneic tumor bearing mice. Mol. Oncol. 2013, 7, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Von Mallinckrodt, B.; Thakur, M.; Weng, A.; Gilabert-Oriol, R.; Durkop, H.; Brenner, W.; Lukas, M.; Beindorff, N.; Melzig, M.F.; Fuchs, H. Dianthin-EGF is an effective tumor targeted toxin in combination with saponins in a xenograft model for colon carcinoma. Futur. Oncol. 2014, 10, 2161–2175. [Google Scholar] [CrossRef] [PubMed]
- Gilabert-Oriol, R.; Weng, A.; Trautner, A.; Weise, C.; Schmid, D.; Bhargava, C.; Niesler, N.; Wookey, P.J.; Fuchs, H.; Thakur, M. Combinatorial approach to increase efficacy of Cetuximab, Panitumumab and Trastuzumab by dianthin conjugation and co-application of SO1861. Biochem. Pharmacol. 2015, 97, 247–255. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, A.; Futaki, S.; Harashima, H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: Ways to overcome endosomal entrapment. AAPS J. 2009, 11, 13–22. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Yang, H.; Lin, Q.; Huang, H. Arg9-peptide facilitates the internalization of an anti-CEA immunotoxin and potentiates its specific cytotoxicity to target cells. Int. J. Biochem. Cell Biol. 2005, 37, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Omata, D.; Negishi, Y.; Hagiwara, S.; Yamamura, S.; Endo-Takahashi, Y.; Suzuki, R.; Maruyama, K.; Nomizu, M.; Aramaki, Y. Bubble liposomes and ultrasound promoted endosomal escape of TAT-PEG liposomes as gene delivery carriers. Mol. Pharm. 2011, 8, 2416–2423. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Cai, K.; Hu, Y.; Li, J.; Ding, X.; Zhang, B.; Xu, D.; Yang, W.; Liu, P. Redox-responsive molecular nanoreservoirs for controlled intracellular anticancer drug delivery based on magnetic nanoparticles. Adv. Mater. 2012, 24, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Lukianova-Hleb, E.Y.; Belyanin, A.; Kashinath, S.; Wu, X.; Lapotko, D.O. Plasmonic nanobubble-enhanced endosomal escape processes for selective and guided intracellular delivery of chemotherapy to drug-resistant cancer cells. Biomaterials 2012, 33, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Weyergang, A.; Berstad, M.E.; Bull-Hansen, B.; Olsen, C.E.; Selbo, P.K.; Berg, K. Photochemical activation of drugs for the treatment of therapy-resistant cancers. Photochem. Photobiol. Sci. 2015, 14, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Weyergang, A.; Selbo, P.K.; Berstad, M.E.; Bostad, M.; Berg, K. Photochemical internalization of tumor-targeted protein toxins. Lasers Surg. Med. 2011, 43, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Norum, O.J.; Selbo, P.K.; Weyergang, A.; Giercksky, K.E.; Berg, K. Photochemical internalization (PCI) in cancer therapy: From bench towards bedside medicine. J. Photochem. Photobiol. B 2009, 96, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.; Nordstrand, S.; Selbo, P.K.; Tran, D.T.; Angell-Petersen, E.; Hogset, A. Disulfonated tetraphenyl chlorin (TPCS2a), a novel photosensitizer developed for clinical utilization of photochemical internalization. Photochem. Photobiol. Sci. 2011, 10, 1637–1651. [Google Scholar] [CrossRef] [PubMed]
- Selbo, P.K.; Weyergang, A.; Hogset, A.; Norum, O.J.; Berstad, M.B.; Vikdal, M.; Berg, K. Photochemical internalization provides time- and space-controlled endolysosomal escape of therapeutic molecules. J. Control. Release 2010, 148, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Bostad, M.; Kausberg, M.; Weyergang, A.; Olsen, C.E.; Berg, K.; Hogset, A.; Selbo, P.K. Light-triggered, efficient cytosolic release of IM7-saporin targeting the putative cancer stem cell marker CD44 by photochemical internalization. Mol. Pharm. 2014, 11, 2764–2776. [Google Scholar] [CrossRef] [PubMed]
- Stratford, E.W.; Bostad, M.; Castro, R.; Skarpen, E.; Berg, K.; Hogset, A.; Myklebost, O.; Selbo, P.K. Photochemical internalization of CD133-targeting immunotoxins efficiently depletes sarcoma cells with stem-like properties and reduces tumorigenicity. Biochim. Biophys. Acta 2013, 1830, 4235–4243. [Google Scholar] [CrossRef] [PubMed]
- Bostad, M.; Olsen, C.E.; Peng, Q.; Berg, K.; Hogset, A.; Selbo, P.K. Light-controlled endosomal escape of the novel CD133-targeting immunotoxin AC133-saporin by photochemical internalization—A minimally invasive cancer stem cell-targeting strategy. J. Control. Release 2015, 206, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Selbo, P.K.; Rosenblum, M.G.; Cheung, L.H.; Zhang, W.; Berg, K. Multi-modality therapeutics with potent anti-tumor effects: Photochemical internalization enhances delivery of the fusion toxin scFvMEL/rGel. PLoS ONE 2009, 4, e6691. [Google Scholar] [CrossRef] [PubMed]
- Berstad, M.B.; Cheung, L.H.; Berg, K.; Peng, Q.; Fremstedal, A.S.; Patzke, S.; Rosenblum, M.G.; Weyergang, A. Design of an EGFR-targeting toxin for photochemical delivery: In vitro and in vivo selectivity and efficacy. Oncogene 2015, 34, 5582–5592. [Google Scholar] [CrossRef] [PubMed]
- Selbo, P.K.; Sivam, G.; Fodstad, O.; Sandvig, K.; Berg, K. Photochemical internalisation increases the cytotoxic effect of the immunotoxin MOC31-gelonin. Int. J. Cancer 2000, 87, 853–859. [Google Scholar] [CrossRef]
- Lund, K.; Bostad, M.; Skarpen, E.; Braunagel, M.; Kiprijanov, S.; Krauss, S.; Duncan, A.; Hogset, A.; Selbo, P.K. The novel EpCAM-targeting monoclonal antibody 3-17I linked to saporin is highly cytotoxic after photochemical internalization in breast, pancreas and colon cancer cell lines. MAbs 2014, 6, 1038–1050. [Google Scholar] [CrossRef] [PubMed]
- Bull-Hansen, B.; Cao, Y.; Berg, K.; Skarpen, E.; Rosenblum, M.G.; Weyergang, A. Photochemical activation of the recombinant HER2-targeted fusion toxin MH3-B1/rGel; Impact of HER2 expression on treatment outcome. J. Control. Release 2014, 182, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Weyergang, A.; Cheung, L.H.; Rosenblum, M.G.; Mohamedali, K.A.; Peng, Q.; Waltenberger, J.; Berg, K. Photochemical internalization augments tumor vascular cytotoxicity and specificity of VEGF(121)/rGel fusion toxin. J. Control. Release 2014, 180, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bull-Hansen, B.; Berstad, M.B.; Berg, K.; Cao, Y.; Skarpen, E.; Fremstedal, A.S.; Rosenblum, M.G.; Peng, Q.; Weyergang, A. Photochemical activation of MH3-B1/rGel: A HER2-targeted treatment approach for ovarian cancer. Oncotarget 2015, 6, 12436–12451. [Google Scholar] [CrossRef] [PubMed]
- Berstad, M.B.; Weyergang, A.; Berg, K. Photochemical internalization (PCI) of HER2-targeted toxins: Synergy is dependent on the treatment sequence. Biochim. Biophys. Acta 2012, 1820, 1849–1858. [Google Scholar] [CrossRef] [PubMed]
- Yip, W.L.; Weyergang, A.; Berg, K.; Tonnesen, H.H.; Selbo, P.K. Targeted delivery and enhanced cytotoxicity of cetuximab-saporin by photochemical internalization in EGFR-positive cancer cells. Mol. Pharm. 2007, 4, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Weyergang, A.; Selbo, P.K.; Berg, K. Photochemically stimulated drug delivery increases the cytotoxicity and specificity of EGF-saporin. J. Control. Release 2006, 111, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Gaumann, A.K.; Kiefer, F.; Alfer, J.; Lang, S.A.; Geissler, E.K.; Breier, G. Receptor tyrosine kinase inhibitors: Are they real tumor killers? Int. J. Cancer 2016, 138, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.; Bachran, C. Targeted tumor therapies at a glance. Curr. Drug Targets 2009, 10, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates-an emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Antibody Drug Conjugates for Cancer Therapy. Pharmacol. Rev. 2016, 68, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Hackenberger, C.P.; Leonhardt, H.; Helma, J. Current Status: Site-Specific Antibody Drug Conjugates. J. Clin. Immunol. 2016, 36 (Suppl. 1), 100–107. [Google Scholar] [CrossRef] [PubMed]
- Yamaizumi, M.; Mekada, E.; Uchida, T.; Okada, Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell 1978, 15, 245–250. [Google Scholar] [CrossRef]
- Shapira, A.; Benhar, I. Toxin-based therapeutic approaches. Toxins 2010, 2, 2519–2583. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Eslami, M.; Sahandi-Zangabad, P.; Mirab, F.; Farajisafiloo, N.; Shafaei, Z.; Ghosh, D.; Bozorgomid, M.; Dashkhaneh, F.; Hamblin, M.R. pH-Sensitive stimulus-responsive nanocarriers for targeted delivery of therapeutic agents. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, B.; Taranejoo, S.; Monemian, S.A.; Salehi Moghaddam, Z.; Daliri, K.; Derakhshankhah, H.; Derakhshani, Z. Classification of stimuli-responsive polymers as anticancer drug delivery systems. Drug Deliv. 2015, 22, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Backendorf, C.; Noteborn, M.H. Apoptin towards safe and efficient anticancer therapies. Adv. Exp. Med. Biol. 2014, 818, 39–59. [Google Scholar] [PubMed]
- Los, M.; Panigrahi, S.; Rashedi, I.; Mandal, S.; Stetefeld, J.; Essmann, F.; Schulze-Osthoff, K. Apoptin, a tumor-selective killer. Biochim. Biophys. Acta 2009, 1793, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Schipper, M.L.; Patel, M.R.; Gambhir, S.S. Evaluation of firefly luciferase bioluminescence mediated photodynamic toxicity in cancer cells. Mol. Imaging Biol. 2006, 8, 218–225. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Enhancer | Toxin | Antigenic Target | Targeted Toxin | Enhancer Concentration | Max. Enhancement Factor | Ref. |
|---|---|---|---|---|---|---|
| Amantadine | ricin A chain | CD5 | T101-RTA | 1 mM | 1180 | [16] |
| saporin | CD5 | OKT1-SAP | 1 mM | 4 | [25] | |
| CD5 | T101-SAP | 1 mM | 4 | [25] | ||
| CD5 | SOT1a-SAP | 1 mM | 4 | [26] | ||
| Ammonium chloride | ricin A chain | CD5 | T101-RTA | 10 mM | 6700 | [16] |
| Thy 1.2 (CD 90) | AT15E-RTA | 10 mM | 6 | [16] | ||
| melanotransferrin | 96.5-RTA | 10 mM | 42 | [16] | ||
| CD7 | WT1-RTA | 6 mM | 100 | [27] | ||
| CD25 | anti-TAC-RTA | 10 mM | 100 | [28] | ||
| cCLLa | CLL2m/RTA | 10 mM | 80 | [29] | ||
| EGFR | EGF-RTA | 10 mM | 12 | [30] | ||
| CD3 | WT32-RTA | 6 mM | Significant increase | [31] | ||
| Chloroquine | gelonin | LH receptor | lutropin-gelonin | 9.6–29 µM | 15 | [19] |
| GD2 | gelonin-14G2a | 10 µM | 10 | [32] | ||
| pokeweed antiviral protein | transferrin receptor | 5E9-11-PAP | 10–100 µM | 65 | [20] | |
| T cells | T3-3A1-PAP | 10–100 µM | 65 | [20] | ||
| CD19 | B43-PAP | 40 µM | Significant increase | [33] | ||
| ricin A chain | CD5 | T101-RTA | 100 µM | 2500 | [16] | |
| saporin | transferrin receptor | saporin-transferrin | 10 µM | Significant increase | [21] | |
| granzyme B | EGFR | Gb(R201K)-scFv1711 | 50 µM | 3 | [34] | |
| Dimethylamine | ricin A chain | CD5 | T101-RTA | 10 mM | 3300 | [16] |
| Lipopolyamines | saporin | urokinase receptor | uPA-saporin | 5 µg/mL | 83 | [23] |
| Methylamine | ricin A chain | CD5 | T101-RTA | 10 mM | 13,300 | [16] |
| Quinacrine | gelonin | LH receptor | lutropin-gelonin | 2.6–7.6 µM | 15 | [19] |
| Trimethylamine | ricin A chain | CD5 | T101-RTA | 10 mM | 80 | [16] |
| β-Glycylphenyl-naphthylamide (GPN) | Pseudomonas exotoxin | transferrin receptor | HB21-PE | 10–20 µg/mL | 9 | [24] |
| EGFR | EGF-PE | 10–20 µg/mL | 6 | [24] |
| Enhancer | Toxin | Antigenic Target | Targeted Toxin | Enhancer Concentration | Max. Enhancement Factor | Ref. |
|---|---|---|---|---|---|---|
| Grisorixin | ricin A chain | CD5 | T101-RTA | 50 nM | 25,000 | [16] |
| Lasalocid | ricin A chain | CD5 | T101-RTA | 1 µM | 33,000 | [16] |
| Monensin | gelonin | LH receptor | lutropin-gelonin | 0.3–2.9 µM | 15 | [19] |
| GD2 | gelonin-14G2a | 100 nM | 10 | [32] | ||
| ricin A chain | CD5 | T101-RTA | 50 nM | 50,000 | [16] | |
| Thy 1.2 | AT15E-RTA | 50 nM | 4 | [16] | ||
| p97 | 96.5-RTA | 50 nM | 420 | [16] | ||
| transferrin receptor | Tfn-RTA | 10–100 nM | 30,000 | [45] | ||
| CD10 | anti-CALLA-RTA | 10–100 nM | Significant increase | [45] | ||
| CD25 | anti-TAC-RTA | 25 nM | 400 | [28] | ||
| CEA | anti-CEA-RTA | 0.5–1 µM | Significant increase | [46] | ||
| p55 | 260F9-rRTA | 100 nM | 34 | [47] | ||
| gp74 | 45-2D9-RTA | 0.5 µM | Significant increase | [48] | ||
| p55 | 260F9-rRTA | 10–100 nM | Significant increase | [38] | ||
| CD7 | 3A1-rRTA | 10–100 nM | Significant increase | [38] | ||
| transferrin receptor | R17-217-rRTA | 10–100 nM | Significant increase | [38] | ||
| transferrin receptor | Tfn-RTA | 900 nM | 500 | [39] | ||
| transferrin receptor | OKT9-RTA | 900 nM | 3300 | [39] | ||
| transferrin receptor | OX26-RTA | 900 nM | 330 | [39] | ||
| N-CAM | SEN36-RTA | 100 nM | 12,000 | [37] | ||
| MUC1 | BrE-3-RTA | 100 nM | 100 | [49] | ||
| transferrin receptor | 454A12MAb-RTA | 10–100 nM | 4 | [50] | ||
| saporin | transferrin receptor | Tfn-So6 | 900 nM | 1250 | [39] | |
| Nigericin | ricin A chain | CD5 | T101-RTA | 10 nM | 6700 | [16] |
| transferrin receptor | Tfn-RTA | 10–100 nM | Significant increase | [45] | ||
| CD10 | anti-CALLA-RTA | 10–100 nM | Significant increase | [45] | ||
| CEA | anti-CEA-RTA | 0.5 µM | Significant increase | [46] |
| Enhancer | Toxin | Antigenic Target | Targeted Toxin | Enhancer Concentration | Max. Enhancement Factor | Ref. |
|---|---|---|---|---|---|---|
| Diltiazem | Pseudomonas exotoxin | transferrin receptor | HB21-PE | 10–20 µg/mL | 6 | [24] |
| EGFR | EGF-PE | 10–20 µg/mL | 8 | [24] | ||
| Indolizine (SR 33287; SR33557) | ricin A chain | CD5 | T101-RTA | 5 µM | 620 | [54] |
| Thy 1.2 | AT15E-RTA | 5 µM | 84 | [54] | ||
| Methoxyverapamil (D-600) | Pseudomonas exotoxin | transferrin receptor | HB21-PE | 5–20 µg/mL | 12 | [24] |
| EGFR | EGF-PE | 5–20 µg/mL | 20 | [24] | ||
| Perhexiline | ricin A chain | CD5 | T101-RTA | 1–5 µM | 2000 | [55] |
| HLA-DR class II | HNC-241-RTA | 1–10 µM | 100 | [55] | ||
| Verapamil | gelonin | LH receptor | lutropin-gelonin | 10–41 µM | 15 | [19] |
| Pseudomonas exotoxin | transferrin receptor | HB21-PE | 2.5–20 µg/mL | 11 | [24] | |
| EGFR | EGF-PE | 10–20 µg/mL | 40 | [24] | ||
| ricin A chain | transferrin receptor | 454A12-rRTA | 20 µg/mL | 25 | [52] | |
| p55 | 260F9-rRTA | 20 µg/mL | 8 | [52] | ||
| HER2 | 454C11-RTA | 20 µg/mL | Significant increase | [52] | ||
| cCLLa | CLL2m/RTA | 20 µg/mL | 80 | [29] | ||
| Verapamil analogs (D792; D595; D528; Sz45) | Pseudomonas exotoxin | transferrin receptor | HB21-PE | 20 µM | 35 | [53] |
| ricin A chain | transferrin receptor | 454A12-rRTA | 1–20 µg/mL | 67 | [53] | |
| p55 | 260F9-rRTA | 20 µM | Significant increase | [53] |
| Enhancer | Toxin | Antigenic Target | Targeted Toxin | Enhancer Concentration | Max. Enhancement Factor | Ref. |
|---|---|---|---|---|---|---|
| Brefeldin-A | ricin A chain | transferrin receptor | 454A12-rRTA | 0.05–0.5 µg/mL | Significant increase | [57] |
| p55 | 260F9-rRTA | 0.025–0.05 µg/mL | Significant increase | [57] | ||
| Cyclosporin A | Pseudomonas exotoxin | EGFR | 425.3PE | 2 µM | Significant increase | [58] |
| MUC1 | BM7PE | 2–4 µM | 40 | [58] | ||
| EpCAM (EGP-2) | MOC31PE | 2 µM | Significant increase | [58] | ||
| ricin A chain | gp70 | 2F10-RTA | 25 mg/kg (in vivo) | 100 | [63] | |
| CD5 | T101-RTA | 4 µmol/mL | 101 | [64] | ||
| Cyclosporin SDZ PSC 833 | ricin A chain | CD5 | T101-RTA | 4 µmol/mL | 105 | [64] |
| Retinoic acid | ricin A chain | transferrin receptor | 454A12-rRTA | 10 µM | 10,000 | [56] |
| transferrin receptor | Tfn-rRTA | 10 µM | 1000 | [56] | ||
| B cells | M6-rRTA | 10 µM | Significant increase | [56] | ||
| p55 | 260F9-rRTA | 10 µM | Significant increase | [56] | ||
| Wortmannin | gelonin | bFGFR | bFGF-gelonin | 1–10 µM | Significant increase | [59] |
| saporin | bFGFR | bFGF-SAP | 1–10 µM | Significant increase | [59] | |
| EGFR | HBEGF-SAP | 1–10 µM | Significant increase | [59] | ||
| bFGFR | 11A8-SAP | 1–10 µM | Significant increase | [59] |
| Enhancer | Application | Toxin | Antigenic Target | Targeted Toxin | Enhancer Concentration | Max. Enhancement Factor | Ref. |
|---|---|---|---|---|---|---|---|
| Adenovirus | whole virus | Pseudomonas exotoxin | EGFR | PE-EGF | 2 × 109 pfu/mL | 10,000 | [78] |
| ricin A chain | CEA | anti-CEA-RTA | 3 × 108 pfu/mL | 33 | [46] | ||
| Penton base protein (adenovirus capsid protein) | whole virus | Pseudomonas exotoxin | EGFR | PE-EGF | 9 × 103 viruses/cell | Significant increase | [79] |
| KFT25 (N-terminus of Protein G) | viral peptides (fusion proteins) | dianthin | transferrin receptor | Tfn-KFT25-DIA | ≤30,000 ng/mL | 3.8 | [80] |
| ricin A chain | transferrin receptor | Tfn-KFT25-RTA | ≤10 pM | 20 | [81] | ||
| HA23 | viral peptides (conjugates and free peptides) | ricin A chain | gp120 | anti-gp120(HIV)-RTA-HA23 | 0–300 µg/ mL | 5 | [82] |
| PreS2-domain of hepatitis-B virus surface antigen (TLM) | viral peptides (fusion protein) | saporin | EGFR | saporin-TLM-EGF | ≤100 nM | 1 (in vitro) 2.2 (in vivo) | [74,83] |
| angiogenin | CD64 | anti-CD64-TLM-angiogenin | ≤100 nM | 20 | [84] |
| Enhancer = Toxin | Antigenic Target | Targeted Toxin | Enhancer = Toxin Concentration (IC50) | Ref. |
|---|---|---|---|---|
| Perforin (N-terminal 34 amino acids) | CTLA-4 (CD152) | hS83P34 | 200–1000 nM | [119] |
| Perforin / Granzyme B | VEGF | VEGF-SEA (D227A mutant) | released by attracted immune cells | [120] |
| Granzyme B | Lewis Y | GzmB-dsFv-B3 | 35–140 nM | [121] |
| CD64 | Gb-H22(scFv) | 1.7–17 nM | [122] | |
| Gb(R201K)-H22(scFv) | 4–7 nM | [124] | ||
| CD30 | Gb(R201K)-Ki4(scFv) | 1.7 nM | [130] | |
| EGFR | Gb(R201K)-scFv1711 | 133 nM | [34] | |
| HER2 | GrB/4D5 | 242–629 nM | [125] | |
| GrB/4D5/26 | 29–93 nM | [125] | ||
| Granzyme M | CD64 | Gm-H22(scFv) | 1.2–6.4 nM | [123] |
| Photosensitizer (Enhancer) | Toxin | Antigenic Target | Targeted Toxin | Photosensitizer (Enhancer) Concentration | Ref. |
|---|---|---|---|---|---|
| AlPcS2a | gelonin | CSPG4 | scFvMEL/rGel | 5 µg/mL | [159] |
| TPCS2a | gelonin | EGFR | rGel/EGF | 0.1–0.4 µg/mL | [160] |
| HER2 | MH3-B1/rGel | 0.1–0.4 µg/mL | [163,165] | ||
| VEGFR | VEGF121/rGel | 0.4 µg/mL | [164] | ||
| saporin | CD133 | CD133/1 (AC133)-saporin | 0.4–1 µg/mL | [157,158] | |
| CD133 | CD133/2 (293C)-saporin | 0.2–1 µg/mL | [157] | ||
| CD44 | IM7-saporin | 0.35–1 µg/mL | [156] | ||
| EpCAM (EGP-2) | 3–17I-saporin | 0.35 µg/mL | [162] | ||
| HER2 | Trastuzumab-saporin | 0.2 µg/mL | [166] | ||
| TPPS2a | gelonin | EpCAM (EGP-2) | MOC31-gelonin | 0.3–1 µg/mL | [161] |
| saporin | EGFR | Cetuximab-saporin | 0.1–1 µg/mL | [167] | |
| EGFR | EGF-saporin | 0.1–0.2 µg/mL | [168] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuchs, H.; Weng, A.; Gilabert-Oriol, R. Augmenting the Efficacy of Immunotoxins and Other Targeted Protein Toxins by Endosomal Escape Enhancers. Toxins 2016, 8, 200. https://doi.org/10.3390/toxins8070200
Fuchs H, Weng A, Gilabert-Oriol R. Augmenting the Efficacy of Immunotoxins and Other Targeted Protein Toxins by Endosomal Escape Enhancers. Toxins. 2016; 8(7):200. https://doi.org/10.3390/toxins8070200
Chicago/Turabian StyleFuchs, Hendrik, Alexander Weng, and Roger Gilabert-Oriol. 2016. "Augmenting the Efficacy of Immunotoxins and Other Targeted Protein Toxins by Endosomal Escape Enhancers" Toxins 8, no. 7: 200. https://doi.org/10.3390/toxins8070200