SPARC Inhibits Metabolic Plasticity in Ovarian Cancer

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
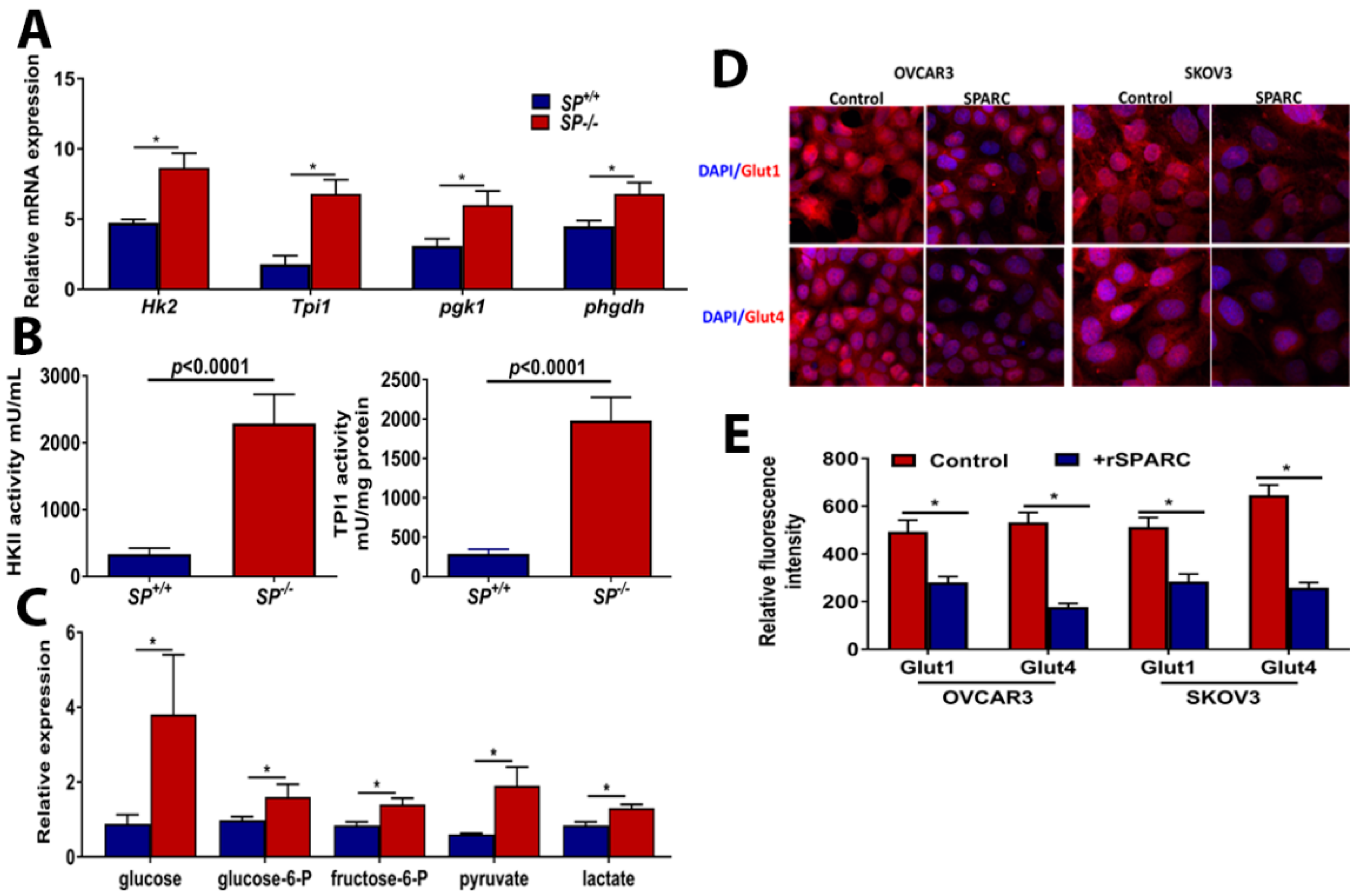
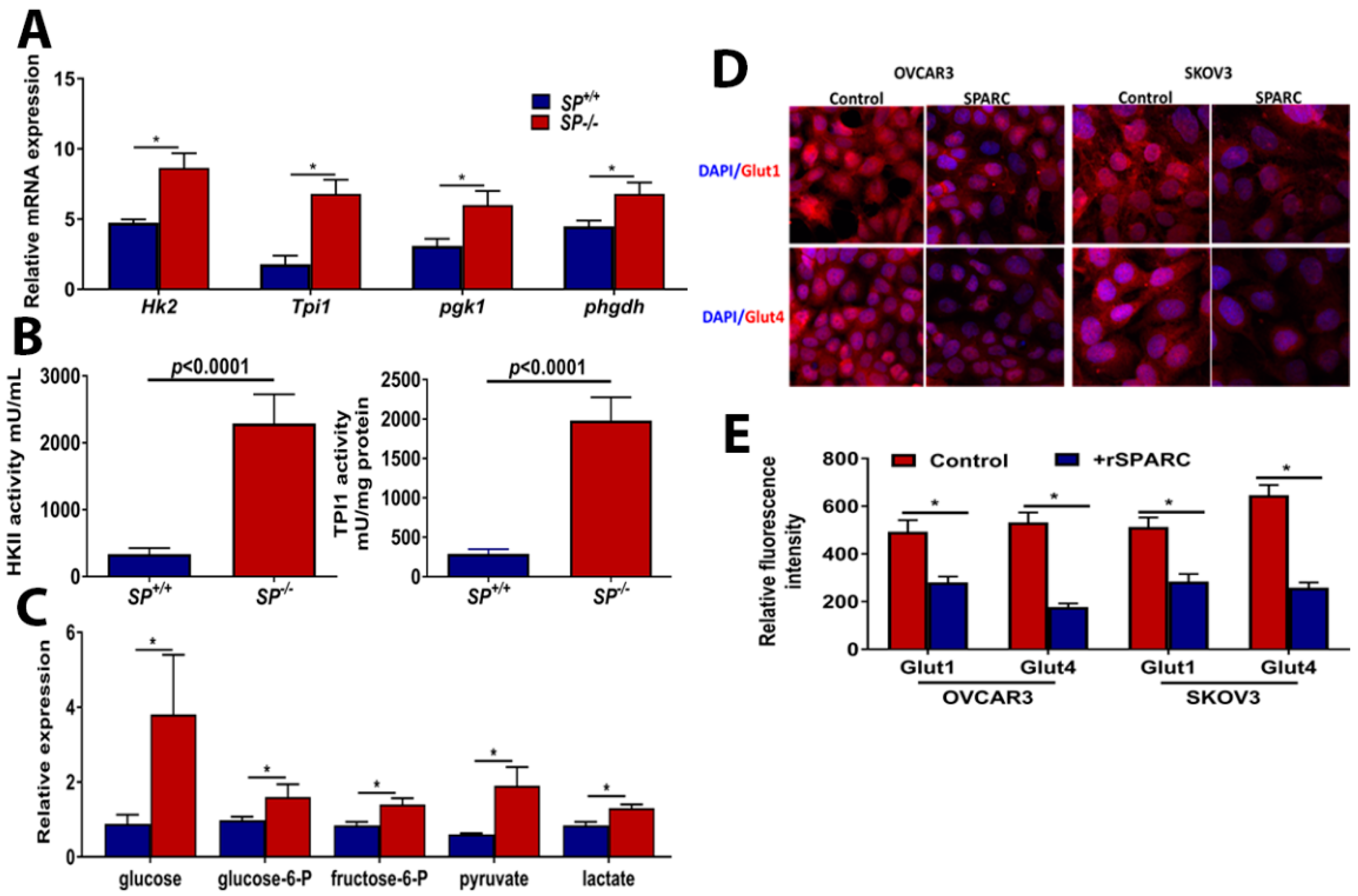
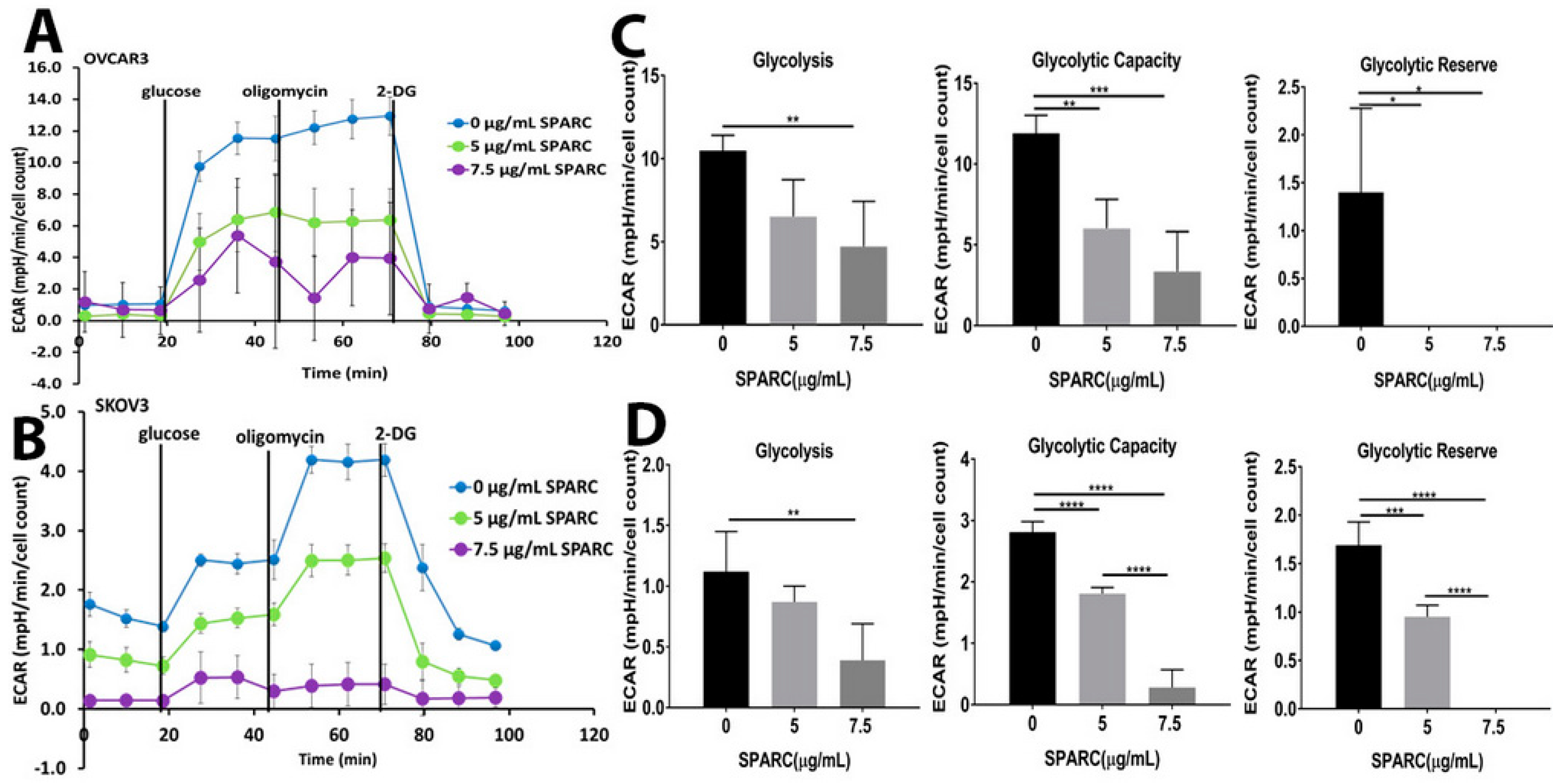
2.1. Loss of Stromal-SPARC Is Associated with Enhanced Glycolysis in Syngeneic ID8 Intraperitoneal Tumors
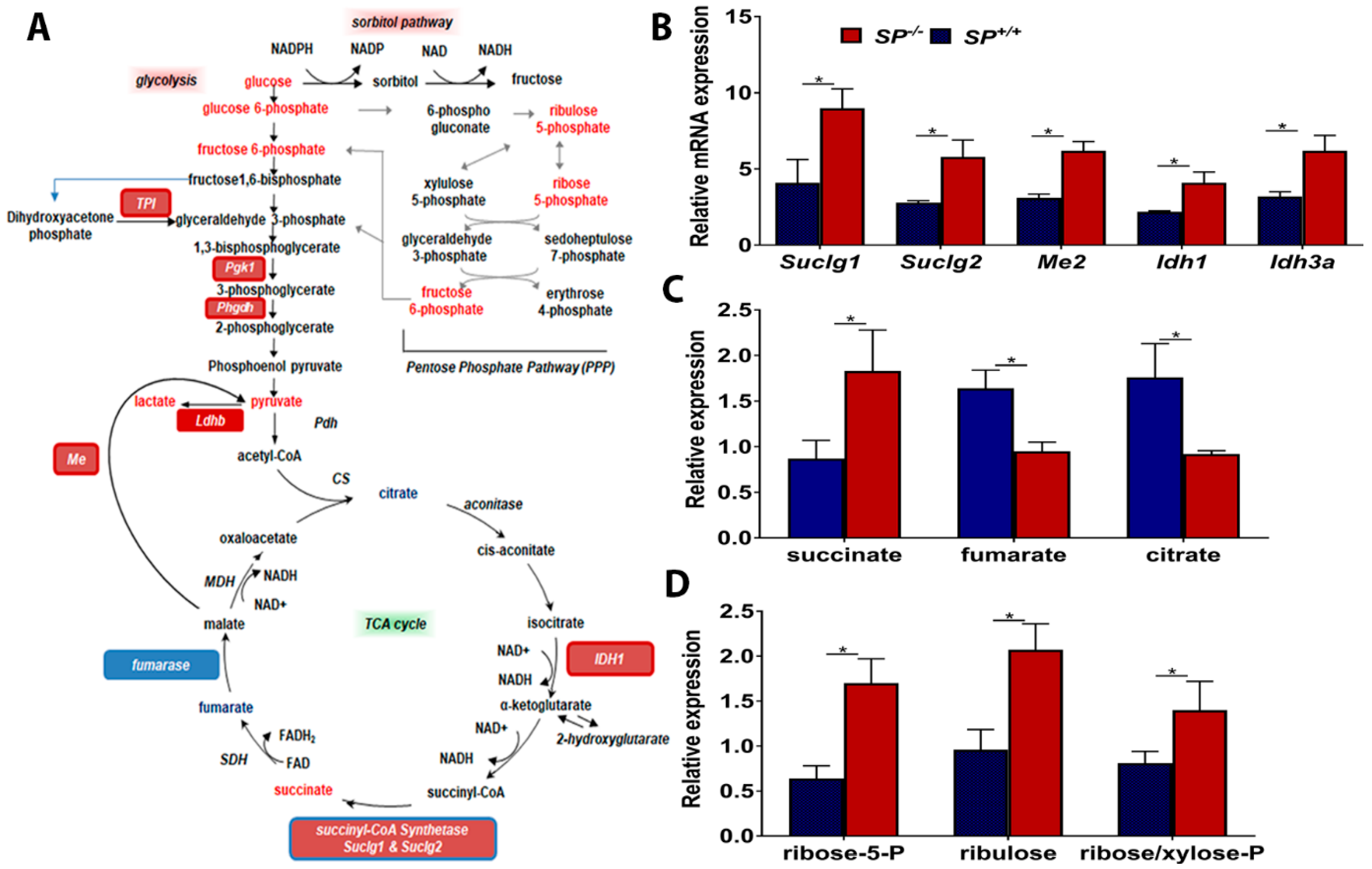
2.2. Effect of Loss of Stromal-SPARC on TCA Cycle and Pentose Phosphate Pathway (PPP)
2.3. Loss of Stromal-Sparc Increases OXPHOS with Overexpression of Mitochondrial ETC Enzymes
2.4. Effect of SPARC on Mitochondrial Functions
2.5. Perturbed Redox Homeostasis in Tumors Dissected from SP−/− Mice
2.6. Targeting Mitochondrial ETC by Phenformin Mitigates the Accelerated Growth of OvCa Syngeneic Tumors
3. Discussion
4. Material and Methods
4.1. Cell Culture and Reagents
4.2. In Vivo Syngeneic Model
4.3. Mouse Therapeutic Trial with Phenformin
4.4. Immunohistochemistry
4.5. Laser Captured Microdissection (LCM)
4.6. RNA Extraction, Amplification, Labeling and Analysis
4.7. Quantitative RT-PCR Analysis
4.8. Metabolomic Profiling Sample Preparation
4.9. Data Extraction, Compound Identification, Metabolite Quantification and Data Normalization
4.10. Intracellular ROS Formation in Tumor Tissues
4.11. Total ATP Measurement, Hexokinase 2 and TPI1 Enzyme Activity Assays
4.12. Transmission Electron Microscopy (TEM)
4.13. Glycolysis and Mitochondrial Stress Assays
4.14. Measurement of Mitochondrial Contents
4.15. Statistical Analysis
4.16. Bioinformatics Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xintaropoulou, C.; Ward, C.; Wise, A.; Queckborner, S.; Turnbull, A.; Michie, C.O.; Williams, A.R.W.; Rye, T.; Gourley, C.; Langdon, S.P. Expression of glycolytic enzymes in ovarian cancers and evaluation of the glycolytic pathway as a strategy for ovarian cancer treatment. BMC Cancer 2018, 18, 636. [Google Scholar] [CrossRef] [PubMed]
- Irie, H.; Banno, K.; Yanokura, M.; Iida, M.; Adachi, M.; Nakamura, K.; Umene, K.; Nogami, Y.; Masuda, K.; Kobayashi, Y.; et al. Metformin: A candidate for the treatment of gynecological tumors based on drug repositioning. Oncol. Lett. 2016, 11, 1287–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehuédé, C.; Dupuy, F.; Rabinovitch, R.; Jones, R.G.; Siegel, P.M. Metabolic Plasticity as a Determinant of Tumor Growth and Metastasis. Cancer Res. 2016, 76, 5201–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iurlaro, R.; León-Annicchiarico, C.L.; Muñoz-Pinedo, C. Chapter Three-Regulation of Cancer Metabolism by Oncogenes and Tumor Suppressors. In Methods in Enzymology; Galluzzi, L., Kroemer, G., Eds.; Academic Press: San Diego, CA, USA, 2014; Volume 542, pp. 59–80. [Google Scholar]
- Said, N.; Socha, M.J.; Olearczyk, J.J.; Elmarakby, A.A.; Imig, J.D.; Motamed, K. Normalization of the ovarian cancer microenvironment by SPARC. Mol. Cancer Res. 2007, 5, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Said, N.; Motamed, K. Absence of host-secreted protein acidic and rich in cysteine (SPARC) augments peritoneal ovarian carcinomatosis. Am. J. Pathol. 2005, 167, 1739–1752. [Google Scholar] [CrossRef]
- Said, N.; Najwer, I.; Motamed, K. Secreted protein acidic and rich in cysteine (SPARC) inhibits integrin-mediated adhesion and growth factor-dependent survival signaling in ovarian cancer. Am. J. Pathol. 2007, 170, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Said, N.A.; Elmarakby, A.A.; Imig, J.D.; Fulton, D.J.; Motamed, K. SPARC ameliorates ovarian cancer-associated inflammation. Neoplasia 2008, 10, 1092–1104. [Google Scholar] [CrossRef] [PubMed]
- Cavill, R.; Kamburov, A.; Ellis, J.K.; Athersuch, T.J.; Blagrove, M.S.C.; Herwig, R.; Ebbels, T.M.D.; Keun, H.C. Consensus-Phenotype Integration of Transcriptomic and Metabolomic Data Implies a Role for Metabolism in the Chemosensitivity of Tumour Cells. PLoS Comput. Biol. 2011, 7, e1001113. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.N.; David, R. Reactions of citric acid cycle. In Lehninger Principles of Biochemistry; W.H. Freeman and Company: New York, NY, USA, 2005; pp. 601–630. [Google Scholar]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liemburg-Apers, D.C.; Willems, P.H.G.M.; Koopman, W.J.H.; Grefte, S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, N.; Frierson, H.F.; Sanchez-Carbayo, M.; Brekken, R.A.; Theodorescu, D. Loss of SPARC in bladder cancer enhances carcinogenesis and progression. J. Clin. Investig. 2013, 123, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione Synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. Overcoming Drug Development Bottlenecks With Repurposing: Repurposing biguanides to target energy metabolism for cancer treatment. Nat. Med. 2014, 20, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Investig. 2007, 117, 1422–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, D.; Abt, E.; Gerken, L.; Vasquez, D.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.; Czernin, J.; Mischel, P.; et al. LKB1 Inactivation Dictates Therapeutic Response of Non-Small Cell Lung Cancer to the Metabolism Drug Phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, A.L.; Sun, W.; Kilgore, J.; Guo, H.; Fang, Z.; Yin, Y.; Jones, H.M.; Gilliam, T.P.; Zhou, C.; Bae-Jump, V.L. Phenformin has anti-tumorigenic effects in human ovarian cancer cells and in an orthotopic mouse model of serous ovarian cancer. Oncotarget 2017, 8, 100113–100127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, M.; Gravel, S.P.; Chenard, V.; Sikstrom, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Alvero, A.B.; Montagna, M.K.; Sumi, N.J.; Joo, W.D.; Graham, E.; Mor, G. Multiple blocks in the engagement of oxidative phosphorylation in putative ovarian cancer stem cells: Implication for maintenance therapy with glycolysis inhibitors. Oncotarget 2014, 5, 8703–8715. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Feron, O. Cancer cell metabolism and mitochondria: Nutrient plasticity for TCA cycle fueling. Biochim. Biophys. Acta 2017, 1868, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herst, P.M.; Berridge, M.V. Cell surface oxygen consumption: A major contributor to cellular oxygen consumption in glycolytic cancer cell lines. Biochim. Biophys. Acta 2007, 1767, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Zhu, W.; Qin, J.; Chen, M.; Gong, L.; Li, L.; Liu, X.; Tao, Y.; Yin, H.; Zhou, H.; et al. Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology 2017, 65, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullarky, E.; Mattaini, K.R.; Vander Heiden, M.G.; Cantley, L.C.; Locasale, J.W. PHGDH amplification and altered glucose metabolism in human melanoma. Pigment Cell Melanoma Res. 2011, 24, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Shashni, B.; Sakharkar, K.R.; Nagasaki, Y.; Sakharkar, M.K. Glycolytic enzymes PGK1 and PKM2 as novel transcriptional targets of PPARgamma in breast cancer pathophysiology. J. Drug Target. 2013, 21, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Park, Y.; Andrabi, S.A.; Shelton, L.M.; Gilkes, D.M.; Semenza, G.L. PHGDH Expression is Required for Mitochondrial Redox Homeostasis, Breast Cancer Stem Cell Maintenance and Lung Metastasis. Cancer Res. 2016, 76, 4430–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [PubMed]
- Cormio, A.; Cormio, G.; Musicco, C.; Sardanelli, A.M.; Gasparre, G.; Gadaleta, M.N. Mitochondrial changes in endometrial carcinoma: Possible role in tumor diagnosis and prognosis (review). Oncol. Rep. 2015, 33, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.J.; Li, S. The prognostic value of metformin for cancer patients with concurrent diabetes: A systematic review and meta-analysis. Diabetes Obes. Metab. 2014, 16, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Said, N.A.; Najwer, I.; Socha, M.J.; Fulton, D.J.; Mok, S.C.; Motamed, K. SPARC inhibits LPA-mediated mesothelial-ovarian cancer cell crosstalk. Neoplasia 2007, 9, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.R.; Chang, H.C.; Chuang, L.Y.; Hung, W.C. The nonsteroidal anti-inflammatory drug NS398 reactivates SPARC expression via promoter demethylation to attenuate invasiveness of lung cancer cells. Exp. Biol. Med. 2008, 233, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef] [PubMed]
- Ihaka, R.; Gentleman, R. R: A language for data analysis and graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnaiyan, P.; Kensicki, E.; Bloom, G.; Prabhu, A.; Sarcar, B.; Kahali, S.; Eschrich, S.; Qu, X.; Forsyth, P.; Gillies, R. The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism. Cancer Res. 2012, 72, 5878–5888. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.K.; Vareed, S.K.; Basu, S.; Putluri, V.; Putluri, N.; Panzitt, K.; Brennan, C.A.; Chinnaiyan, A.M.; Vergara, I.A.; Erho, N.; et al. Metabolomic profiling identifies biochemical pathways associated with castration-resistant prostate cancer. J. Proteome Res. 2014, 13, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.P.; Rajendiran, T.M.; Ateeq, B.; Asangani, I.A.; Athanikar, J.N.; Yocum, A.K.; Mehra, R.; Siddiqui, J.; Palapattu, G.; Wei, J.T.; et al. The role of sarcosine metabolism in prostate cancer progression. Neoplasia 2013, 15, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Poisson, L.M.; Sreekumar, A.; Chinnaiyan, A.M.; Ghosh, D. Pathway-directed weighted testing procedures for the integrative analysis of gene expression and metabolomic data. Genomics 2012, 99, 265–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Somashekar, B.S.; Ponnusamy, M.; Gursky, A.; Dailey, S.; Kunju, P.; Lee, C.T.; Chinnaiyan, A.M.; Rajendiran, T.M.; Ramamoorthy, A. HR-MAS NMR tissue metabolomic signatures cross-validated by mass spectrometry distinguish bladder cancer from benign disease. J. Proteome Res. 2013, 12, 3519–3528. [Google Scholar] [CrossRef] [PubMed]
- Cheville, N.F.; Stasko, J. Techniques in Electron Microscopy of Animal Tissue. Vet. Pathol. 2014, 51, 28–41. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naczki, C.; John, B.; Patel, C.; Lafferty, A.; Ghoneum, A.; Afify, H.; White, M.; Davis, A.; Jin, G.; Kridel, S.; et al. SPARC Inhibits Metabolic Plasticity in Ovarian Cancer. Cancers 2018, 10, 385. https://doi.org/10.3390/cancers10100385
Naczki C, John B, Patel C, Lafferty A, Ghoneum A, Afify H, White M, Davis A, Jin G, Kridel S, et al. SPARC Inhibits Metabolic Plasticity in Ovarian Cancer. Cancers. 2018; 10(10):385. https://doi.org/10.3390/cancers10100385
Chicago/Turabian StyleNaczki, Christine, Bincy John, Chirayu Patel, Ashlyn Lafferty, Alia Ghoneum, Hesham Afify, Michael White, Amanda Davis, Guangxu Jin, Steven Kridel, and et al. 2018. "SPARC Inhibits Metabolic Plasticity in Ovarian Cancer" Cancers 10, no. 10: 385. https://doi.org/10.3390/cancers10100385
APA StyleNaczki, C., John, B., Patel, C., Lafferty, A., Ghoneum, A., Afify, H., White, M., Davis, A., Jin, G., Kridel, S., & Said, N. (2018). SPARC Inhibits Metabolic Plasticity in Ovarian Cancer. Cancers, 10(10), 385. https://doi.org/10.3390/cancers10100385