Molecular Scoring of Hepatocellular Carcinoma for Predicting Metastatic Recurrence and Requirements of Systemic Chemotherapy

 ,
,  , ,
, , 

Abstract
:1. Introduction
2. Results
2.1. Mutations of CTNNB1 and TP53 Genes and TERT Promoter, in HCCs from Hepatic Resection
2.2. Regional Hypermethylation at TSG Promoters and Global Hypomethylation in HCCs from Hepatic Resection
2.3. Degree of Chromosomal Alterations in HCC
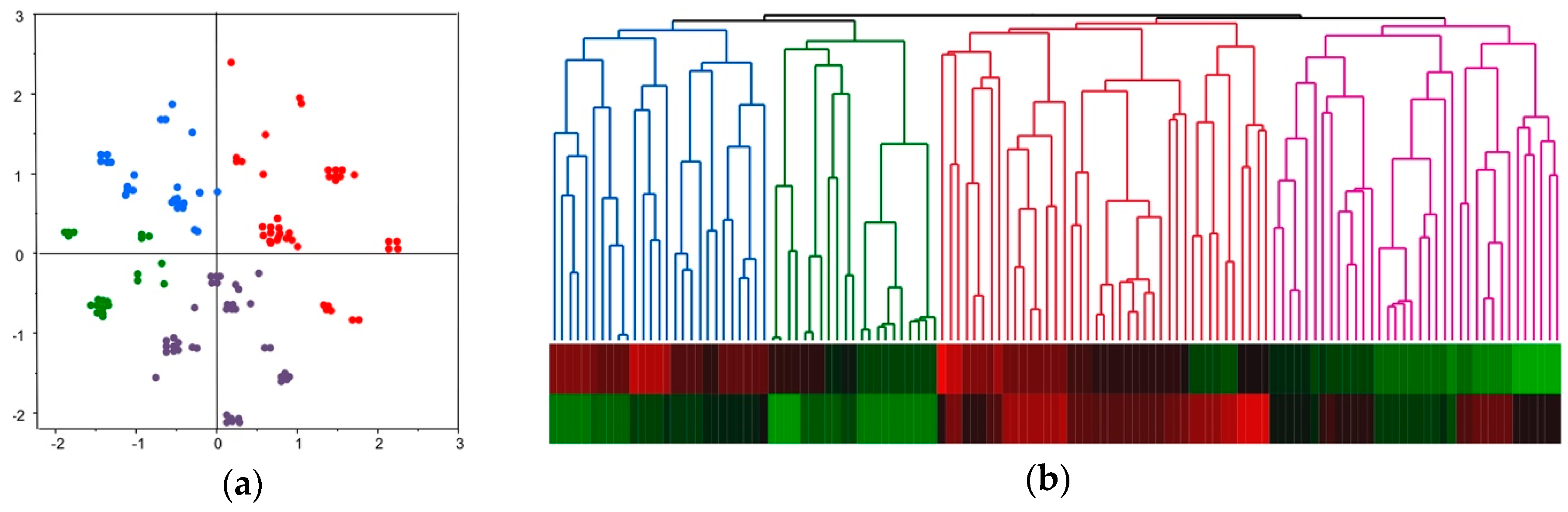
2.4. Molecular Classification of HCC Based on Comprehensive Analyses of Mutation, DNA Methylation, and Chromosomal Alterations
2.5. Molecular Classification and Clinical Feature of HCC
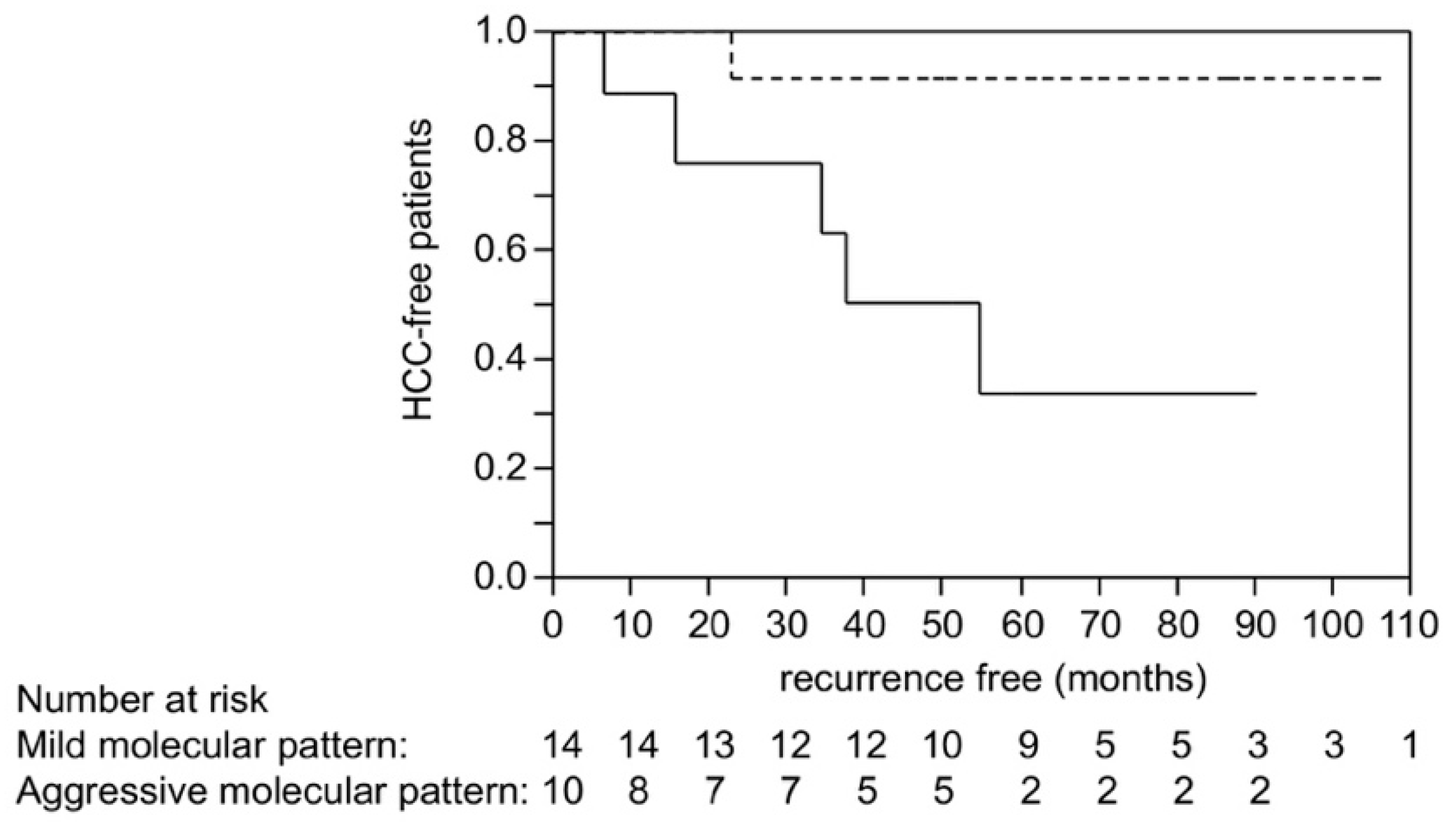
2.6. Scoring Using Molecular Risk Factors and Recurrence after Liver Transplantation in HCC
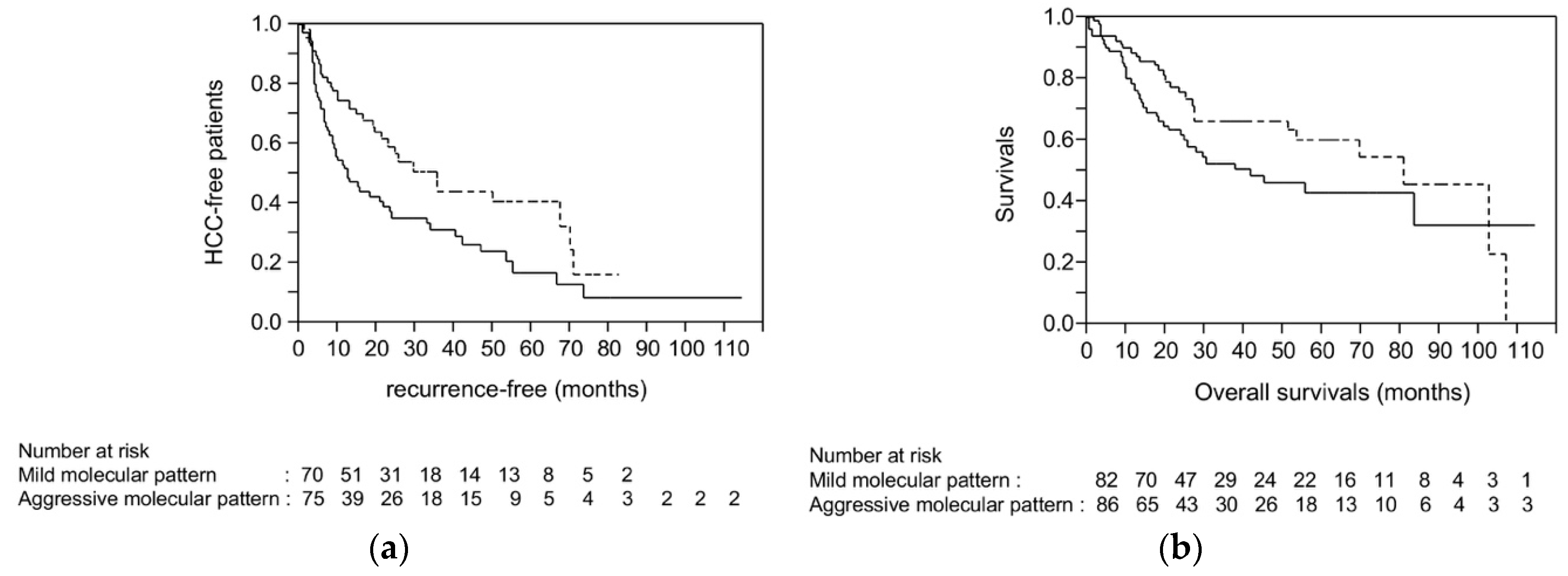
2.7. Survival Analysis Using Dataset from TCGA
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Mutational Analysis of Cancer-Related Genes in HCC
4.3. Detection of Regional Promoter Methylation of Tumor Suppressor Genes in HCC
4.4. Detection of Global Hypomethylation in HCC
4.5. Quantification of Altered Chromosomal Region in HCC
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Fernandez-Sevilla, E.; Allard, M.A.; Selten, J.; Golse, N.; Vibert, E.; Sa Cunha, A.; Cherqui, D.; Castaing, D.; Adam, R. Recurrence of hepatocellular carcinoma after liver transplantation: Is there a place for resection? Liver Transpl. 2017, 23, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Kudo, M. A New Era of Systemic Therapy for Hepatocellular Carcinoma with Regorafenib and Lenvatinib. Liver Cancer 2017, 6, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Zhu, A.X.; Baron, A.D.; Malfertheiner, P.; Kudo, M.; Kawazoe, S.; Pezet, D.; Weissinger, F.; Brandi, G.; Barone, C.A.; Okusaka, T.; et al. Ramucirumab as Second-Line Treatment in Patients with Advanced Hepatocellular Carcinoma: Analysis of REACH Trial Results by Child-Pugh Score. JAMA Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Nishida, N.; Kudo, M. Immune checkpoint blockade for the treatment of human hepatocellular carcinoma. Hepatol. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed]
- Thorgeirsson, S.S.; Grisham, J.W. Molecular pathogenesis of human hepatocellular carcinoma. Nat. Genet. 2002, 31, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Fukuda, Y.; Komeda, T.; Ito, T.; Nishimura, T.; Minata, M.; Kuno, M.; Katsuma, H.; Ikai, I.; Yamaoka, Y.; et al. Prognostic impact of multiple allelic losses on metastatic recurrence in hepatocellular carcinoma after curative resection. Oncology 2002, 62, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Nishimura, T.; Ito, T.; Komeda, T.; Fukuda, Y.; Nakao, K. Chromosomal instability and human hepatocarcinogenesis. Histol. Histopathol. 2003, 18, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Kudo, M.; Nishimura, T.; Arizumi, T.; Takita, M.; Kitai, S.; Yada, N.; Hagiwara, S.; Inoue, T.; Minami, Y.; et al. Unique association between global DNA hypomethylation and chromosomal alterations in human hepatocellular carcinoma. PLoS ONE 2013, 8, e72312. [Google Scholar] [CrossRef] [PubMed]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Kudo, M.; Nagasaka, T.; Ikai, I.; Goel, A. Characteristic patterns of altered DNA methylation predict emergence of human hepatocellular carcinoma. Hepatology 2012, 56, 994–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, N.; Chishina, H.; Arizumi, T.; Takita, M.; Kitai, S.; Yada, N.; Hagiwara, S.; Inoue, T.; Minami, Y.; Ueshima, K.; et al. Identification of epigenetically inactivated genes in human hepatocellular carcinoma by integrative analyses of methylation profiling and pharmacological unmasking. Dig. Dis. 2014, 32, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Kudo, M. Alteration of Epigenetic Profile in Human Hepatocellular Carcinoma and Its Clinical Implications. Liver Cancer 2014, 3, 417–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, N.; Nishimura, T.; Nakai, T.; Chishina, H.; Arizumi, T.; Takita, M.; Kitai, S.; Yada, N.; Hagiwara, S.; Inoue, T.; et al. Genome-wide profiling of DNA methylation and tumor progression in human hepatocellular carcinoma. Dig. Dis. 2014, 32, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Iwanishi, M.; Minami, T.; Chishina, H.; Arizumi, T.; Takita, M.; Kitai, S.; Yada, N.; Ida, H.; Hagiwara, S.; et al. Hepatic DNA Methylation Is Affected by Hepatocellular Carcinoma Risk in Patients with and without Hepatitis Virus. Dig. Dis. 2015, 33, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Nishimura, T.; Nagasaka, T.; Ikai, I.; Goel, A.; Boland, C.R. Extensive methylation is associated with beta-catenin mutations in hepatocellular carcinoma: Evidence for two distinct pathways of human hepatocarcinogenesis. Cancer Res. 2007, 67, 4586–4594. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Goel, A. Genetic and epigenetic signatures in human hepatocellular carcinoma: A systematic review. Curr. Genom. 2011, 12, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Kudo, M. Clinical Significance of Epigenetic Alterations in Human Hepatocellular Carcinoma and Its Association with Genetic Mutations. Dig. Dis. 2016, 34, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Toffanin, S.; Lachenmayer, A.; Villanueva, A.; Minguez, B.; Llovet, J.M. Molecular classification and novel targets in hepatocellular carcinoma: Recent advancements. Semin. Liver Dis. 2010, 30, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Fukuda, Y.; Kokuryu, H.; Toguchida, J.; Yandell, D.W.; Ikenega, M.; Imura, H.; Ishizaki, K. Role and mutational heterogeneity of the p53 gene in hepatocellular carcinoma. Cancer Res. 1993, 53, 368–372. [Google Scholar] [PubMed]
- Rebouissou, S.; Couchy, G.; Libbrecht, L.; Balabaud, C.; Imbeaud, S.; Auffray, C.; Roskams, T.; Bioulac-Sage, P.; Zucman-Rossi, J. The beta-catenin pathway is activated in focal nodular hyperplasia but not in cirrhotic FNH-like nodules. J. Hepatol. 2008, 49, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebouissou, S.; Franconi, A.; Calderaro, J.; Letouze, E.; Imbeaud, S.; Pilati, C.; Nault, J.C.; Couchy, G.; Laurent, A.; Balabaud, C.; et al. Genotype-phenotype correlation of CTNNB1 mutations reveals different ss-catenin activity associated with liver tumor progression. Hepatology 2016, 64, 2047–2061. [Google Scholar] [CrossRef] [PubMed]
- Desert, R.; Rohart, F.; Canal, F.; Sicard, M.; Desille, M.; Renaud, S.; Turlin, B.; Bellaud, P.; Perret, C.; Clement, B.; et al. Human hepatocellular carcinomas with a periportal phenotype have the lowest potential for early recurrence after curative resection. Hepatology 2017, 66, 1502–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyault, S.; Rickman, D.S.; de Reynies, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Herault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, N.; Nagasaka, T.; Nishimura, T.; Ikai, I.; Boland, C.R.; Goel, A. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology 2008, 47, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Calderaro, J.; Di Tommaso, L.; Balabaud, C.; Zafrani, E.S.; Bioulac-Sage, P.; Roncalli, M.; Zucman-Rossi, J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology 2014, 60, 1983–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, D.Y.; Villanueva, A.; Hoshida, Y.; Peix, J.; Newell, P.; Minguez, B.; LeBlanc, A.C.; Donovan, D.J.; Thung, S.N.; Sole, M.; et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008, 68, 6779–6788. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentilini, D.; Scala, S.; Gaudenzi, G.; Garagnani, P.; Capri, M.; Cescon, M.; Grazi, G.L.; Bacalini, M.G.; Pisoni, S.; Dicitore, A.; et al. Epigenome-wide association study in hepatocellular carcinoma: Identification of stochastic epigenetic mutations through an innovative statistical approach. Oncotarget 2017, 8, 41890–41902. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, T.; Nishida, N.; Itoh, T.; Kuno, M.; Minata, M.; Komeda, T.; Fukuda, Y.; Ikai, I.; Yamaoka, Y.; Nakao, K. Comprehensive allelotyping of well-differentiated human hepatocellular carcinoma with semiquantitative determination of chromosomal gain or loss. Genes Chromosomes Cancer 2002, 35, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Nishida, N.; Itoh, T.; Komeda, T.; Fukuda, Y.; Ikai, I.; Yamaoka, Y.; Nakao, K. Discrete breakpoint mapping and shortest region of overlap of chromosome arm 1q gain and 1p loss in human hepatocellular carcinoma detected by semiquantitative microsatellite analysis. Genes Chromosomes Cancer 2005, 42, 34–43. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Backgrounds | CTNNB1 Mutation | TP53 Mutation | TERT-p Mutation 1 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| + 2 | − 3 | p Value | + | − | p Value | + | − | p Value | ||
| Sex | male | 28 | 62 | 0.0088 | 22 | 68 | 0.2153 | 60 | 30 | 0.6083 |
| female | 3 | 32 | 5 | 30 | 25 | 10 | ||||
| Age | >60 years old | 21 | 45 | 0.0546 | 13 | 53 | 0.5845 | 48 | 18 | 0.2308 |
| ≤60 years old | 10 | 49 | 14 | 45 | 37 | 22 | ||||
| HBV 4 | positive | 8 | 21 | 0.6918 | 11 | 18 | 0.0147 | 16 | 13 | 0.0911 |
| negative | 23 | 73 | 16 | 81 | 69 | 27 | ||||
| HCV 5 | positive | 22 | 55 | 0.2162 | 15 | 62 | 0.4658 | 61 | 16 | 0.0007 |
| negative | 9 | 39 | 12 | 36 | 24 | 24 | ||||
| NBNC 6 | yes | 2 | 19 | 0.0755 | 3 | 18 | 0.3719 | 10 | 11 | 0.0282 |
| no | 29 | 75 | 24 | 80 | 75 | 29 | ||||
| Serum AFP level | ≥200 ng/mL | 4 | 42 | 0.0015 | 16 | 30 | 0.0063 | 27 | 19 | 0.0888 |
| <200 ng/mL | 27 | 52 | 11 | 69 | 58 | 21 | ||||
| Tumor size | ≥3.0 cm | 24 | 62 | 0.2607 | 21 | 65 | 0.2831 | 61 | 25 | 0.2532 |
| <3.0 cm | 7 | 31 | 6 | 32 | 23 | 15 | ||||
| Vascular invasion | presence | 9 | 49 | 0.0194 | 19 | 39 | 0.0029 | 37 | 21 | 0.4097 |
| absence | 22 | 43 | 7 | 58 | 46 | 19 | ||||
| Tumor number | solitary | 17 | 38 | 0.3067 | 10 | 45 | 0.5765 | 39 | 16 | 0.8219 |
| multiple | 13 | 45 | 13 | 45 | 40 | 18 | ||||
| Differentiation | well | 16 | 20 | 0.0018 | 3 | 33 | 0.0235 | 28 | 8 | 0.1354 |
| moderately/poorly | 15 | 71 | 23 | 63 | 55 | 31 | ||||
| Clinicopathological Backgrounds | Hypermethylation of Tumor Suppressor Gene 1 | Significant Global Hypomethylation 2 | FAL (%) 3 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| + 2 | − 3 | p Value | + | − | p Value | + | − | p Value | ||
| Sex | male | 61 | 29 | 0.2636 | 51 | 39 | 0.2703 | 47 | 43 | 0.6204 |
| female | 20 | 15 | 16 | 19 | 20 | 15 | ||||
| Age | >60 years old | 43 | 23 | 0.9306 | 32 | 34 | 0.2252 | 34 | 32 | 0.6211 |
| ≤60 years old | 38 | 21 | 35 | 24 | 33 | 26 | ||||
| HBV 4 | positive | 17 | 12 | 0.4266 | 20 | 9 | 0.0583 | 19 | 10 | 0.1420 |
| negative | 64 | 32 | 47 | 49 | 48 | 48 | ||||
| HCV 5 | positive | 55 | 22 | 0.0494 | 36 | 41 | 0.0519 | 38 | 39 | 0.2276 |
| negative | 26 | 22 | 31 | 17 | 29 | 19 | ||||
| NBNC 6 | yes | 11 | 10 | 0.1914 | 13 | 8 | 0.4028 | 12 | 9 | 0.7212 |
| no | 70 | 34 | 54 | 50 | 55 | 49 | ||||
| Serum AFP level | ≥200 ng/mL | 30 | 16 | 0.9406 | 30 | 16 | 0.0469 | 38 | 8 | <0.0001 |
| <200 ng/mL | 51 | 28 | 37 | 42 | 29 | 50 | ||||
| Tumor size | ≥3.0 cm | 58 | 28 | 0.3057 | 48 | 28 | 0.3849 | 49 | 37 | 0.2079 |
| <3.0 cm | 22 | 16 | 18 | 20 | 17 | 21 | ||||
| Vascular invasion | presence | 40 | 18 | 0.3004 | 37 | 21 | 0.0216 | 39 | 19 | 0.0043 |
| absence | 39 | 26 | 28 | 37 | 27 | 38 | ||||
| Tumor number | solitary | 38 | 17 | 0.3321 | 28 | 27 | 0.7871 | 30 | 25 | 0.6622 |
| multiple | 35 | 23 | 31 | 27 | 34 | 24 | ||||
| Differentiation | well | 24 | 12 | 0.7748 | 18 | 18 | 0.6386 | 14 | 22 | 0.0393 |
| moderately/poorly | 55 | 31 | 47 | 39 | 51 | 35 | ||||
| Characteristics of Backgrounds | A1 (%) 1 | A2 (%) | B1 (%) | B2 (%) | p Value |
|---|---|---|---|---|---|
| (n = 27) | (n = 21) | (n = 41) | (n = 36) | ||
| Molecular events | |||||
| CTNNB1 mutation | |||||
| positive (n = 31) | 1 (4) | 0 (0) | 13 (32) | 17 (47) | <0.0001 |
| negative (n = 94) | 26 | 21 | 28 | 19 | |
| TP53 mutation | |||||
| positive (n = 27) | 3 (11) | 0 (0) | 23 (56) | 1 (3) | <0.0001 |
| negative (n = 98) | 24 | 21 | 18 | 35 | |
| TERT promoter mutation | |||||
| positive (n = 85) | 5 (19) | 15 (71) | 31 (76) | 34 (94) | <0.0001 |
| negative (n = 40) | 22 | 6 | 10 | 2 | |
| TSG promoter hypermethylation | |||||
| presence (n = 81) | 6 (22) | 2 (10) | 39 (95) | 34 (94) | <0.0001 |
| absence (n = 44) | 21 | 19 | 2 | 2 | |
| Significant global hypomethylation | |||||
| presence (n = 67) | 16 (59) | 2 (10) | 38 (93) | 11 (31) | <0.0001 |
| absence (n = 58) | 11 | 19 | 3 | 25 | |
| Chromosomal alterations | |||||
| FAL ≥ 21% (n = 67) | 19 (70) | 3 (14) | 37 (90) | 8 (22) | <0.0001 |
| FAL < 21% (n = 58) | 8 | 18 | 4 | 28 | |
| Clinicopathological backgrounds | |||||
| Age (years old) | |||||
| ≤60 (n = 59) | 16 | 7 | 21 | 15 | 0.2720 |
| >60 (n = 66) | 11 | 14 | 20 | 21 | |
| Sex | |||||
| Male (n = 90) | 20 | 12 | 30 | 28 | 0.3496 |
| Female (n = 35) | 7 | 9 | 11 | 8 | |
| HBsAg | |||||
| Positive (n = 29) | 10 | 2 | 12 | 5 | 0.0525 |
| Negative (n = 96) | 17 | 19 | 29 | 31 | |
| HCVAb | |||||
| Positive (n = 77) | 10 (37) | 15 (71) | 25 (61) | 27 (75) | 0.0149 |
| Negative (n = 48) | 17 | 6 | 16 | 9 | |
| NBNC | |||||
| yes (n = 21) | 7 | 4 | 6 | 4 | 0.4478 |
| no (n = 104) | 20 | 17 | 35 | 32 | |
| Serum AFP level (ng/mL) | |||||
| ≥200 (n = 46) | 14 (52) | 4 (19) | 23 (56) | 5 (14) | 0.0002 |
| <200 (n = 79) | 13 | 17 | 18 | 31 | |
| Tumor size (cm) | |||||
| ≥3.0 (n = 86) | 18 | 12 | 30 | 26 | 0.5117 |
| <3.0 (n = 38) | 9 | 9 | 10 | 10 | |
| Vascular invasion | |||||
| Presence (n = 58) | 17 (63) | 5 (24) | 25 (64) | 11 (31) | 0.0013 |
| Absence (n = 65) | 10 | 16 | 14 | 25 | |
| Number of tumors | |||||
| Multiple (n = 58) | 14 | 10 | 19 | 15 | 0.7273 |
| Solitary (n = 55) | 10 | 8 | 18 | 19 | |
| Differentiation | |||||
| Moderately/poorly (n = 86) | 22 | 12 | 31 | 21 | 0.0820 |
| Well (n = 36) | 5 | 8 | 8 | 15 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishida, N.; Nishimura, T.; Kaido, T.; Minaga, K.; Yamao, K.; Kamata, K.; Takenaka, M.; Ida, H.; Hagiwara, S.; Minami, Y.; et al. Molecular Scoring of Hepatocellular Carcinoma for Predicting Metastatic Recurrence and Requirements of Systemic Chemotherapy. Cancers 2018, 10, 367. https://doi.org/10.3390/cancers10100367
Nishida N, Nishimura T, Kaido T, Minaga K, Yamao K, Kamata K, Takenaka M, Ida H, Hagiwara S, Minami Y, et al. Molecular Scoring of Hepatocellular Carcinoma for Predicting Metastatic Recurrence and Requirements of Systemic Chemotherapy. Cancers. 2018; 10(10):367. https://doi.org/10.3390/cancers10100367
Chicago/Turabian StyleNishida, Naoshi, Takafumi Nishimura, Toshimi Kaido, Kosuke Minaga, Kentaro Yamao, Ken Kamata, Mamoru Takenaka, Hiroshi Ida, Satoru Hagiwara, Yasunori Minami, and et al. 2018. "Molecular Scoring of Hepatocellular Carcinoma for Predicting Metastatic Recurrence and Requirements of Systemic Chemotherapy" Cancers 10, no. 10: 367. https://doi.org/10.3390/cancers10100367
APA StyleNishida, N., Nishimura, T., Kaido, T., Minaga, K., Yamao, K., Kamata, K., Takenaka, M., Ida, H., Hagiwara, S., Minami, Y., Sakurai, T., Watanabe, T., & Kudo, M. (2018). Molecular Scoring of Hepatocellular Carcinoma for Predicting Metastatic Recurrence and Requirements of Systemic Chemotherapy. Cancers, 10(10), 367. https://doi.org/10.3390/cancers10100367