3.1. Graded Niche Fabrication
We fabricated scaffolds in the SZ2080 photoresist with 1% concentration of Irg photoinitiator. Previously we had fabricated scaffolds in the same SZ2080 photoresist but with Michler’s ketone (Bis) photoinitiator [
17], which showed strong autofluorescence, hindering the characterization of the fluorescent markers for cell proliferation (
Figure 1). The Irg-based photoresist used here was found to be biocompatible [
20] similarly to the previously studied Bis-SZ2080, but with greatly reduced auto-fluorescence. These results are in agreement with previous work showing that it is the addition of the photoinitiator which influences both the photosensitivity and fluorescence of the photoresist [
18,
21]. It should also be noted that 2PP is possible in SZ2080 without any photoinitiator, but at the expense ofa significantly smaller processing window for the fabrication of high quality 3D microstructures [
18].
Figure 1.
Comparison of autofluorescence in scaffolds formed with Bis (top row) and Irg (bottom row) photoinitiators in SZ2080 with (a, d) blue (4′,6-diamidino-2-phenylindole—DAPI), (b, e) green (fluorescein isothiocyanate—FITC) and (c, f) red (Tetramethylrhodamineisothiocyanate—TRITC) fluorescent markers.
Figure 1.
Comparison of autofluorescence in scaffolds formed with Bis (top row) and Irg (bottom row) photoinitiators in SZ2080 with (a, d) blue (4′,6-diamidino-2-phenylindole—DAPI), (b, e) green (fluorescein isothiocyanate—FITC) and (c, f) red (Tetramethylrhodamineisothiocyanate—TRITC) fluorescent markers.
We obtained significant improvements in niche fabrication with respect to our previous results, owing to a novel Yb-based writing laser, enabling us to also vary the repetition rate in searching for optimum processing conditions. At 200-kHz repetition rate and 1.9-nJ pulse energy (27-TW/cm
2 peak intensity) after the lens, we found an ideal processing window with 1-mm/s scan speed, representing a nearly three order of magnitude improvement in fabrication speed compared to our prior work [
17]. The polymerized lines in Irg-based SZ2080 were of 5 μm × 2.5 μm thickness, twice as thick as our previous results in Bis-based SZ2080 [
22], which we attribute partially to the 50-fold higher average laser fluence in the present work. This processing window provides, on the one hand, a very good mechanical stability of the structures, and, on the other hand, a very high processing speed, thus making the structuring of large surfaces feasible. For example, this faster processing speed allowed us to study the influence of the microscaffold’s relative distance on MSC behavior. As shown in
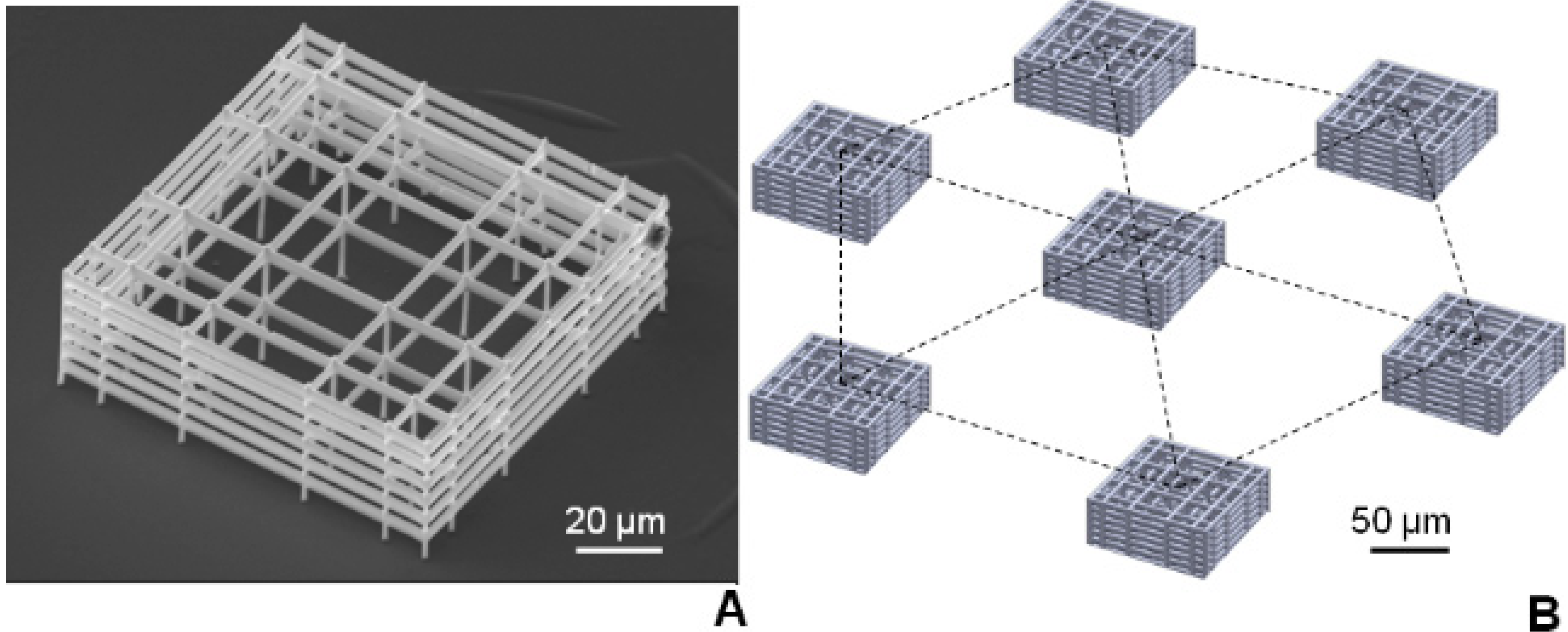
Figure 2, we fabricated seven microscaffolds on each cover glass, arranged in a hexagonal pattern with six scaffolds at the vertices and one at the center. The hexagon side was varied between 200 and 600 μm at 100 μm steps.
The niche geometry was selected from eight previously tested ones, as the one most favoring spontaneous MSC homing and proliferation [
17]. This graded geometry (
Figure 2) allows easy penetration of the cells from the larger central pores and a higher surface-to-volume ratio at the borders of the scaffold, where the external walls confine the cells. Individual microscaffolds were 30 µm high and 90 µm × 90 µm in transverse dimensions and consisted of a lattice of interconnected lines, with a graded spacing between 10 and 30 μm in the transverse direction and with a uniform spacing of 15 μm in the vertical direction. The 3D scaffold was surrounded by four outer confinement walls formed by horizontal rods spaced by 7.5 μm resulting in small gaps of 2 μm, which enable nutrients to pass through to the cells in the scaffold but prevent cells, which have entered to the niche, from escaping through the sides.
Figure 2.
(A) SEM of the synthetic niche formed by 2PP. (B) Layout of scaffolds on each coverglass:seven niches were arranged in a hexagonal pattern and their relative distance was set at 200, 300, 400, 500, or 600 µm.
Figure 2.
(A) SEM of the synthetic niche formed by 2PP. (B) Layout of scaffolds on each coverglass:seven niches were arranged in a hexagonal pattern and their relative distance was set at 200, 300, 400, 500, or 600 µm.
3.2. Stem Cell Proliferation and Colonization on Flat Surfaces
We first studied the response of stem cells on flat microscope slides without any niches. Cells adhered extensively to the substrate in a few hours, migrated and colonized all regions of the samples in a few days. The behaviour of cells cultured on unwritten microscopy slides did not differ significantly in any aspect from the behavior of cells localized, on the 2PP-written samples, in the regions far from the niches.
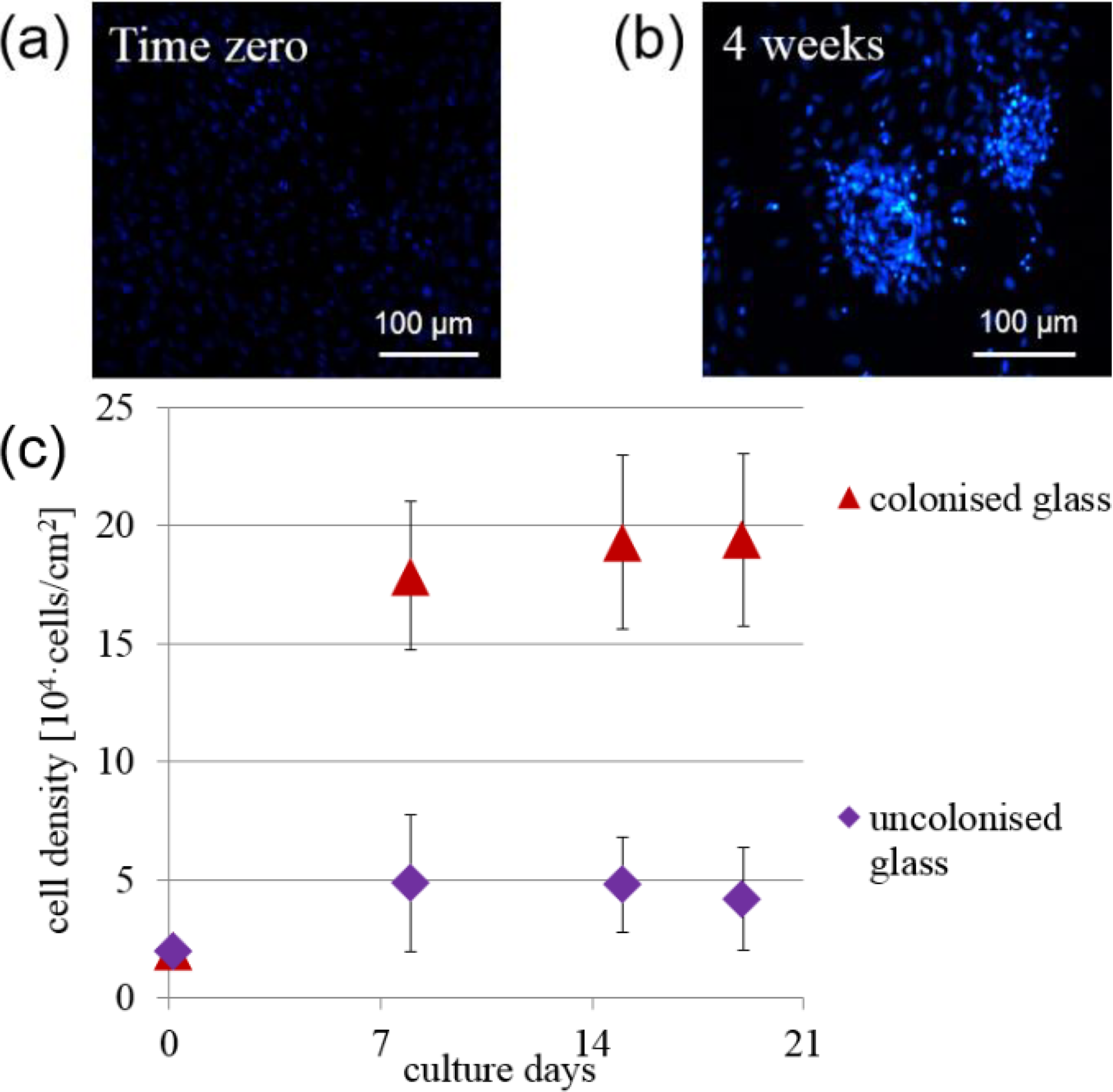
Figure 3 shows the time evolution of cell density in the colonies spontaneously formed on the glass substrate, and also in sample areas not colonized. Clearly, the cell density increased with time inside the colonies. Far from the colonies, the cell density does not increase significantly with time up to a period of three weeks. Also shown in
Figure 3 are fluorescence microscope pictures at time zero and after four weeks, demonstrating the formation of colonies.
Figure 3.
Fluorescence microcopy view of cell nuclei on flat glass at (a) time zero and at (b) four weeks; (c) Evolution of mean cell density versus time for cells in colonies and inother areas of the glass substrate. Values are given as mean and standard deviation of n = 15 measurements.
Figure 3.
Fluorescence microcopy view of cell nuclei on flat glass at (a) time zero and at (b) four weeks; (c) Evolution of mean cell density versus time for cells in colonies and inother areas of the glass substrate. Values are given as mean and standard deviation of n = 15 measurements.
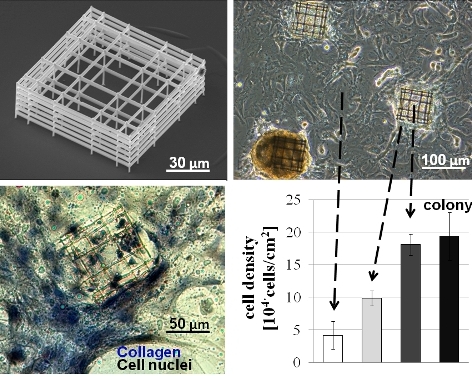
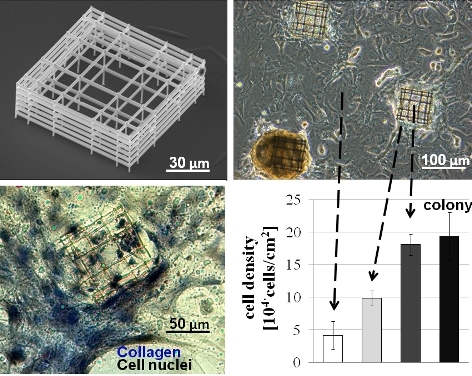
Cell clonogenicity was found, with a nearly constant density of about 1000 colonies/cm
2 measured over the 3 week time period studied. The colonies were observed to change shape with time from culture day 7, with colonies raising up from the glass surface and progressively forming spherical cell aggregates at the two week point, as described in
Figure 4A (top row). The average colony diameter (
Figure 4B) initially increased as the colony formed and spread on the flat glass but with increased time, the diameter decreased as the colony raised up to form the spherical cell aggregate. The average distance between spontaneously formed colonies was about 400 µm (
Figure 4C), which did not vary significantly with time from 0 to 21 days.
Figure 4.
(A) Diagram of cell aggregate formation on flat glass (top row) and on niche (bottom row), (B) colony diameter evolution with time, (C) inter-colony distance versus time. Values are given as mean and standard deviation of n = 30 measurements.
Figure 4.
(A) Diagram of cell aggregate formation on flat glass (top row) and on niche (bottom row), (B) colony diameter evolution with time, (C) inter-colony distance versus time. Values are given as mean and standard deviation of n = 30 measurements.
3.3. Stem Cell Proliferation and Colonization within Niches
Cell aggregates were also observed to growon laser-formed niches, with a similar shape evolution as on the flat glass. The individual cells which take on rounder shapes when they adhere inside the niches (
Figure 5A) eventually form an aggregate of cells (
Figure 5B) with its bottom part within the niche. A pictorial description of this evolution is shown in
Figure 4A (bottom).
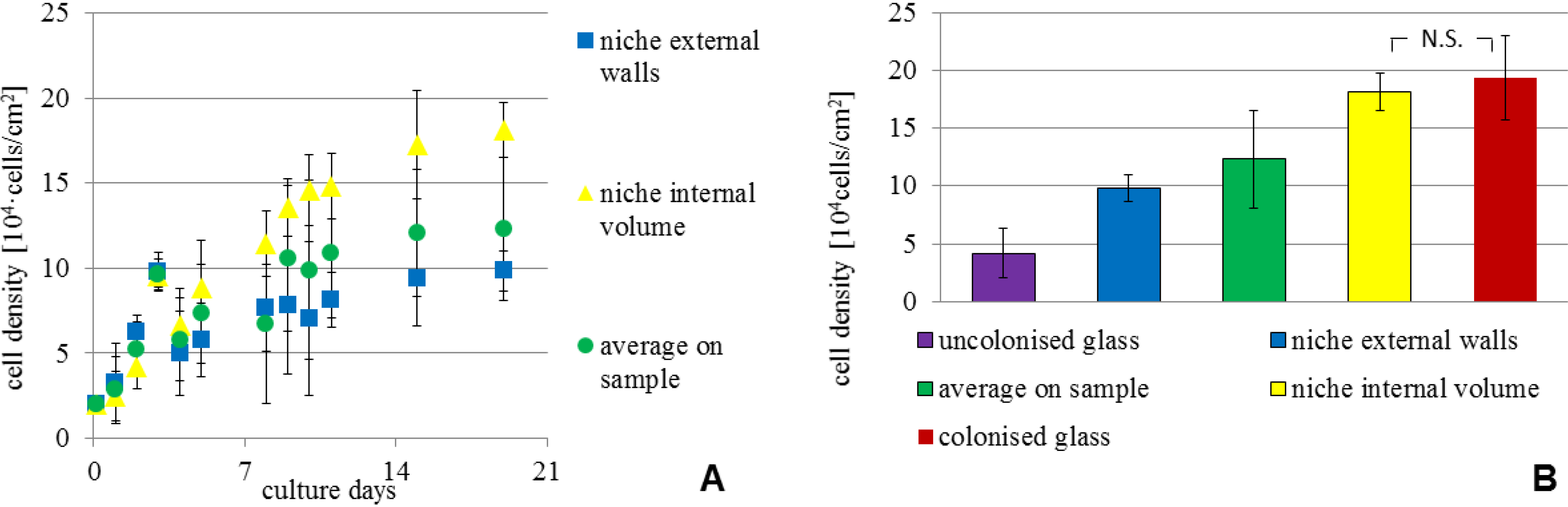
Figure 6A shows the cell density
versus time for stem cells adhered inside the niches, on the external walls and also the average value on the entire sample for comparison. Stem cell density was highest inside the niches and increased with time, similar to the trend for cells in spontaneous colonies formed on the flat glass substrate (
Figure 3).
To help in comparing the cell density in the various locations,
Figure 6B shows the cell density at the three week mark. With respect to initial seeding density, the increase was by six-fold on the culture substrate on average, but by almost 10-fold in colonies, which formed both within the region covered by the structural niches and at random locations on the surrounding glass. When accounting for the cells also adhered to the external niche walls, the laser-formed niches had nearly three times higher cell density compared to the sample average, demonstrating that the artificial niches provide an environment favoring a local increase in cell density.
Figure 5.
(A) SEM micrographs showing cells adhered to the internal lattice of the niche, with spherical morphology (arrows) and (B) spherical cell aggregates.
Figure 5.
(A) SEM micrographs showing cells adhered to the internal lattice of the niche, with spherical morphology (arrows) and (B) spherical cell aggregates.
Figure 6.
Cell morphology and proliferation in niches. (A) Growth curves measured for MSCs cultured on the synthetic niches; (B) cell density at culture day 21, measured at the various locations on the samples. All measurements are given as mean and standard deviation of n = 15 measurements; p < 0.01 for all pair-wise comparisons, except for * p > 0.05. N.S. indicates not statistically significant.
Figure 6.
Cell morphology and proliferation in niches. (A) Growth curves measured for MSCs cultured on the synthetic niches; (B) cell density at culture day 21, measured at the various locations on the samples. All measurements are given as mean and standard deviation of n = 15 measurements; p < 0.01 for all pair-wise comparisons, except for * p > 0.05. N.S. indicates not statistically significant.
We showed previously [
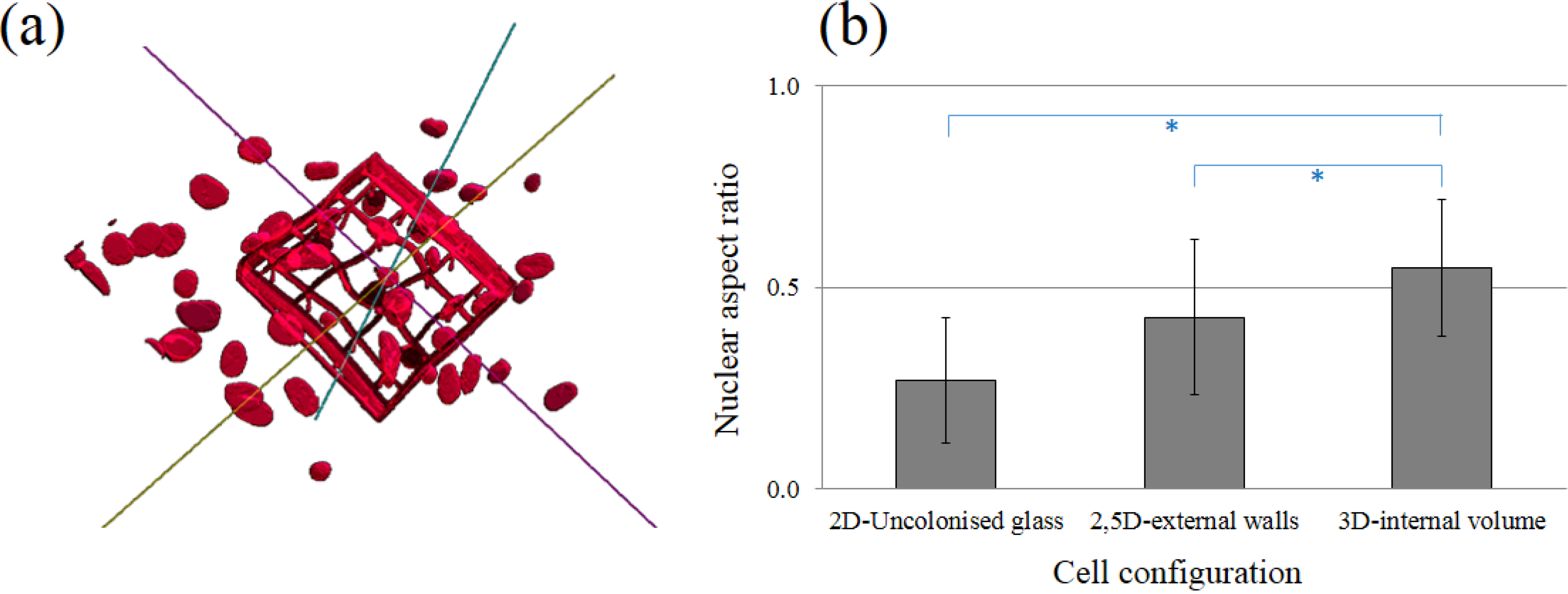
17] that cell morphology evolves from flat, when the cells are on the substrate and on the outer niche walls, to a more round shape once they are inside the niches, as they are able to bind in all spatial directions to the network of polymerized lines. A more quantitative validation of this important observation is shown in
Figure 7, which shows a 3D reconstruction of solid models of cell nuclei from sequential images of cell-seeded niches, acquired with confocal microscopy on DAPI-stained cells. In
Figure 7b, the nuclear aspect ratio, calculated as the ratio between shortest and longest semi-axes of the best fitting ellipsoid for the reconstructed nuclei, is shown at the various niche locations. A nuclear aspect ratio of 0 and 1 represent polarized and spherical nuclei, respectively. A two-fold more symmetric ellipsoidal shape is seen for the cells adhered inside the niche compared to those on the glass substrate.
Figure 7.
Cell nuclear morphology. (a) 3D reconstruction of solid models of cell nuclei from sequential images of cell-seeded niches. (b) Nuclear aspect ratio at various niche locations. Values are given as mean and standard deviation of n = 10 measurements, with * p < 0.01 for all pair-wise comparisons.
Figure 7.
Cell nuclear morphology. (a) 3D reconstruction of solid models of cell nuclei from sequential images of cell-seeded niches. (b) Nuclear aspect ratio at various niche locations. Values are given as mean and standard deviation of n = 10 measurements, with * p < 0.01 for all pair-wise comparisons.
3.3. Stem Cell Aggregation with Multiple Niches: Effect of Niche Separation
We next studied multiple niches oriented in a hexagonal geometry as depicted in
Figure 2B. The niches were found to be structurally stable in culture up to three weeks. However, in certain conditions, we observed niche detachment from the glass substrate, attributed to the traction exerted by the growing cell colonies (
Figure 8E). In previous work we estimated local substrate stresses induced by cell contraction [
23]. The forces exerted by single cardiac progenitors were in the order of 10 nN. Other authors also estimated that the axons of neural progenitors can exert forces in the order of 3 nN, able to bend 2PP-fabricated polymeric ridges [
24]. Previously we showed that the average distance between spontaneous colonies is about 400 µm on a substrate without niches (
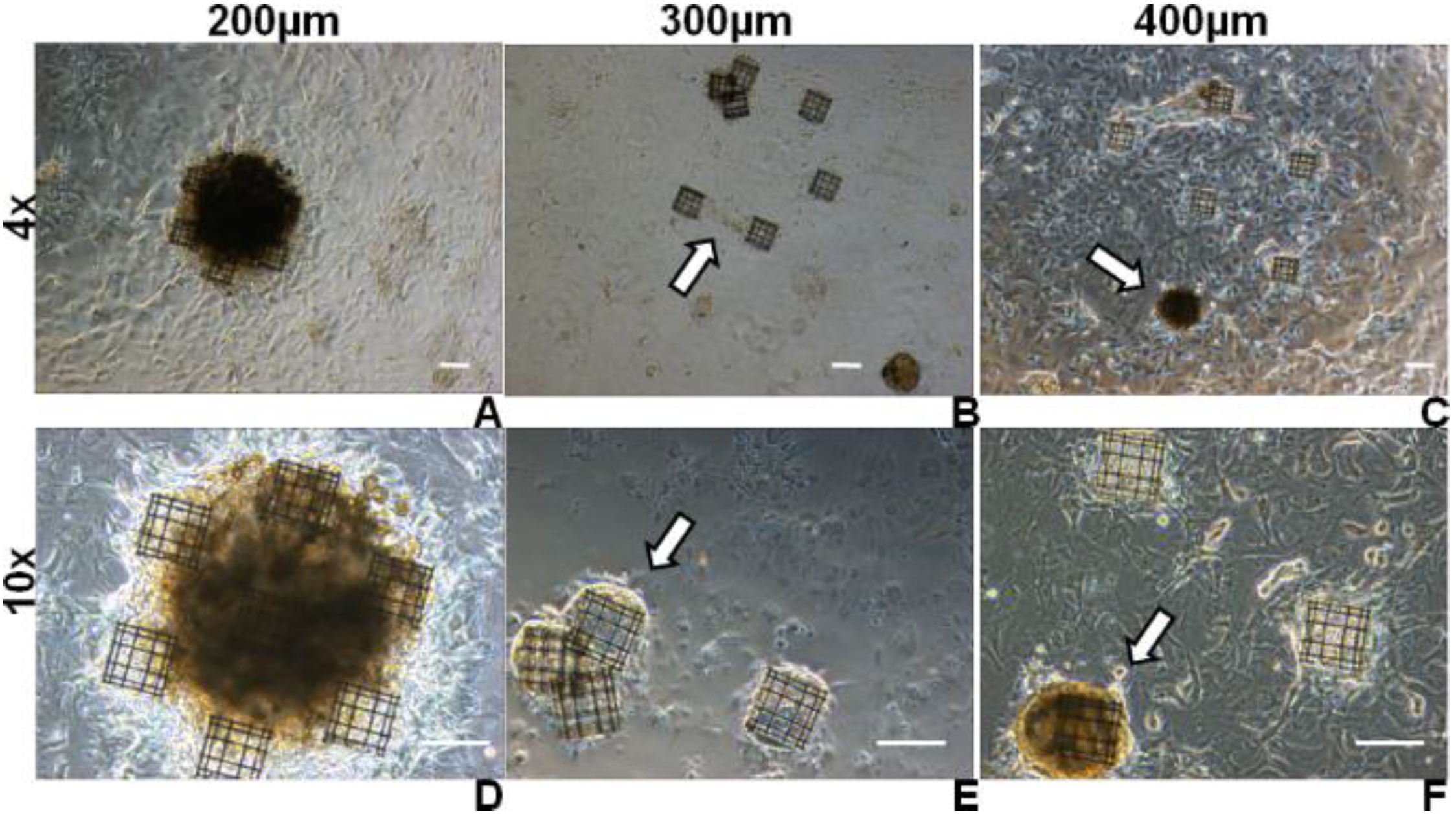
Figure 4C). When the scaffolds were at a distance much lower than this value, the cells on neighbouring scaffolds could easily interact with each other and a single large aggregate formed on top of the niche ensemble, as in the case of 200 μm inter-niche separation (
Figure 8A,D); when the distance between the scaffolds was in the same order as the spontaneous separation between colonies (400 μm), colonies could not bridge the gap and individual aggregates formed on top of each niche (arrows in
Figure 8C,F); when the distance between the scaffolds was intermediate (300 μm), the cells tended to pull the various scaffolds together to form a single colony(arrow in
Figure 8B) and occasionally caused the detachment of the structures (arrow in
Figure 8E). These detached niches did not fragment however, demonstrating satisfactory mechanical properties in relation to their application in advanced cell culture substrates.
Figure 8.
At three weeks, phase contrast images acquired on MSC-seeded niche systems with different inter-niche relative distance of 200 µm (A, D), 300 μm (B, E), and 400 μm (C, F). Scale bars represent 100 µm. In (C, F), aggregates forming on top of each niche are indicated by arrows. In (B), an arrow shows the crosstalk between cells on adjacent niches which tended to pull the niches together and in some cases result in detachment, as indicated by the arrow in (E).
Figure 8.
At three weeks, phase contrast images acquired on MSC-seeded niche systems with different inter-niche relative distance of 200 µm (A, D), 300 μm (B, E), and 400 μm (C, F). Scale bars represent 100 µm. In (C, F), aggregates forming on top of each niche are indicated by arrows. In (B), an arrow shows the crosstalk between cells on adjacent niches which tended to pull the niches together and in some cases result in detachment, as indicated by the arrow in (E).
3.4. Stem Cell Renewal and Differentiation
Our study allowed us to directly compare, on the same sample, significantly different culture configurations: (a) flat glass (2D), (b) 3D-patterned vertical niche walls (engineered, 2.5D), (c) niche internal scaffold structure (engineered, 3D) and (d) cell micro-aggregates (spontaneous, 3D). Using this model, we studied differences in the spontaneous tendency of cells to renew and differentiate in the various configurations (a)–(d), without inducing interferences in the cross-talk between these various populations (
i.e., through paracrine mechanisms), which may impact proliferation and differentiation. In aggregates on the flat glass substrate, we found that only a few cells were positive to proliferative markers at the aggregate base (
Figure 9D–F),
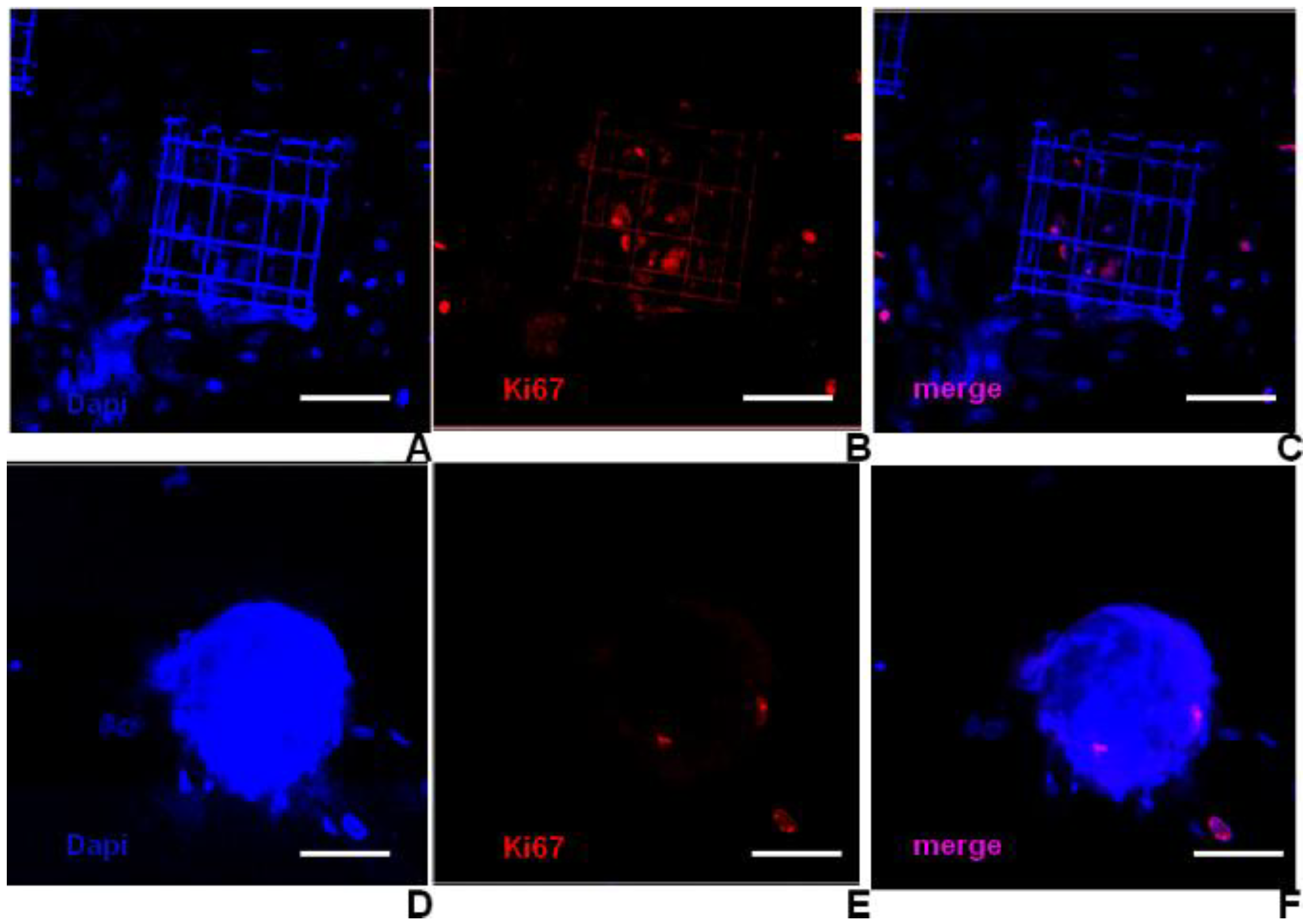
i.e., at the interface between the aggregate and the flat glass surface, and to chondrogenic markers thoroughly. On the contrary, in aggregates growing on top of the niches, the proliferation markers were positive for the cells in the niches (
Figure 9A–C), again located under the aggregates. Thus, structural niches were able not only to direct stem cell homing and colony formation, but also to guide aggregate formation, providing increased surface-to-volume ratios and space for stem cells to adhere and renew, respectively.
Cell differentiation showed quite different trends depending on the culture location (
Figure 10 and
Figure 11). It should be emphasized that chondrogenic, osteogenic, and adipogenic markers were stained in separate samples. The spherical cell aggregates were positive for early markers of chondrogenesis (aspecific collagen, GAG and collagen type 1) and negative for collagen type 2 and for all the other osteogenic and adipogenic markers tested. We are sure of this negativity because the positivity of the Ki67 marker was not masked by the autofluorescence of the structures. Cells adhering to the flat glass in regions far from the colonies were positive for early markers of osteogenic differentiation (collagen type 1 and mineral deposits) and negative for osteocalcin and all the other chondrogenic and adipogenic markers tested. Inside the niches however, cells were systematically negative for all the differentiation markers tested (
Figure 10A–C and
Figure 11B,C). In
Figure 10A–C, we show the histological results in niches when the spacing is 200μm, to render possible the visualization of the internal niche areas, and in
Figure 10D–F single aggregates. These results are likely correlated to the cell nuclear morphology, estimated from 3D image reconstruction, which varied significantly between the three regions of flat glass external niche walls, and internal niche lattice (
Figure 7). Nuclear deformation was highest on flat glass and lowest inside structural niches.
Figure 9.
Results of proliferation analysis using immunostaining for the Ki67 antigen. Images are z-stack projections of confocal acquisitions made over a thickness of 30 μm above the glass surface. The observation is performed in a region of the sample with a scaffold (A)–(C) and in a region with just flat glass (D)–(F). In both cases, a cell aggregate was present on top of the imaged region. (A), (D) Show the nuclei distribution; (B), (E) the proliferating cells; (C), (F) a merged image of the two previous ones. Proliferating cells are preferentially located in the central regions of the niches (B),while only a few active cells are observed at the bottom of cell aggregates spontaneously formed on flat glass (E). Scale is 50 µm.
Figure 9.
Results of proliferation analysis using immunostaining for the Ki67 antigen. Images are z-stack projections of confocal acquisitions made over a thickness of 30 μm above the glass surface. The observation is performed in a region of the sample with a scaffold (A)–(C) and in a region with just flat glass (D)–(F). In both cases, a cell aggregate was present on top of the imaged region. (A), (D) Show the nuclei distribution; (B), (E) the proliferating cells; (C), (F) a merged image of the two previous ones. Proliferating cells are preferentially located in the central regions of the niches (B),while only a few active cells are observed at the bottom of cell aggregates spontaneously formed on flat glass (E). Scale is 50 µm.
Figure 10.
Results of histology staining. (A), (D) Results of Von Kossa staining, in which cell nuclei stain pink and mineral deposits stain black. Sparse deposits are visible on the flat glass surface, in regions external to the niches and between aggregates; (B), (E) results of Masson Trichrome staining, in which cell nuclei stain black and collagen stains blue. Collagen is visualized in regions external to the niche and within spherical aggregates; (C, F) results of Safranin staining, in which nuclei stain pink and GAG stains orange. GAG is visualized within spherical aggregates. Scale bars are 50 µm.
Figure 10.
Results of histology staining. (A), (D) Results of Von Kossa staining, in which cell nuclei stain pink and mineral deposits stain black. Sparse deposits are visible on the flat glass surface, in regions external to the niches and between aggregates; (B), (E) results of Masson Trichrome staining, in which cell nuclei stain black and collagen stains blue. Collagen is visualized in regions external to the niche and within spherical aggregates; (C, F) results of Safranin staining, in which nuclei stain pink and GAG stains orange. GAG is visualized within spherical aggregates. Scale bars are 50 µm.
Figure 11.
MSC differentiation. Results of immunostaining for collagen type 1. (A)–(C) Collagen type 1 is typically visualized on the flat culture surface, externally to niches; (D)–(F) in cell aggregates, collagen type 1 is typically visualized on the spherical periphery of the aggregate. Scale bars are 50 µm.
Figure 11.
MSC differentiation. Results of immunostaining for collagen type 1. (A)–(C) Collagen type 1 is typically visualized on the flat culture surface, externally to niches; (D)–(F) in cell aggregates, collagen type 1 is typically visualized on the spherical periphery of the aggregate. Scale bars are 50 µm.
This interpretation must be supported by a more in-depth characterization with specific markers, as well as with experiments which could establish a cause–effect relationship between the cell nuclear morphology and cell fate. However, cells adhered and grew in the structural niches as single cells and not aggregates, coherently with their 3D adhesion configuration, likely inducing an isotropic cytoskeletal tension state that mimics the one present in the native niche [
25]. The combination of cytoskeletal isotropy and hypoxia (inevitably occurring in static culture at increasing cell densities, and presumably maximal at the bottom of the highly-consuming cell aggregates forming on top of the niches), is also emerging as a critical determinant of multipotency maintenance in stem cells [
26]. The absence of positivity of all the tested differentiation markers in the niches may be a sign of maintenance of the cell multipotency, especially because it was also correlated to increased proliferation. However, further studies to detect the simultaneous presence of known membrane markers of MSC stemness (including CD29, CD44, CD90, CD105) are necessary to corroborate the hypothesis of maintenance of stemness inside the niches.
3.5. Potential Clinical Implications
If the observations reported here for animal cells are confirmed for human cells in our future experiments, our “structurally” biomimetic niche system could find potential applications in two main directions.
By extrapolation from our cell density measurements, showing no difference in the cell density occurring in glass colonies with respect to niche colonies at three culture weeks, we can hypothesize that a macroscopic culture plate, completely covered by niches, would induce a homogeneous cell density approaching colony density,
i.e., 0.2 million cells/cm
2, a cell density 67% greater than the average 0.12 million cells/cm
2 measured in standard 2D culture (
Figure 6, “average on sample” curve). Thus, one potential application of our niche system could be to produce therapeutic MSC cells in large, pharmaceutically relevant scales.
Secondly, we could exploit the ability of these niches to increase cell renewal while maintaining cell multipotency in the structured layer. MSCs from patients could be expanded and maintained in culture for longer periods, until the formation of niche aggregates, which could be easily harvested from the culture substrate and delivered as a customized cell therapy for chronic disorders, on a repeated basis. The cells with higher “stemness” found within niches could be maintained in culture after aggregate removal, and the process of aggregate fabrication would spontaneously restart production of a further therapeutic cell dose. In our observations, the aggregate cells consistently committed towards the chondrogenic lineage, thus they contained cells potentially useful for cartilage regeneration therapies.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










