Identification of the Transcription Factor Relationships Associated with Androgen Deprivation Therapy Response and Metastatic Progression in Prostate Cancer

 , , , , ,
, , , , , 

Abstract
:1. Introduction
2. Results
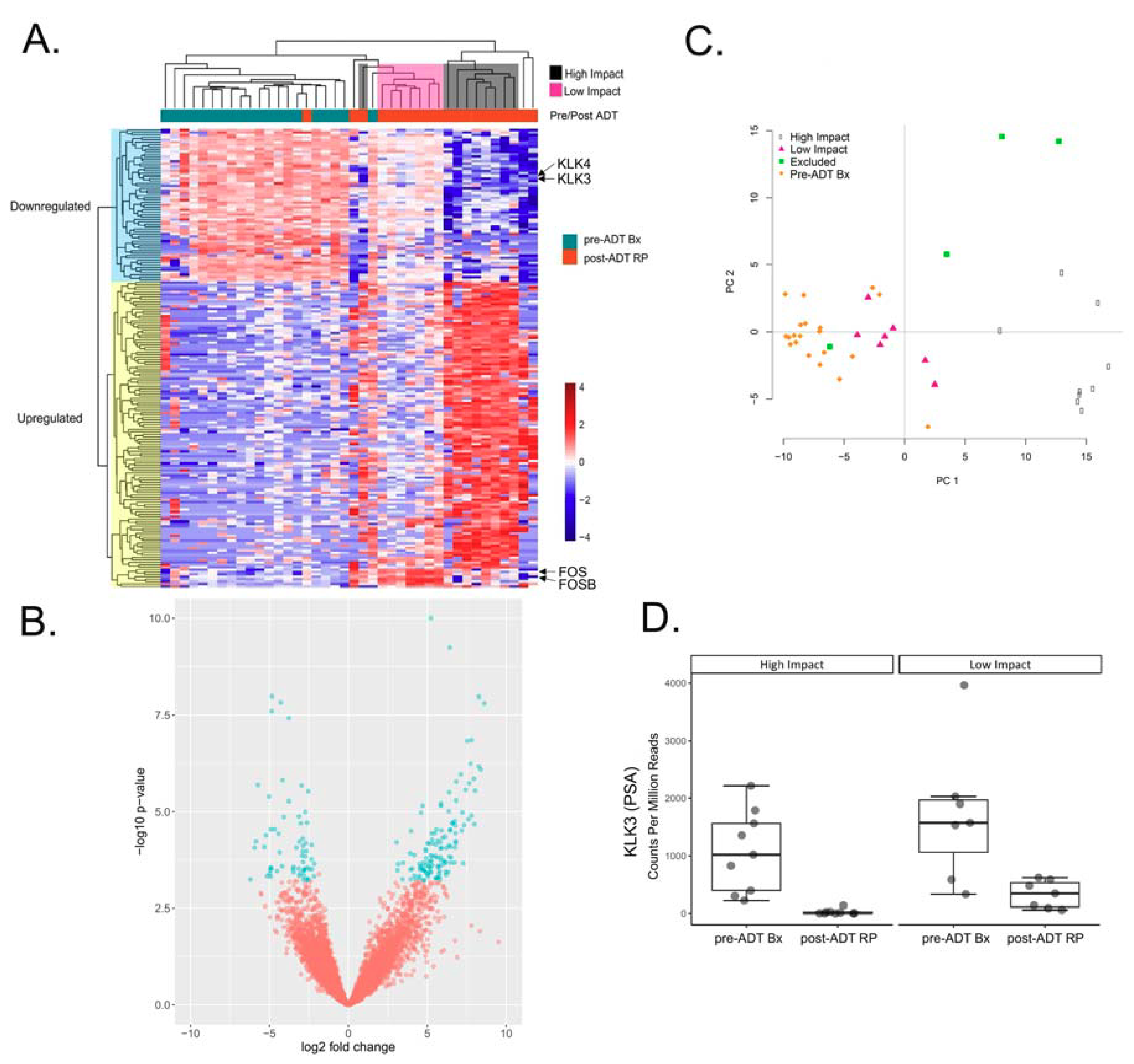
2.1. Differential Expression Analysis Reveals Two Distinct Transcriptional Responses to ADT
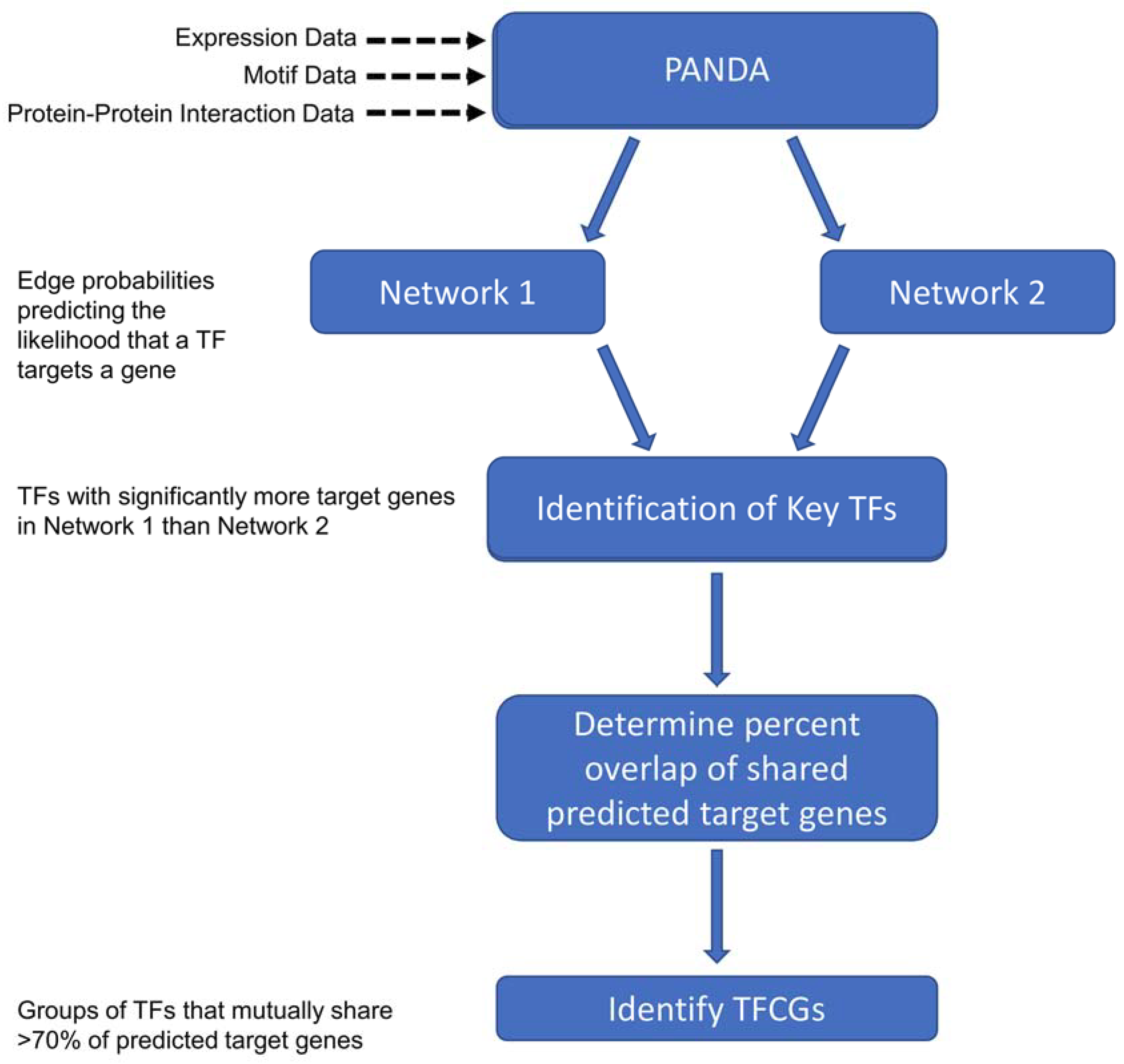
2.2. Identifying Transcription Factors (TFs) Enriched for Unique Targets in the High Impact Network
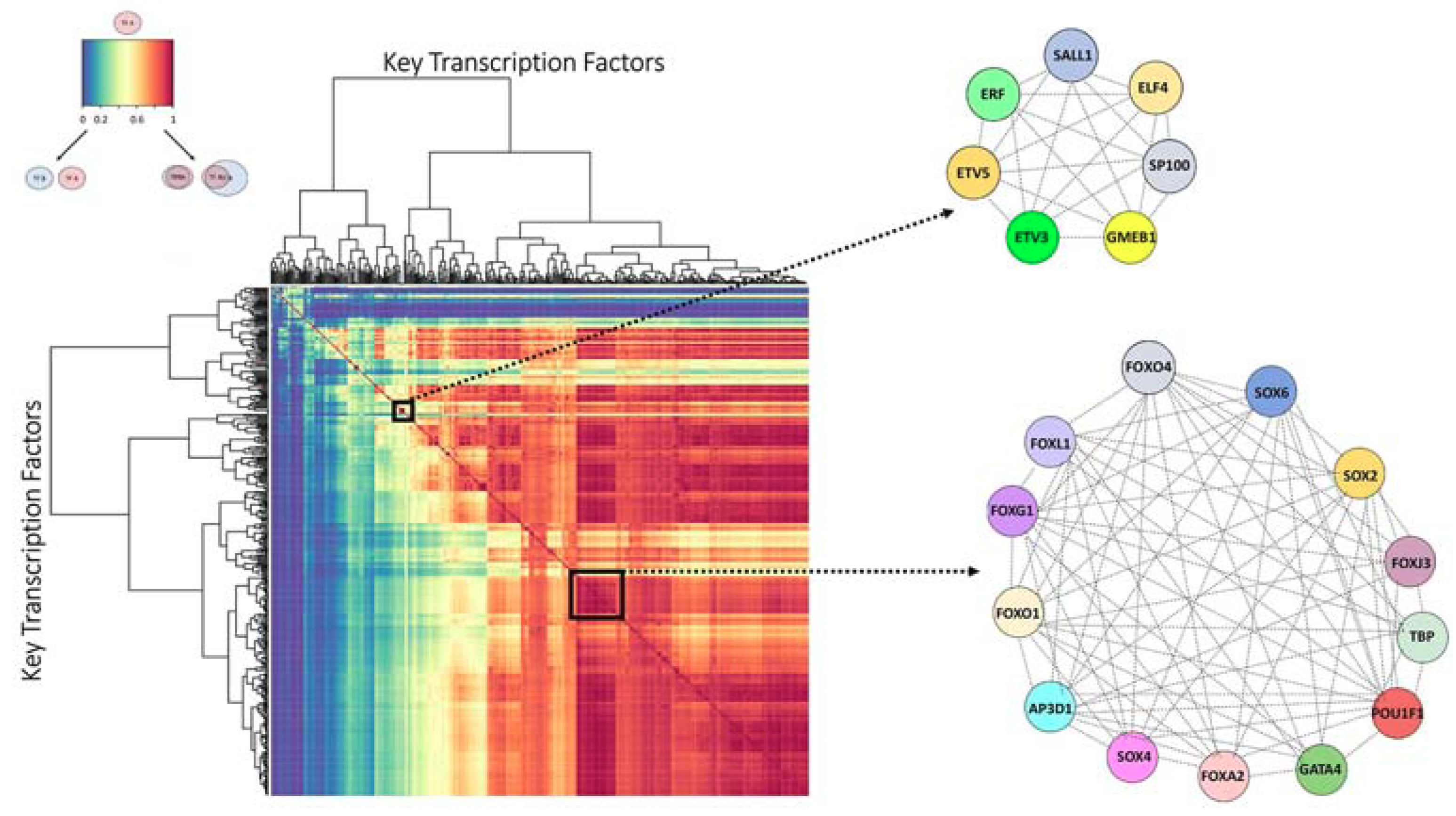
2.3. Finding the Transcription Factor Coordinated Groups (TCFGs) in the High Impact Network
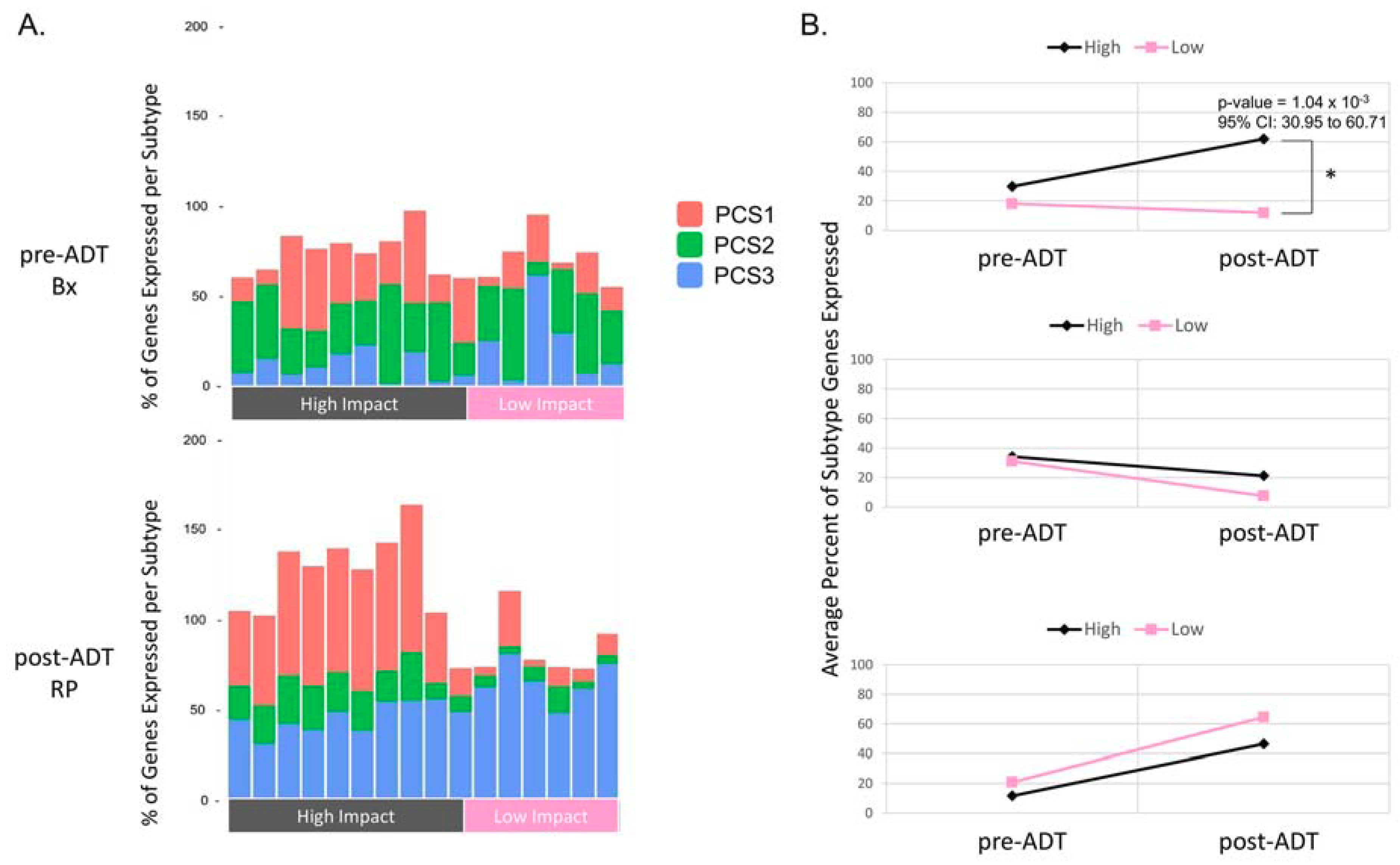
2.4. Comparison of the Metastatic PCS1 Network and High Impact Group Network Reveals Common TFCGs
3. Discussion
4. Materials and Methods
4.1. Tissue Specimens
4.2. RNA-Sequencing and Differential Gene Expression Analysis of Pre/Post ADT Patient Samples
4.3. Constructing Transcriptional Networks Using PANDA
4.4. Protein-Protein Interaction Data
4.5. Motif Data
4.6. Expression Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef] [PubMed]
- Bolla, M.; Collette, L.; Blank, L.; Warde, P.; Dubois, J.B.; Mirimanoff, R.O.; Storme, G.; Bernier, J.; Kuten, A.; Sternberg, C.; et al. Long-term results with immediate androgen suppression and external irradiation in patients with locally advanced prostate cancer (an eortc study): A phase iii randomised trial. Lancet 2002, 360, 103–106. [Google Scholar] [CrossRef]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.B.; Dits, N.F.; Erkens-Schulze, S.; van Weerden, W.M.; Jenster, G. Bypass mechanisms of the androgen receptor pathway in therapy-resistant prostate cancer cell models. PLoS ONE 2010, 5, e13500. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.G.; Li, F.X.; Pan, S.; Xu, C.B.; Dai, J.Q.; Zhao, X.H. Identification of genes associated with castrationresistant prostate cancer by gene expression profile analysis. Mol. Med. Rep. 2017, 16, 6803–6813. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [PubMed]
- Holzbeierlein, J.; Lal, P.; LaTulippe, E.; Smith, A.; Satagopan, J.; Zhang, L.; Ryan, C.; Smith, S.; Scher, H.; Scardino, P.; et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am. J. Pathol. 2004, 164, 217–227. [Google Scholar] [CrossRef]
- Longo, D.L. New therapies for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 479–481. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Qi, Y.; Zhang, G.; Xu, D.; Zhan, Y.; Alvarez, X.; Guo, Z.; Fu, X.; Plymate, S.R.; Sartor, O.; et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget 2014, 5, 1646–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Li, W.; Zhang, Y.; Yuan, X.; Xu, K.; Yu, J.; Chen, Z.; Beroukhim, R.; Wang, H.; Lupien, M.; et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009, 138, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Faivre, E.J.; Wilcox, D.; Lin, X.; Hessler, P.; Torrent, M.; He, W.; Uziel, T.; Albert, D.H.; McDaniel, K.; Kati, W.; et al. Exploitation of castration-resistant prostate cancer transcription factor dependencies by the novel bet inhibitor abbv-075. Mol. Cancer Res. 2017, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 162, 454. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, S.; Knudsen, B.S.; Erho, N.; Alshalalfa, M.; Takhar, M.; Al-Deen Ashab, H.; Davicioni, E.; Karnes, R.J.; Klein, E.A.; Den, R.B.; et al. Integrated classification of prostate cancer reveals a novel luminal subtype with poor outcome. Cancer Res. 2016, 76, 4948–4958. [Google Scholar] [CrossRef] [PubMed]
- Glass, K.; Huttenhower, C.; Quackenbush, J.; Yuan, G.C. Passing messages between biological networks to refine predicted interactions. PLoS ONE 2013, 8, e64832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, K.; Quackenbush, J.; Spentzos, D.; Haibe-Kains, B.; Yuan, G.C. A network model for angiogenesis in ovarian cancer. BMC Bioinform. 2015, 16, 115. [Google Scholar] [CrossRef] [PubMed]
- Favata, M.F.; Horiuchi, K.Y.; Manos, E.J.; Daulerio, A.J.; Stradley, D.A.; Feeser, W.S.; Van Dyk, D.E.; Pitts, W.J.; Earl, R.A.; Hobbs, F.; et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998, 273, 18623–18632. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ivanov, A.A.; Su, R.; Gonzalez-Pecchi, V.; Qi, Q.; Liu, S.; Webber, P.; McMillan, E.; Rusnak, L.; Pham, C.; et al. The oncoppi network of cancer-focused protein-protein interactions to inform biological insights and therapeutic strategies. Nat. Commun. 2017, 8, 14356. [Google Scholar] [CrossRef] [PubMed]
- Peri, S.; Navarro, J.D.; Amanchy, R.; Kristiansen, T.Z.; Jonnalagadda, C.K.; Surendranath, V.; Niranjan, V.; Muthusamy, B.; Gandhi, T.K.; Gronborg, M.; et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003, 13, 2363–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kel, A.E.; Gossling, E.; Reuter, I.; Cheremushkin, E.; Kel-Margoulis, O.V.; Wingender, E. Match: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003, 31, 3576–3579. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Tsai, Y.C.; Siu, M.K.; Yeh, H.L.; Chen, C.L.; Yin, J.J.; Huang, J.; Liu, Y.N. Inhibition of the androgen receptor induces a novel tumor promoter, zbtb46, for prostate cancer metastasis. Oncogene 2017, 36, 6213–6224. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Ramachandran, S.; Ali Seyed, M.; Scharer, C.D.; Laycock, N.; Dalton, W.B.; Williams, H.; Karanam, S.; Datta, M.W.; Jaye, D.L.; et al. Sex-determining region y box 4 is a transforming oncogene in human prostate cancer cells. Cancer Res. 2006, 66, 4011–4019. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, P.; Carrillo-de Santa Pau, E.; Cox, T.; Sainz, B., Jr.; Dusetti, N.; Greenhalf, W.; Rinaldi, L.; Costello, E.; Ghaneh, P.; Malats, N.; et al. Gata6 regulates emt and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer. Gut 2017, 66, 1665–1676. [Google Scholar] [CrossRef] [PubMed]
- Juang, Y.L.; Jeng, Y.M.; Chen, C.L.; Lien, H.C. Prrx2 as a novel tgf-beta-induced factor enhances invasion and migration in mammary epithelial cell and correlates with poor prognosis in breast cancer. Mol. Carcinog. 2016, 55, 2247–2259. [Google Scholar] [CrossRef] [PubMed]
- Bassey-Archibong, B.I.; Kwiecien, J.M.; Milosavljevic, S.B.; Hallett, R.M.; Rayner, L.G.; Erb, M.J.; Crawford-Brown, C.J.; Stephenson, K.B.; Bedard, P.A.; Hassell, J.A.; et al. Kaiso depletion attenuates transforming growth factor-beta signaling and metastatic activity of triple-negative breast cancer cells. Oncogenesis 2016, 5, e208. [Google Scholar] [CrossRef] [PubMed]
- Pozner, A.; Terooatea, T.W.; Buck-Koehntop, B.A. Cell-specific kaiso (zbtb33) regulation of cell cycle through cyclin d1 and cyclin e1. J. Biol. Chem. 2016, 291, 24538–24550. [Google Scholar] [CrossRef] [PubMed]
- Iljin, K.; Wolf, M.; Edgren, H.; Gupta, S.; Kilpinen, S.; Skotheim, R.I.; Peltola, M.; Smit, F.; Verhaegh, G.; Schalken, J.; et al. Tmprss2 fusions with oncogenic ets factors in prostate cancer involve unbalanced genomic rearrangements and are associated with hdac1 and epigenetic reprogramming. Cancer Res. 2006, 66, 10242–10246. [Google Scholar] [CrossRef] [PubMed]
- Bouafia, A.; Corre, S.; Gilot, D.; Mouchet, N.; Prince, S.; Galibert, M.D. P53 requires the stress sensor usf1 to direct appropriate cell fate decision. PLoS Genet. 2014, 10, e1004309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diepenbruck, M.; Waldmeier, L.; Ivanek, R.; Berninger, P.; Arnold, P.; van Nimwegen, E.; Christofori, G. Tead2 expression levels control the subcellular distribution of yap and taz, zyxin expression and epithelial-mesenchymal transition. J. Cell Sci. 2014, 127, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Hsieh, C.L.; Omwancha, J.; Zheng, Z.; Chen, S.Y.; Baert, J.L.; Shemshedini, L. Etv1 is a novel androgen receptor-regulated gene that mediates prostate cancer cell invasion. Mol. Endocrinol. 2007, 21, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network. Prostate Cancer (Version 2.2017). Available online: https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf (accessed on 1 September 2017).
- Tomlins, S.A.; Mehra, R.; Rhodes, D.R.; Cao, X.; Wang, L.; Dhanasekaran, S.M.; Kalyana-Sundaram, S.; Wei, J.T.; Rubin, M.A.; Pienta, K.J.; et al. Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 2007, 39, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Yeow, W.S.; Ertel, A.; Coleman, I.; Clegg, N.; Thangavel, C.; Morrissey, C.; Zhang, X.; Comstock, C.E.; Witkiewicz, A.K.; et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Investig. 2010, 120, 4478–4492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Dawood, S.; Holmes, M.D.; Collins, L.C.; Schnitt, S.J.; Cole, K.; Marotti, J.D.; Hankinson, S.E.; Colditz, G.A.; Tamimi, R.M. Androgen receptor expression and breast cancer survival in postmenopausal women. Clin. Cancer Res. 2011, 17, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. Ar-v7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Zong, Y.; Memarzadeh, S.; Xin, L.; Huang, J.; Witte, O.N. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 2610–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soufi, A.; Garcia, M.F.; Jaroszewicz, A.; Osman, N.; Pellegrini, M.; Zaret, K.S. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 2015, 161, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Obinata, D.; Takayama, K.; Takahashi, S.; Inoue, S. Crosstalk of the androgen receptor with transcriptional collaborators: Potential therapeutic targets for castration-resistant prostate cancer. Cancers (Basel) 2017, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Almendro, V.; Marusyk, A.; Polyak, K. Cellular heterogeneity and molecular evolution in cancer. Annu. Rev. Pathol. 2013, 8, 277–302. [Google Scholar] [CrossRef] [PubMed]
- Deplancke, B.; Alpern, D.; Gardeux, V. The genetics of transcription factor DNA binding variation. Cell 2016, 166, 538–554. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, C.; Lenkiewicz, E.; Evers, L.; Holley, T.; Robeson, A.; Kiefer, J.; Demeure, M.J.; Hollingsworth, M.A.; Shen, M.; Prunkard, D.; et al. Advancing a clinically relevant perspective of the clonal nature of cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 12054–12059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Cato, A.C.; Hittmair, A.; Radmayr, C.; Eberle, J.; Bartsch, G.; Klocker, H. Mutant androgen receptor detected in an advanced-stage prostatic carcinoma is activated by adrenal androgens and progesterone. Mol. Endocrinol. 1993, 7, 1541–1550. [Google Scholar] [PubMed]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Alvarez, M.J.; Talos, F.; Dhruv, H.; Rieckhof, G.E.; Iyer, A.; Diefes, K.L.; Aldape, K.; Berens, M.; Shen, M.M.; et al. Identification of causal genetic drivers of human disease through systems-level analysis of regulatory networks. Cell 2014, 159, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.A.; Gutman, D.A.; Chisolm, C.; Appin, C.; Kong, J.; Rong, Y.; Kurc, T.; Van Meir, E.G.; Saltz, J.H.; Moreno, C.S.; et al. The tumor microenvironment strongly impacts master transcriptional regulators and gene expression class of glioblastoma. Am. J. Pathol. 2012, 180, 2108–2119. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shan, F.; Xiong, G.; Chen, X.; Guan, X.; Wang, J.M.; Wang, W.L.; Xu, X.; Bai, Y. Egf-induced c/ebpbeta participates in emt by decreasing the expression of mir-203 in esophageal squamous cell carcinoma cells. J. Cell Sci. 2014, 127, 3735–3744. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Wu, W.J.; Wang, W.J.; Huang, H.Y.; Li, W.M.; Yeh, B.W.; Wu, T.F.; Shiue, Y.L.; Sheu, J.J.; Wang, J.M.; et al. Cebpd amplification and overexpression in urothelial carcinoma: A driver of tumor metastasis indicating adverse prognosis. Oncotarget 2015, 6, 31069–31084. [Google Scholar] [CrossRef] [PubMed]
- Huan, H.; Wen, X.; Chen, X.; Wu, L.; Liu, W.; Habib, N.A.; Bie, P.; Xia, F. C/ebpalpha short-activating rna suppresses metastasis of hepatocellular carcinoma through inhibiting egfr/beta-catenin signaling mediated emt. PLoS ONE 2016, 11, e0153117. [Google Scholar] [CrossRef] [PubMed]
- Barakat, D.J.; Zhang, J.; Barberi, T.; Denmeade, S.R.; Friedman, A.D.; Paz-Priel, I. Ccaat/enhancer binding protein beta controls androgen-deprivation-induced senescence in prostate cancer cells. Oncogene 2015, 34, 5912–5922. [Google Scholar] [CrossRef] [PubMed]
- Bilir, B.; Osunkoya, A.O.; Wiles, W.G.T.; Sannigrahi, S.; Lefebvre, V.; Metzger, D.; Spyropoulos, D.D.; Martin, W.D.; Moreno, C.S. Sox4 is essential for prostate tumorigenesis initiated by pten ablation. Cancer Res. 2016, 76, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Roe, J.S.; Hwang, C.I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer reprogramming promotes pancreatic cancer metastasis. Cell 2017, 170, 875–888 e820. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Kobayashi, N.; Ruscetti, M.; Zhi, A.; Tran, L.M.; Huang, J.; Gleave, M.; Wu, H. Pten loss and ras/mapk activation cooperate to promote emt and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012, 72, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen receptor pathway-independent prostate cancer is sustained through fgf signaling. Cancer Cell 2017, 32, 474–489 e476. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Star: Ultrafast universal rna-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor rna-seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at ucsc. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. Transfac and its module transcompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group Name | Transcription Factors | Kegg Pathway Annotation and/or Reference |
|---|---|---|
| oTFCG1 | NR2F2-SMAD9-PAX2-TAL1-ELK4-ELK3-KLF12-ETV6-SMAD7-MAFA-TCF7L2-ETV4-SREBF2-GATA3-MYBL2-MYB-YBX1-ERG-FLI1-RFX1-SREBF1-HSF4-ZEB1-GABPA-ELF1-ELF5 | Transcriptional misregulation in cancer; TGF-β signaling |
| oTFCG2 | MYF5-TCF4-MYOG-TCF12-NR2C2-NF1A-SMAD5-PAX4-ELK1-SPIB-MYOD1-TCF3-GATA1-NFIX-KLF4-PURA-KLF6-GEN1-E2F3-TFDP1-GTF2I-HIC1-WT1-E2F4 | Pathways regulating pluripotency of stem cells |
| oTFCG3 | JUN-SOX10-SOX18-JUND-JUNB-SMAD3-FOS-RXRA-BRCA1-SMAD2-NR3C1-ETS2-GATA2-YY1-TCF7L1-FOSL2-FOSB-FOSL1 | MAPK signaling; osteoclast differentiation; IL-17 signaling pathway; Wnt signaling; TGF-β signaling |
| oTFCG4 | NFKB1-MTF1-ZIC3-TFCP2-ZBTB7A-MZF1-BCL6B-SP4-SP3-ZIC1-SP2-TP73-TP63 | MicroRNAs in cancer |
| oTFCG5 | HES1-IKZF1-TFAP2C-PAX8-RUNX3-ETV7-THAP1 | Pathways in Cancer |
| oTFCG6 | HSF1-HNF1A-SOX17-FOXM1-IRF4-NKX2-5 | Wnt Signaling |
| oTFCG7 | NR1H3-RARB-NR1I2-RARG-NR1H2-NR1I3 | Insulin resistance; Small cell lung cancer; Non-small cell lung cancer |
| oTFCG8 | GATA5-SRY-SOX8-POU2F1-CUX1 | |
| oTFCG9 | GATA6-FOXA1-POU3F3-FOXD3 | EMT in pancreatic cancer [26] |
| oTFCG10 | EGR1-KLF13-EGR2-HIC2 | GnRH signaling; Human T-cell leukemia virus 1 infection |
| oTFCG11 | ERF-ETV5-ETV3-ELF4 | Transcriptional misregulation in cancer; Prostate Cancer |
| oTFCG12 | EP300-SPI1-SMAD4-E2F1 | Pathways in Cancer; Human T-cell leukemia virus 1 infection; TGF-beta signaling; Prostate Cancer; Wnt signaling; Cell cycle |
| oTFCG13 | SOX4-FOXA2-GATA4 | Prostate cancer oncogene [25] |
| oTFCG14 | E2F7-E2F5-E2F2 | Gastric Cancer; Prostate Cancer; Bladder Cancer |
| oTFCG15 | HOXB2-PRRX2-PDX1 | TGF-beta signaling induced invasion in breast cancer [27] |
| oTFCG16 | ARNT-TFAP2A-TFAP2B | Cushing Syndrome; HIF-1 signaling; Renal Cell Carcinoma |
| oTFCG17 | NFYC-NFYA-NFYB | Antigen processing and presentation |
| oTFCG18 | MAFK-CEBPG-CEBPE | Transcriptional misregulation in cancer; Acute myeloid leukemia |
| oTFCG19 | MAZ-ARHGEF7-CD40 | Regulation of actin cytoskeleton |
| oTFCG20 | MAX-EGR3-ZIC2 | C-type lectin receptor signaling; Small cell lung cancer; Transcriptional misregulation in cancer; MAPK signaling |
| oTFCG21 | GLI3-GLI2 | Hedgehog signaling; Basal Cell Carcinoma; Hippo signaling |
| oTFCG22 | ZBTB33-PLAGL1 | Metastasis and TGF-β signaling in triple negative breast cancer [28]; Cell cycle [29] |
| oTFCG23 | HDAC1-UBP1 | Epigenetic reprogramming in cancer (HDAC) [30] |
| oTFCG24 | FOXL1-TBP | Huntington disease; Basal transcription factors |
| oTFCG25 | KLF2-RREB1 | MAPK Signaling; FOXO signaling |
| oTFCG26 | USF2-USF1 | Inhibition of cell cycle [31] |
| oTFCG27 | CEBPB-CEBPD | TNF Signaling pathway; Transcriptional misregulation in cancer |
| oTFCG28 | CHURC1-TEAD2 | EMT in breast cancer [32] |
| oTFCG29 | ETV1-HIF1A | HIF1-signaling; Angiogenesis; Prostate cancer invasion [33] |
| oTFCG30 | ATM-GTF2IRD1 | FoxO signaling; Cell cycle; NF-kappa β signaling |
| oTFCG31 | MYC-RXRB | Gastric Cancer; Thyroid hormone signaling; Small cell lung cancer; PPAR signaling |
| oTFCG32 | SP1-TP53 | Endocrine resistance; Huntington disease; Breast cancer; Transcriptional misregulation in cancer; Endocrine resistance |
| oTFCG33 | NR4A2-TFAP4 | MAPK Signaling; osteoclast differentiation; IL-17 signaling; Wnt signaling; TGF-β signaling |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, N.V.; Pellegrini, K.L.; Ouellet, V.; Giuste, F.O.; Ramalingam, S.; Watanabe, K.; Adam-Granger, E.; Fossouo, L.; You, S.; Freeman, M.R.; et al. Identification of the Transcription Factor Relationships Associated with Androgen Deprivation Therapy Response and Metastatic Progression in Prostate Cancer. Cancers 2018, 10, 379. https://doi.org/10.3390/cancers10100379
Sharma NV, Pellegrini KL, Ouellet V, Giuste FO, Ramalingam S, Watanabe K, Adam-Granger E, Fossouo L, You S, Freeman MR, et al. Identification of the Transcription Factor Relationships Associated with Androgen Deprivation Therapy Response and Metastatic Progression in Prostate Cancer. Cancers. 2018; 10(10):379. https://doi.org/10.3390/cancers10100379
Chicago/Turabian StyleSharma, Nitya V., Kathryn L. Pellegrini, Veronique Ouellet, Felipe O. Giuste, Selvi Ramalingam, Kenneth Watanabe, Eloise Adam-Granger, Lucresse Fossouo, Sungyong You, Michael R. Freeman, and et al. 2018. "Identification of the Transcription Factor Relationships Associated with Androgen Deprivation Therapy Response and Metastatic Progression in Prostate Cancer" Cancers 10, no. 10: 379. https://doi.org/10.3390/cancers10100379
APA StyleSharma, N. V., Pellegrini, K. L., Ouellet, V., Giuste, F. O., Ramalingam, S., Watanabe, K., Adam-Granger, E., Fossouo, L., You, S., Freeman, M. R., Vertino, P., Conneely, K., Osunkoya, A. O., Trudel, D., Mes-Masson, A.-M., Petros, J. A., Saad, F., & Moreno, C. S. (2018). Identification of the Transcription Factor Relationships Associated with Androgen Deprivation Therapy Response and Metastatic Progression in Prostate Cancer. Cancers, 10(10), 379. https://doi.org/10.3390/cancers10100379