Fenofibrate Interferes with the Diapedesis of Lung Adenocarcinoma Cells through the Interference with Cx43/EGF-Dependent Intercellular Signaling

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
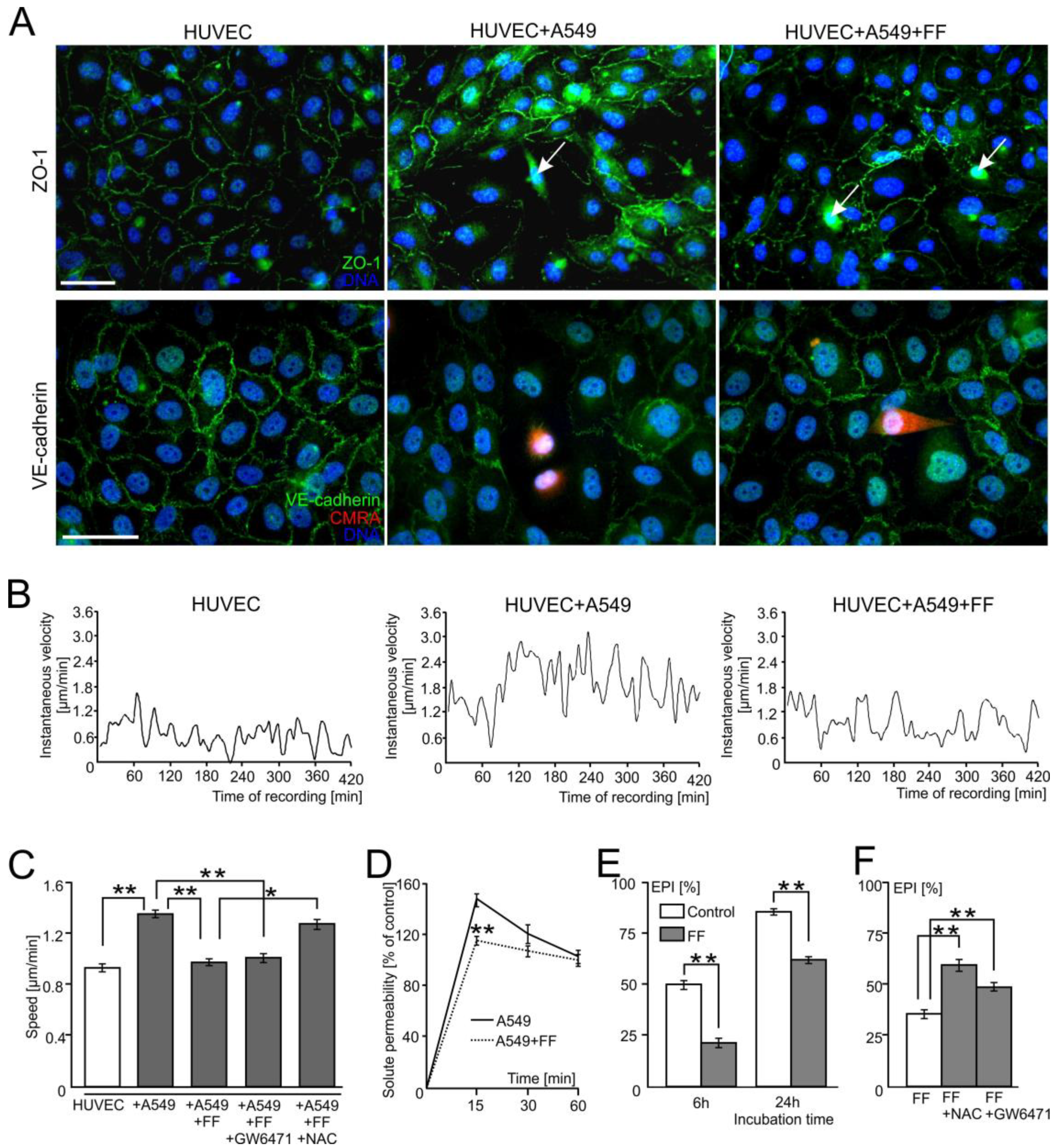
2.1. FF Interferes with the Diapedesis of A549 Cells in a PPARα-Independent Manner
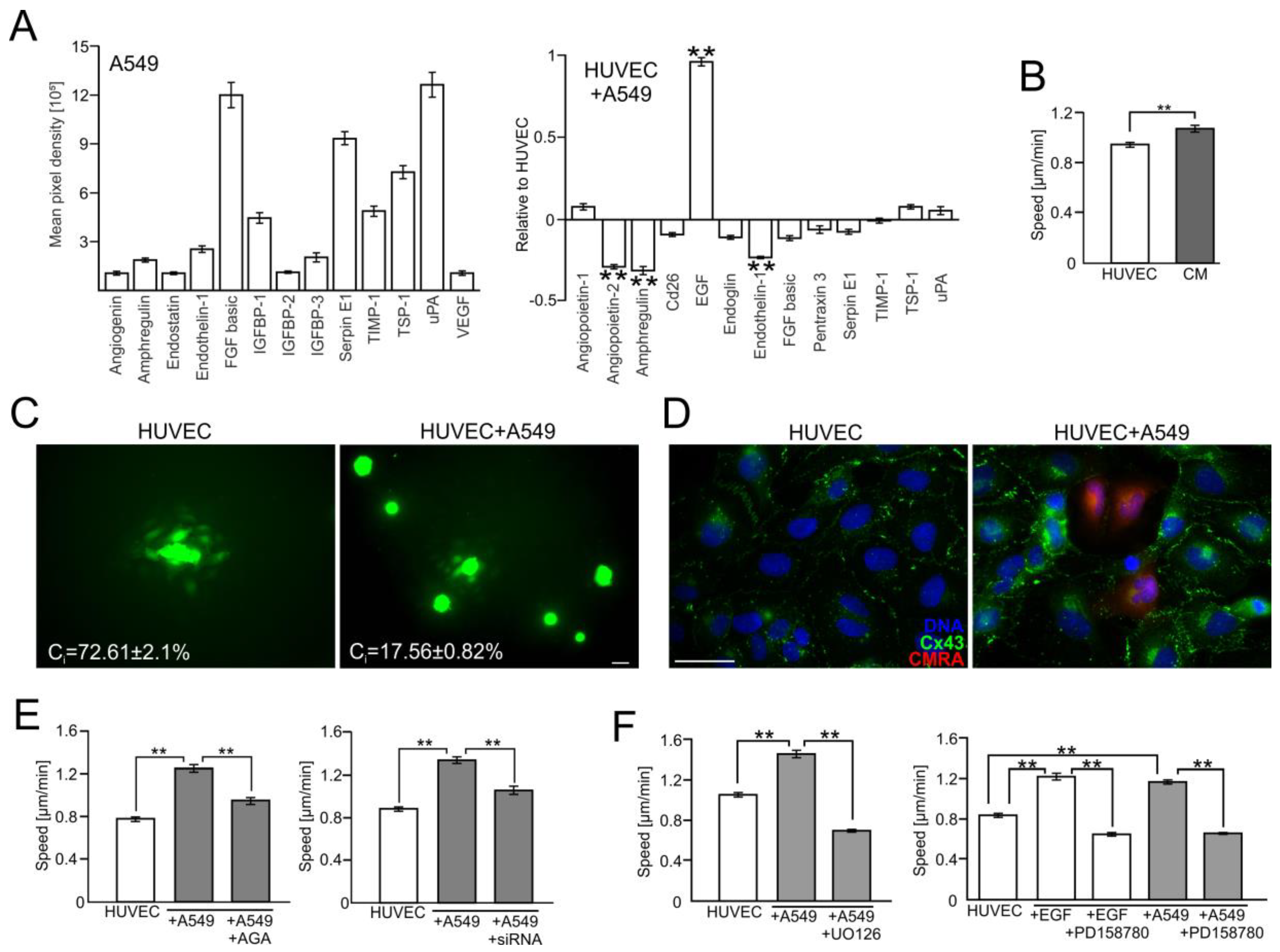
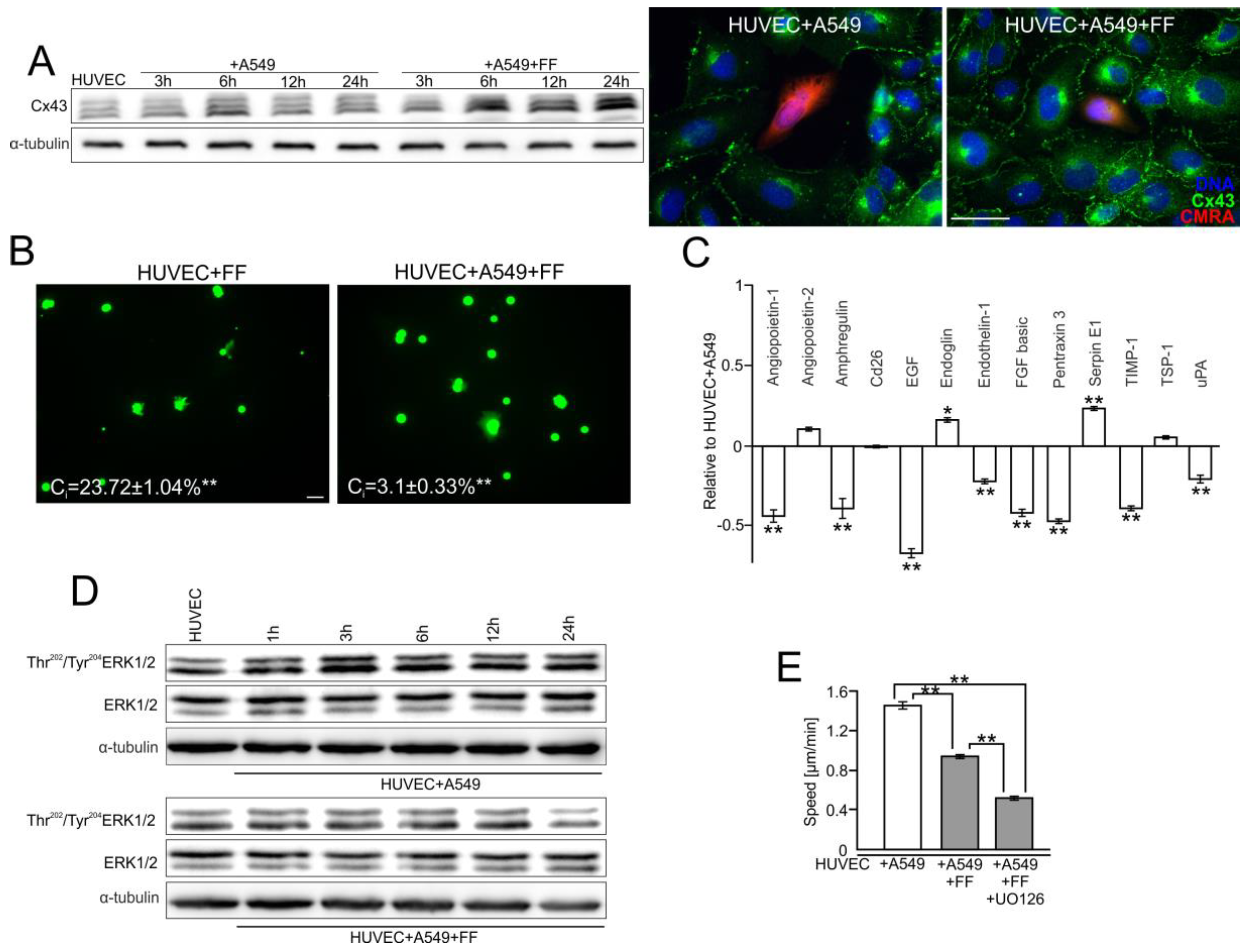
2.2. A549 Cells Impair Endothelial Barrier Function via Intercellular Cx43/EGF/ERK1/2-Dependent Signaling
2.3. FF Interferes with Cx43/EGF-Mediated Signaling between HUVECs and A549 Cells
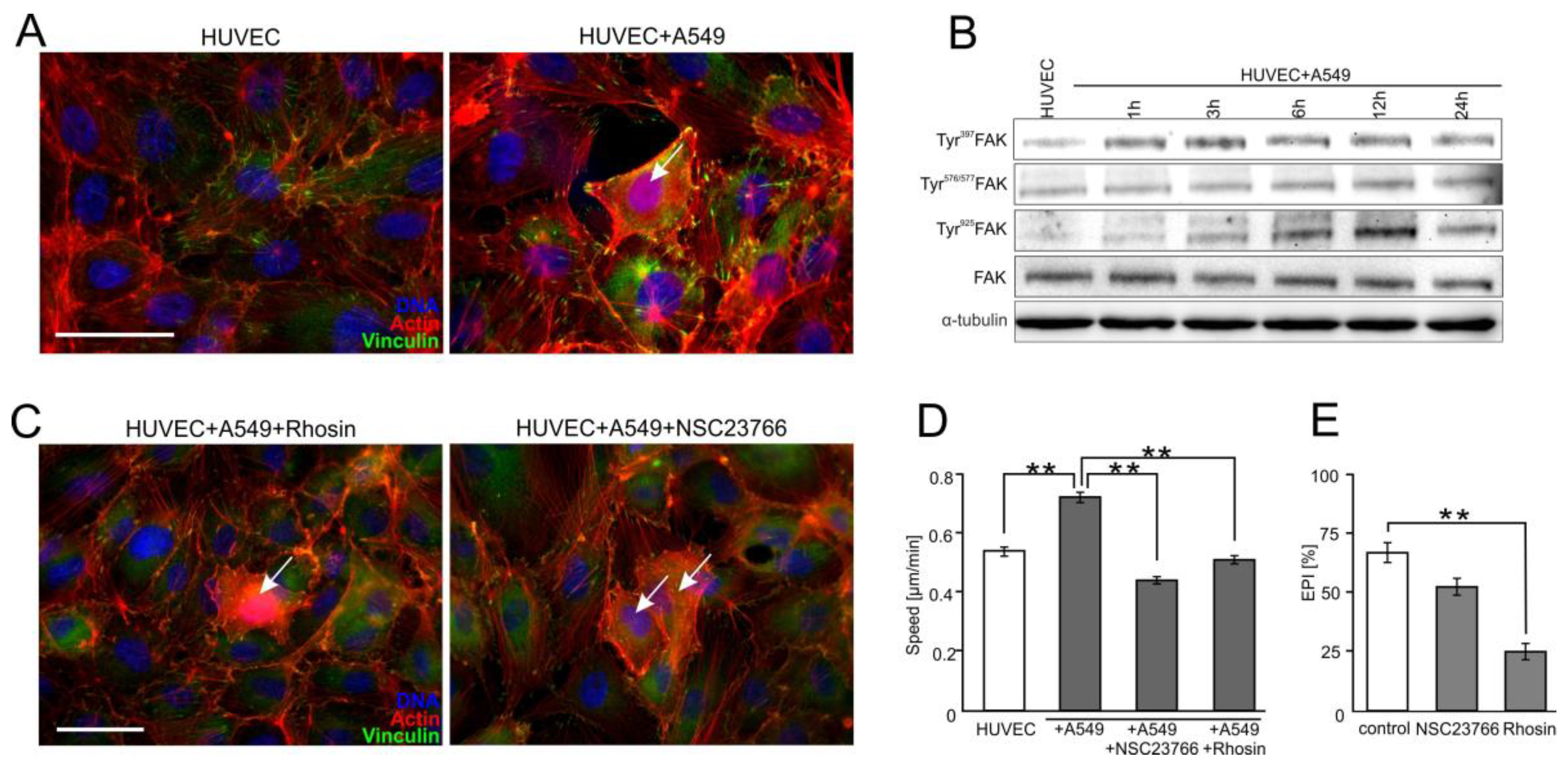
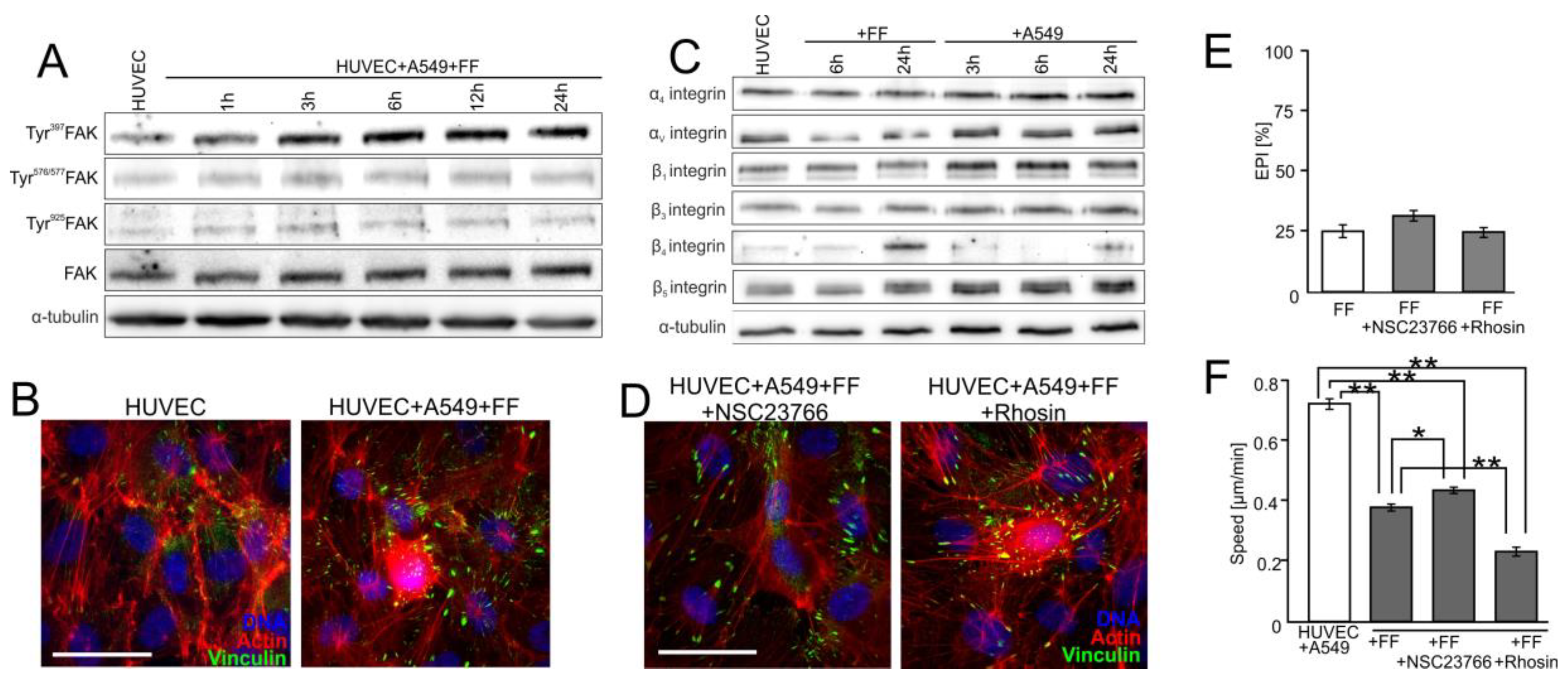
2.4. FAK/Rac1/RhoA-Dependent Signaling Participates in the Activation of HUVECs by A549 Cells
2.5. FF Directly Attenuates HUVEC Susceptibility to the RhoA/Rac1-Dependent Signaling
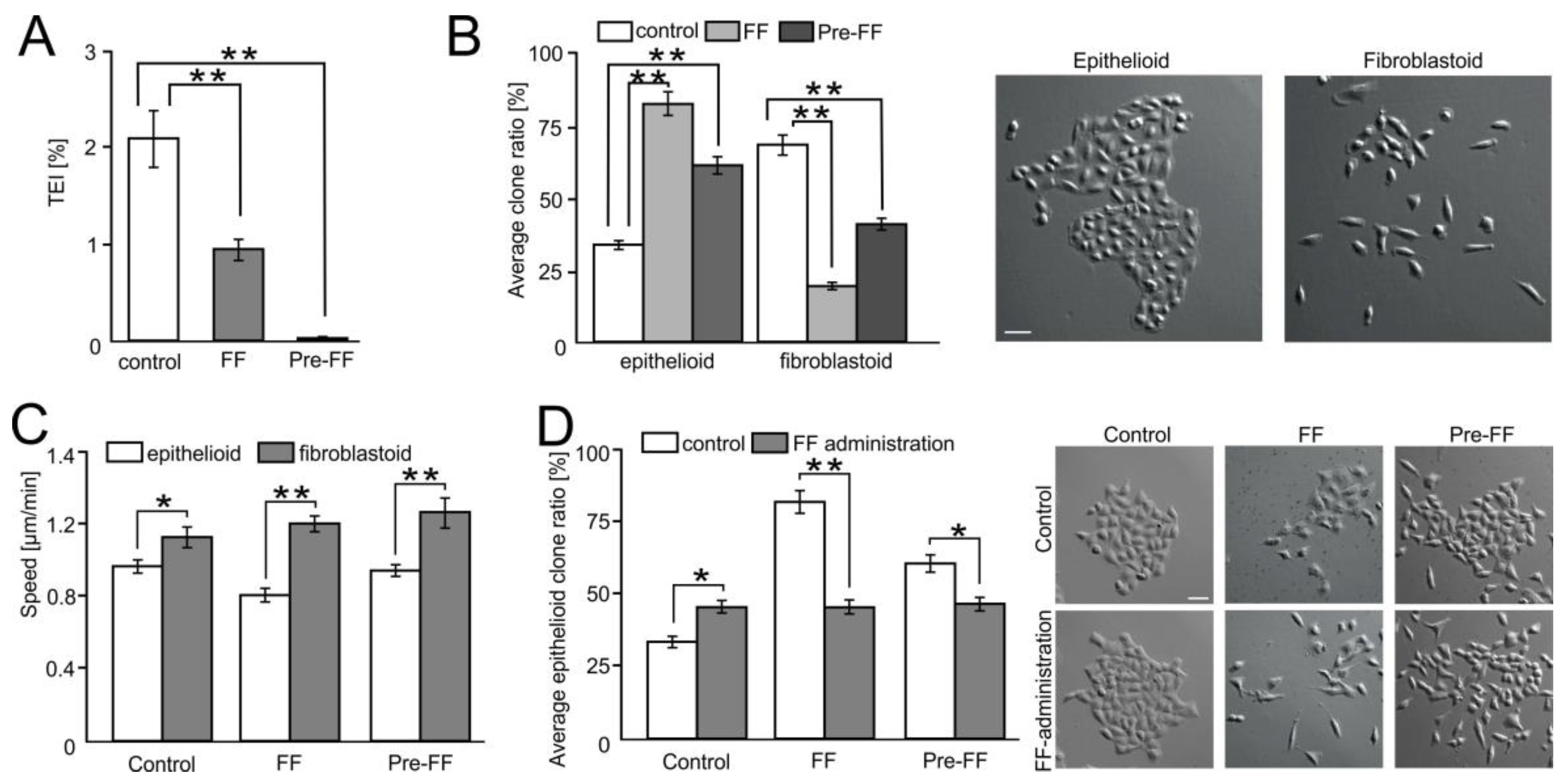
2.6. FF Selectively Impairs Transendothelial Migration of FibroblastoidA549 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Immunofluorescence and Fluorescence Microscopy
4.3. Cell Motility
4.4. Transendothelial Penetration and Permeability Analyses
4.5. Immunoblots and Array Analyses
4.6. Calcein Transfer Assay
4.7. Cx43 Silencing by siRNA
4.8. Transmigration and Microclone Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung Cancer Statistics. Adv. Exp. Med. Biol. 2016, 893, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P. The epithelial-mesenchymal transition (EMT) phenomenon. Ann. Oncol. 2010, 21 (Suppl. 7), vii89–vii92. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ferreira, A.M.; Shao, Q.; Laird, D.W.; Sandig, M. Beta3 integrins facilitate matrix interactions during transendothelial migration of PC3 prostate tumor cells. Prostate 2005, 63, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Woodward, J. Crossing the endothelium: E-selectin regulates tumor cell migration under flow conditions. Cell Adhes. Migr. 2008, 2, 151–152. [Google Scholar] [CrossRef]
- Strell, C.; Niggemann, B.; Voss, M.J.; Powe, D.G.; Zanker, K.S.; Entschladen, F. Norepinephrine promotes the beta1-integrin-mediated adhesion of MDA-MB-231 cells to vascular endothelium by the induction of a GROalpha release. Mol. Cancer Res. 2012, 10, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Reymond, N.; d’Agua, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Piwowarczyk, K.; Wybieralska, E.; Baran, J.; Borowczyk, J.; Rybak, P.; Kosinska, M.; Wlodarczyk, A.J.; Michalik, M.; Siedlar, M.; Madeja, Z.; et al. Fenofibrate enhances barrier function of endothelial continuum within the metastatic niche of prostate cancer cells. Expert Opin. Ther. Targets 2014, 19, 163–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czyz, J.; Piwowarczyk, K.; Paw, M.; Luty, M.; Wrobel, T.; Catapano, J.; Madeja, Z.; Ryszawy, D. Connexin-dependent intercellular stress signaling in tissue homeostasis and tumor development. Acta Biochim. Pol. 2017, 64, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Piwowarczyk, K.; Paw, M.; Ryszawy, D.; Rutkowska-Zapala, M.; Madeja, Z.; Siedlar, M.; Czyz, J. Connexin43high prostate cancer cells induce endothelial connexin43 up-regulation through the activation of intercellular ERK1/2-dependent signaling axis. Eur. J. Cell Biol. 2017, 96, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Goetze, S.; Eilers, F.; Bungenstock, A.; Kintscher, U.; Stawowy, P.; Blaschke, F.; Graf, K.; Law, R.E.; Fleck, E.; Grafe, M. PPAR activators inhibit endothelial cell migration by targeting Akt. Biochem. Biophys. Res. Commun. 2002, 293, 1431–1437. [Google Scholar] [CrossRef]
- Varet, J.; Vincent, L.; Mirshahi, P.; Pille, J.V.; Legrand, E.; Opolon, P.; Mishal, Z.; Soria, J.; Li, H.; Soria, C. Fenofibrate inhibits angiogenesis in vitro and in vivo. Cell. Mol. Life Sci. 2003, 60, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Meissner, M.; Stein, M.; Urbich, C.; Reisinger, K.; Suske, G.; Staels, B.; Kaufmann, R.; Gille, J. PPARalpha activators inhibit vascular endothelial growth factor receptor-2 expression by repressing Sp1-dependent DNA binding and transactivation. Circ. Res. 2004, 94, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Kaipainen, A.; Huang, S.; Butterfield, C.E.; Barnes, C.M.; Fannon, M.; Laforme, A.M.; Chaponis, D.M.; Folkman, J.; Kieran, M.W. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Shang, B.; Zhang, G.; Miele, L.; Sarkar, F.H.; Wang, Z.; Zhou, Q. Tumor cell-mediated neovascularization and lymphangiogenesis contrive tumor progression and cancer metastasis. Biochim. Biophys. Acta 2013, 1836, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Hida, K.; Ohga, N.; Akiyama, K.; Maishi, N.; Hida, Y. Heterogeneity of tumor endothelial cells. Cancer Sci. 2013, 104, 1391–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robison, N.J.; Campigotto, F.; Chi, S.N.; Manley, P.E.; Turner, C.D.; Zimmerman, M.A.; Chordas, C.A.; Werger, A.M.; Allen, J.C.; Goldman, S.; et al. A phase II trial of a multi-agent oral antiangiogenic (metronomic) regimen in children with recurrent or progressive cancer. Pediatr. Blood Cancer 2014, 61, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Reiss, K. Anticancer Properties of PPARα-Effects on Cellular Metabolism and Inflammation. PPAR Res. 2008, 2008, 930705. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Pierzchalska, M.; Reiss, K. Peroxisome proliferator activated receptor α ligands as anticancer drugs targeting mitochondrial metabolism. Curr. Pharm. Biotechnol. 2013, 14, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G. LDL reduction: How low should we go and is it safe? Curr. Cardiol. Rep. 2008, 10, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Adeghate, E.; Adem, A.; Hasan, M.Y.; Tekes, K.; Kalasz, H. Medicinal Chemistry and Actions of Dual and Pan PPAR Modulators. Open. Med. Chem. J. 2011, 5, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, P.; Rohilla, A.; Mahadevan, N. Pleiotropic actions of fenofibrate on the heart. Pharmacol. Res. 2011, 63, 8–12. [Google Scholar] [CrossRef] [PubMed]
- McKeage, K.; Keating, G.M. Fenofibrate: A review of its use in dyslipidaemia. Drugs 2011, 71, 1917–1946. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Plonka, P.M.; Urbanska, K.; Reiss, K. Peroxisome proliferator-activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin. Cancer Res. 2006, 12, 3028–3036. [Google Scholar] [CrossRef] [PubMed]
- Thuillier, P.; Anchiraico, G.J.; Nickel, K.P.; Maldve, R.E.; Gimenez-Conti, I.; Muga, S.J.; Liu, K.L.; Fischer, S.M.; Belury, M.A. Activators of peroxisome proliferator-activated receptor-alpha partially inhibit mouse skin tumor promotion. Mol. Carcinog. 2000, 29, 134–142. [Google Scholar] [CrossRef]
- Saidi, S.A.; Holland, C.M.; Charnock-Jones, D.S.; Smith, S.K. In vitro and in vivo effects of the PPAR-α agonists fenofibrate and retinoic acid in endometrial cancer. Mol. Cancer 2006, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Kaipainen, A.; Kieran, M.W.; Huang, S. PPARs: A Double-Edged Sword in Cancer Therapy? PPAR Res. 2008, 2008, 350351. [Google Scholar] [CrossRef] [PubMed]
- Drukala, J.; Urbanska, K.; Wilk, A.; Grabacka, M.; Wybieralska, E.; Del Valle, L.; Madeja, Z.; Reiss, K. ROS accumulation and IGF-IR inhibition contribute to fenofibrate/PPARα -mediated inhibition of Glioma cell notility in vitro. Mol. Cancer 2010, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.L.; Zhao, B.L. Cytotoxic effect of peroxisome proliferator fenofibrate on human HepG2 hepatoma cell line and relevant mechanisms. Toxicol. Appl. Pharmacol. 2002, 185, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R.; Bottoni, P.; Giardina, B. Mitochondria, PPARs, and Cancer: Is Receptor-Independent Action of PPAR Agonists a Key? PPAR Res. 2008, 2008, 256251. [Google Scholar] [CrossRef] [PubMed]
- Czyz, J.; Szpak, K.; Madeja, Z. The role of connexins in prostate cancer promotion and progression. Nat. Rev. Urol. 2012, 9, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Piwowarczyk, K.; Department of Cell Biology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Gronostajowa 7, 30-387 Kraków, Poland. Personal observation, 2011.
- Bechyne, I.; Szpak, K.; Madeja, Z.; Czyz, J. Functional heterogeneity of non-small lung adenocarcinoma cell sub-populations. Cell Biol. Int. 2011, 36, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Doser, K.; Guserle, R.; Nitsche, V.; Arnold, G. Comparative steady state study with 2 fenofibrate 250 mg slow release capsules. An example of bioequivalence assessment with a highly variable drug. Int. J. Clin. Pharmacol. Ther. 1996, 34, 345–348. [Google Scholar] [PubMed]
- Kajosaari, L.I.; Backman, J.T.; Neuvonen, M.; Laitila, J.; Neuvonen, P.J. Lack of effect of bezafibrate and fenofibrate on the pharmacokinetics and pharmacodynamics of repaglinide. Br. J. Clin. Pharmacol. 2004, 58, 390–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uetake, D.; Ohno, I.; Ichida, K.; Yamaguchi, Y.; Saikawa, H.; Endou, H.; Hosoya, T. Effect of fenofibrate on uric acid metabolism and urate transporter 1. Intern. Med. 2010, 49, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Huveneers, S.; Danen, E.H. Adhesion signaling–crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Kwak, B.R.; Mulhaupt, F.; Veillard, N.; Gros, D.B.; Mach, F. Altered pattern of vascular connexin expression in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Macaluso, M. Fenofibrate triggers apoptosis of glioblastoma cells in vitro: New insights for therapy. Cell. Cycle 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, D.; Kawabe, N.; Nakamura, H.; Tachibana, K.; Ishimoto, K.; Tanaka, T.; Aburatani, H.; Sakai, J.; Hamakubo, T.; Kodama, T.; et al. Fenofibrate suppresses growth of the human hepatocellular carcinoma cell via PPARalpha-independent mechanisms. Eur. J. Cell Biol. 2011, 90, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Urbanska, K.; Pannizzo, P.; Grabacka, M.; Croul, S.; Del Valle, L.; Khalili, K.; Reiss, K. Activation of PPARalpha inhibits IGF-I-mediated growth and survival responses in medulloblastoma cell lines. Int. J. Cancer 2008, 123, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Placha, W.; Plonka, P.M.; Pajak, S.; Urbanska, K.; Laidler, P.; Slominski, A. Inhibition of melanoma metastases by fenofibrate. Arch. Dermatol. Res. 2004, 296, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Wybieralska, E.; Szpak, K.; Gorecki, A.; Bonarek, P.; Miekus, K.; Drukala, J.; Majka, M.; Reiss, K.; Madeja, Z.; Czyz, J. Fenofibrate attenuates contact-stimulated cell motility and gap junctional coupling in DU-145 human prostate cancer cell populations. Oncol. Rep. 2011, 26, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Purdom, S.; Chen, Q.M. Epidermal growth factor receptor-dependent and -independent pathways in hydrogen peroxide-induced mitogen-activated protein kinase activation in cardiomyocytes and heart fibroblasts. J. Pharmacol. Exp. Ther. 2005, 312, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Zimolag, E.; Borowczyk-Michalowska, J.; Kedracka-Krok, S.; Skupien-Rabian, B.; Karnas, E.; Lasota, S.; Sroka, J.; Drukala, J.; Madeja, Z. Electric field as a potential directional cue in homing of bone marrow-derived mesenchymal stem cells to cutaneous wounds. Biochim. Biophys. Acta 2017, 1864, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horne, M.M.; Guadagno, T.M. A requirement for MAP kinase in the assembly and maintenance of the mitotic spindle. J. Cell Biol. 2003, 161, 1021–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piwowarczyk, K.; Sarna, M.; Ryszawy, D.; Czyz, J. Invasive Cx43high sub-line of human prostate DU145 cells displays increased nanomechanical deformability. Acta Biochim. Pol. 2017, 64, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Ryszawy, D.; Sarna, M.; Rak, M.; Szpak, K.; Kedracka-Krok, S.; Michalik, M.; Siedlar, M.; Zuba-Surma, E.; Burda, K.; Korohoda, W.; et al. Functional links between Snail-1 and Cx43 account for the recruitment of Cx43-positive cells into the invasive front of prostate cancer. Carcinogenesis 2014, 35, 1920–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szpak, K.; Wybieralska, E.; Niedzialkowska, E.; Rak, M.; Bechyne, I.; Michalik, M.; Madeja, Z.; Czyz, J. DU-145 prostate carcinoma cells that selectively transmigrate narrow obstacles express elevated levels of CX43. Cell. Mol. Biol. Lett. 2011, 2011 16, 625–637. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piwowarczyk, K.; Kwiecień, E.; Sośniak, J.; Zimoląg, E.; Guzik, E.; Sroka, J.; Madeja, Z.; Czyż, J. Fenofibrate Interferes with the Diapedesis of Lung Adenocarcinoma Cells through the Interference with Cx43/EGF-Dependent Intercellular Signaling. Cancers 2018, 10, 363. https://doi.org/10.3390/cancers10100363
Piwowarczyk K, Kwiecień E, Sośniak J, Zimoląg E, Guzik E, Sroka J, Madeja Z, Czyż J. Fenofibrate Interferes with the Diapedesis of Lung Adenocarcinoma Cells through the Interference with Cx43/EGF-Dependent Intercellular Signaling. Cancers. 2018; 10(10):363. https://doi.org/10.3390/cancers10100363
Chicago/Turabian StylePiwowarczyk, Katarzyna, Edyta Kwiecień, Justyna Sośniak, Eliza Zimoląg, Emiliana Guzik, Jolanta Sroka, Zbigniew Madeja, and Jarosław Czyż. 2018. "Fenofibrate Interferes with the Diapedesis of Lung Adenocarcinoma Cells through the Interference with Cx43/EGF-Dependent Intercellular Signaling" Cancers 10, no. 10: 363. https://doi.org/10.3390/cancers10100363
APA StylePiwowarczyk, K., Kwiecień, E., Sośniak, J., Zimoląg, E., Guzik, E., Sroka, J., Madeja, Z., & Czyż, J. (2018). Fenofibrate Interferes with the Diapedesis of Lung Adenocarcinoma Cells through the Interference with Cx43/EGF-Dependent Intercellular Signaling. Cancers, 10(10), 363. https://doi.org/10.3390/cancers10100363