Electrotransfer of Different Control Plasmids Elicits Different Antitumor Effectiveness in B16.F10 Melanoma

 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Results
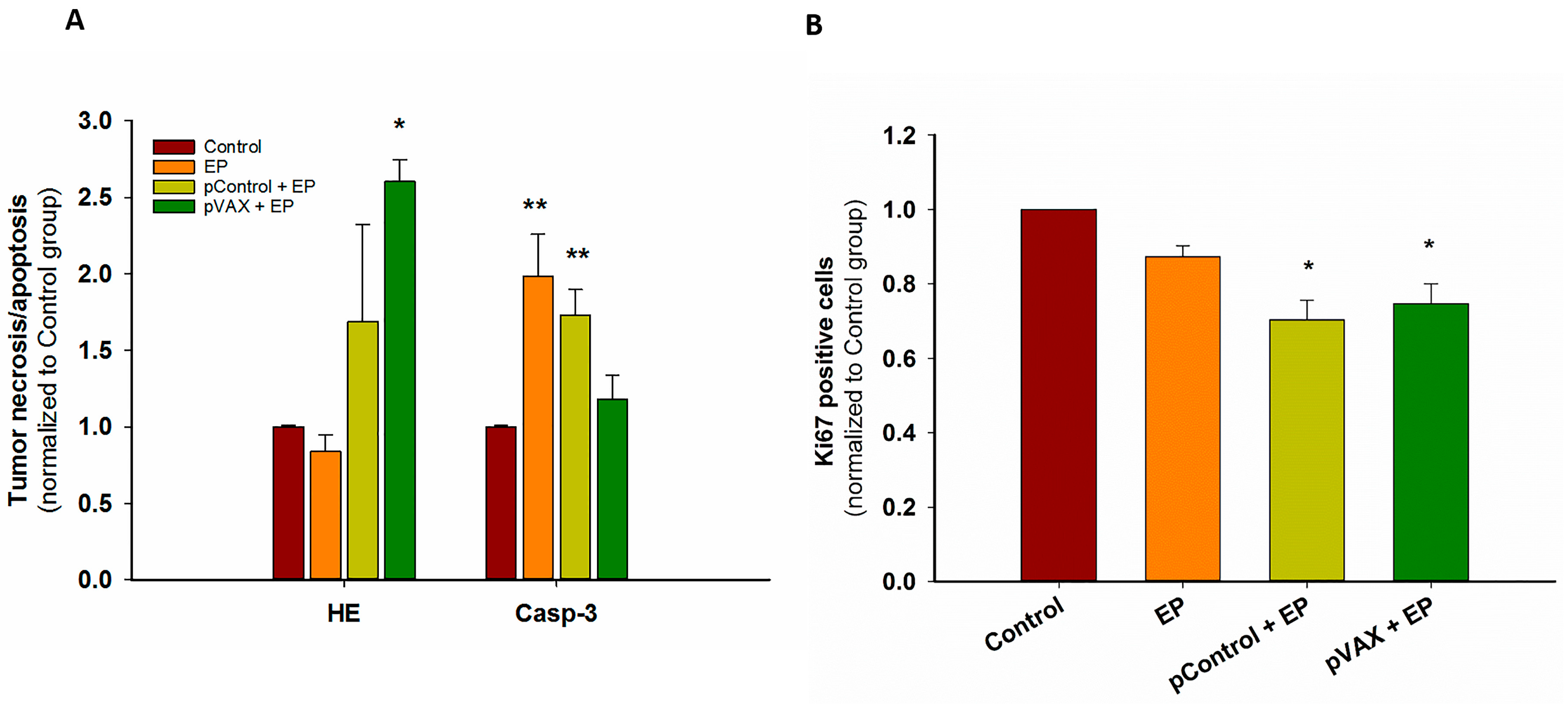
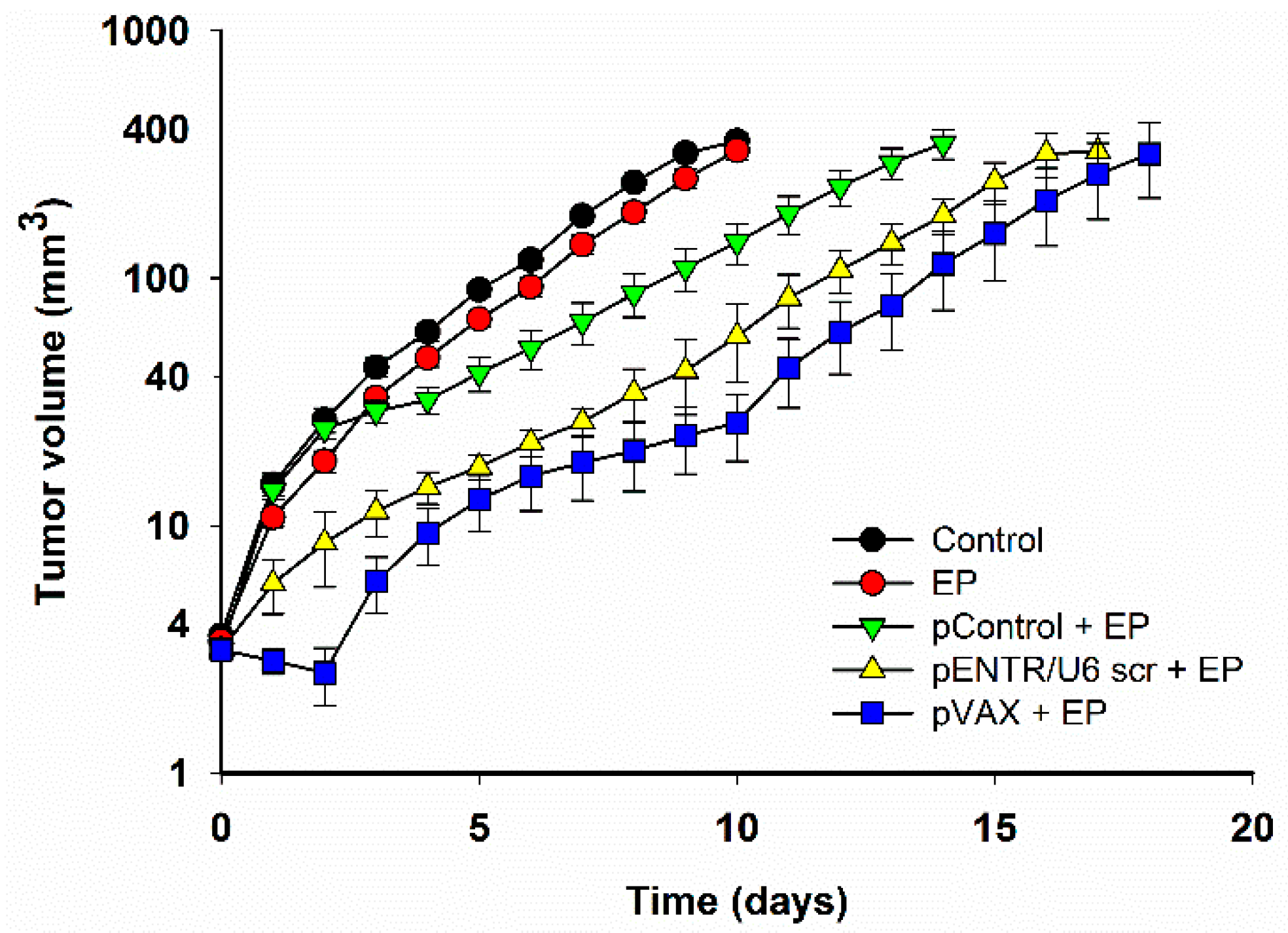
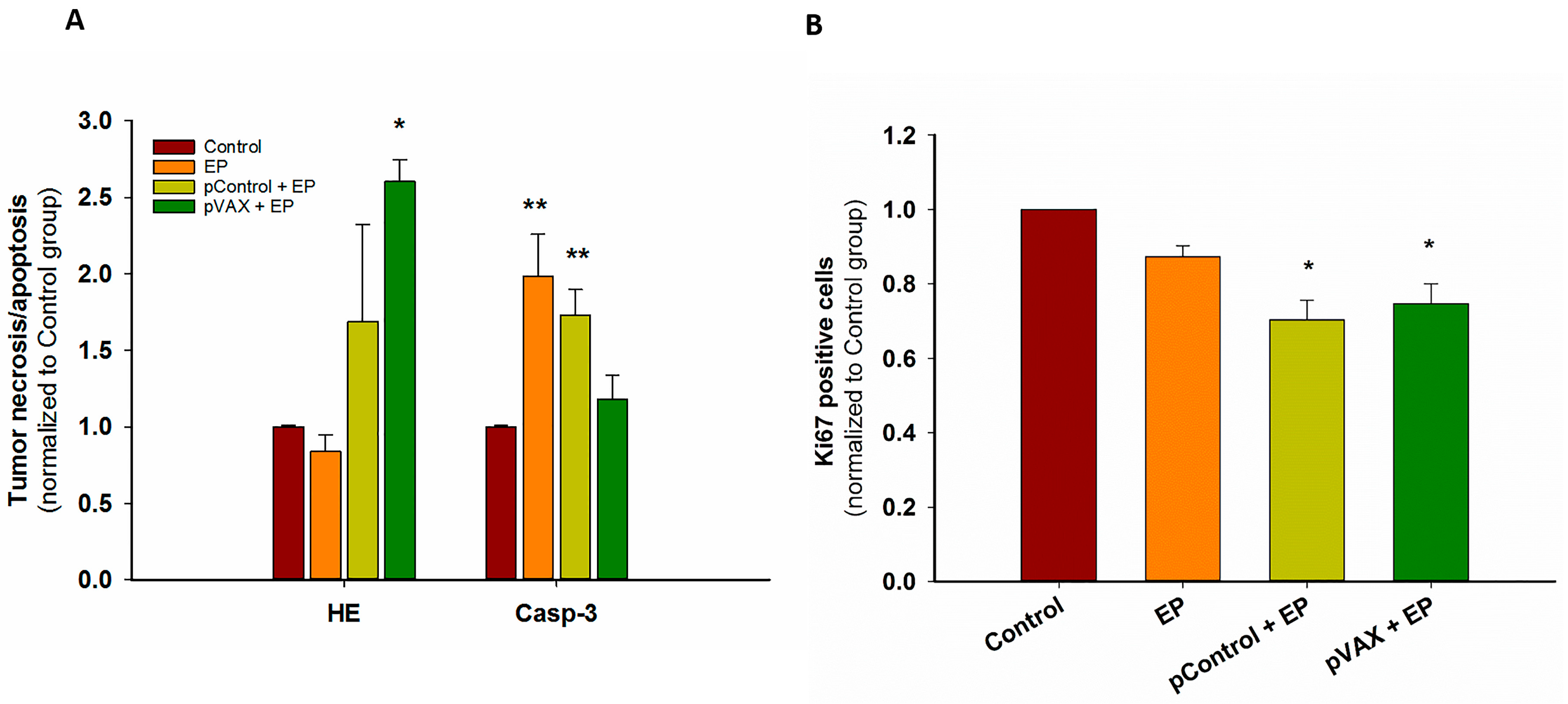
2.1. In Vivo Gene Electrotransfer of Control Plasmids Resulted in Significant Tumor Growth Delay and Complete Tumor Regression. These Effects Correlated with an Increased Expression of Cytokine mRNAs
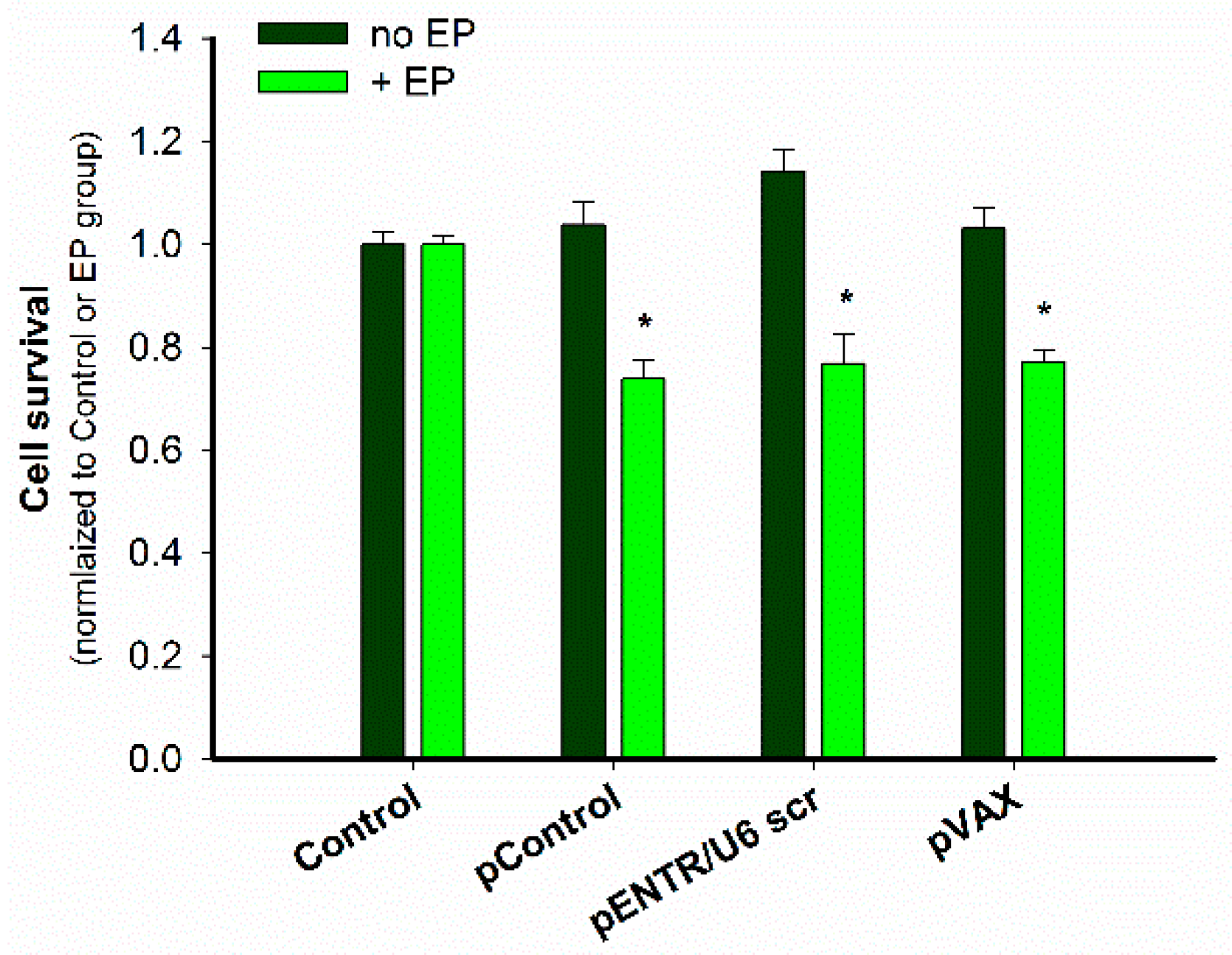
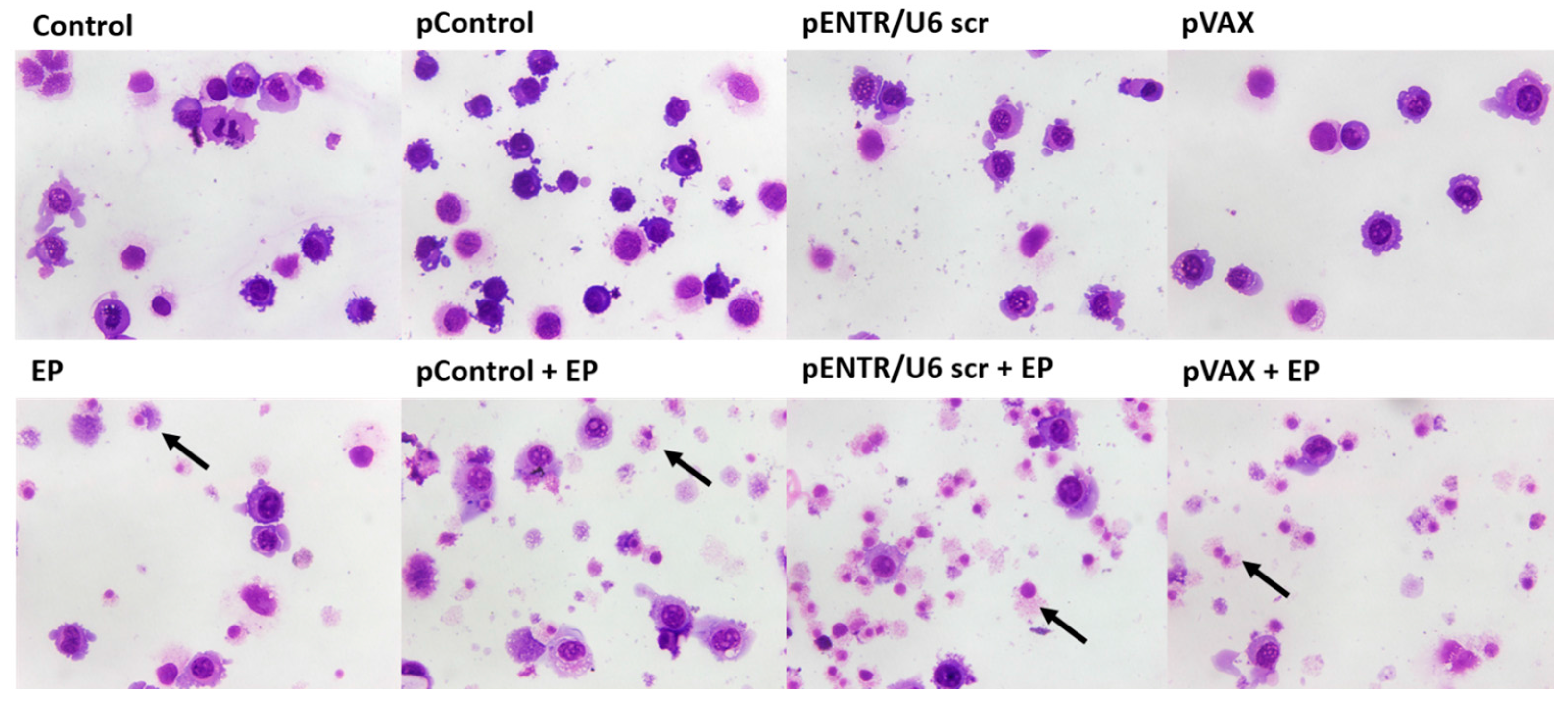
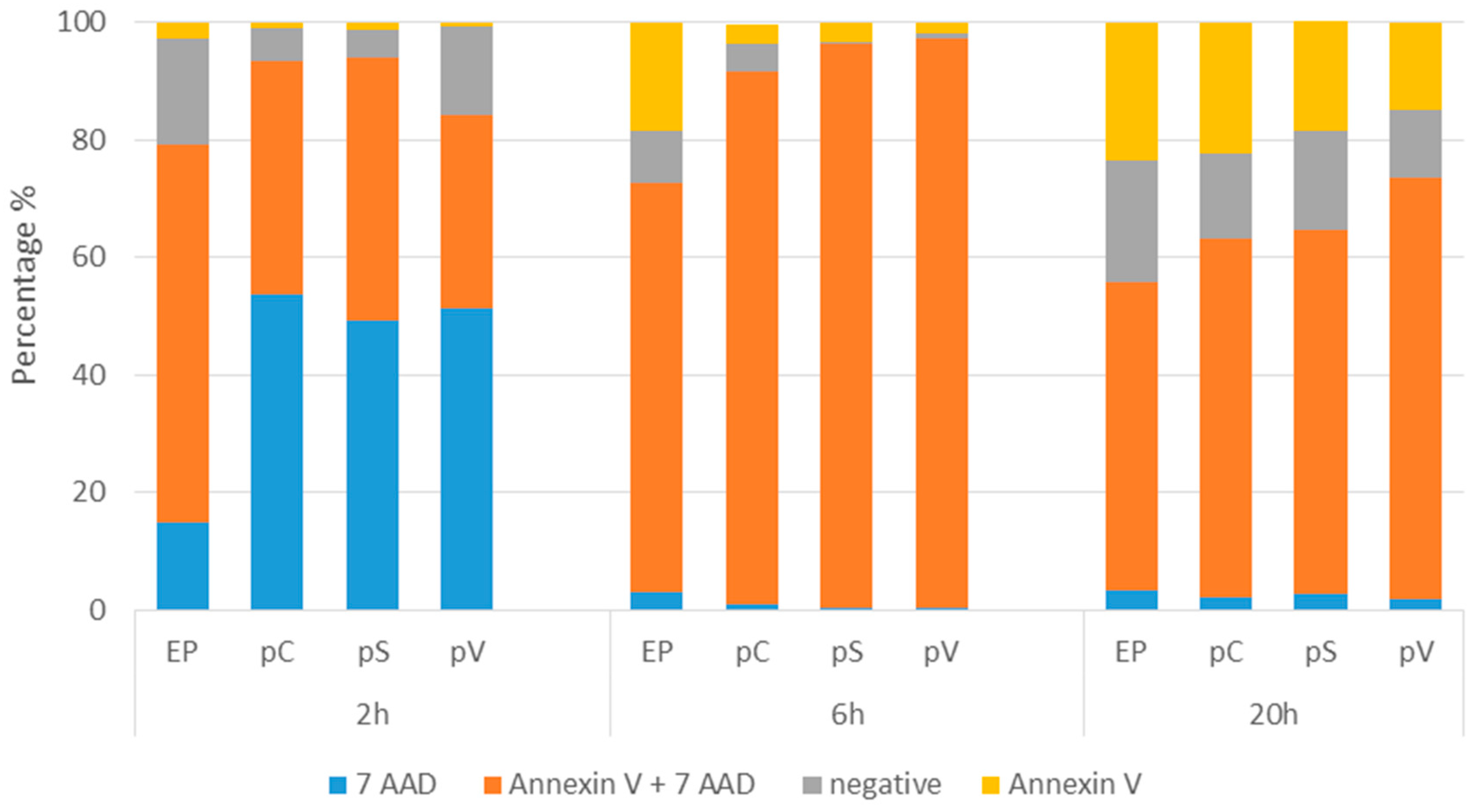
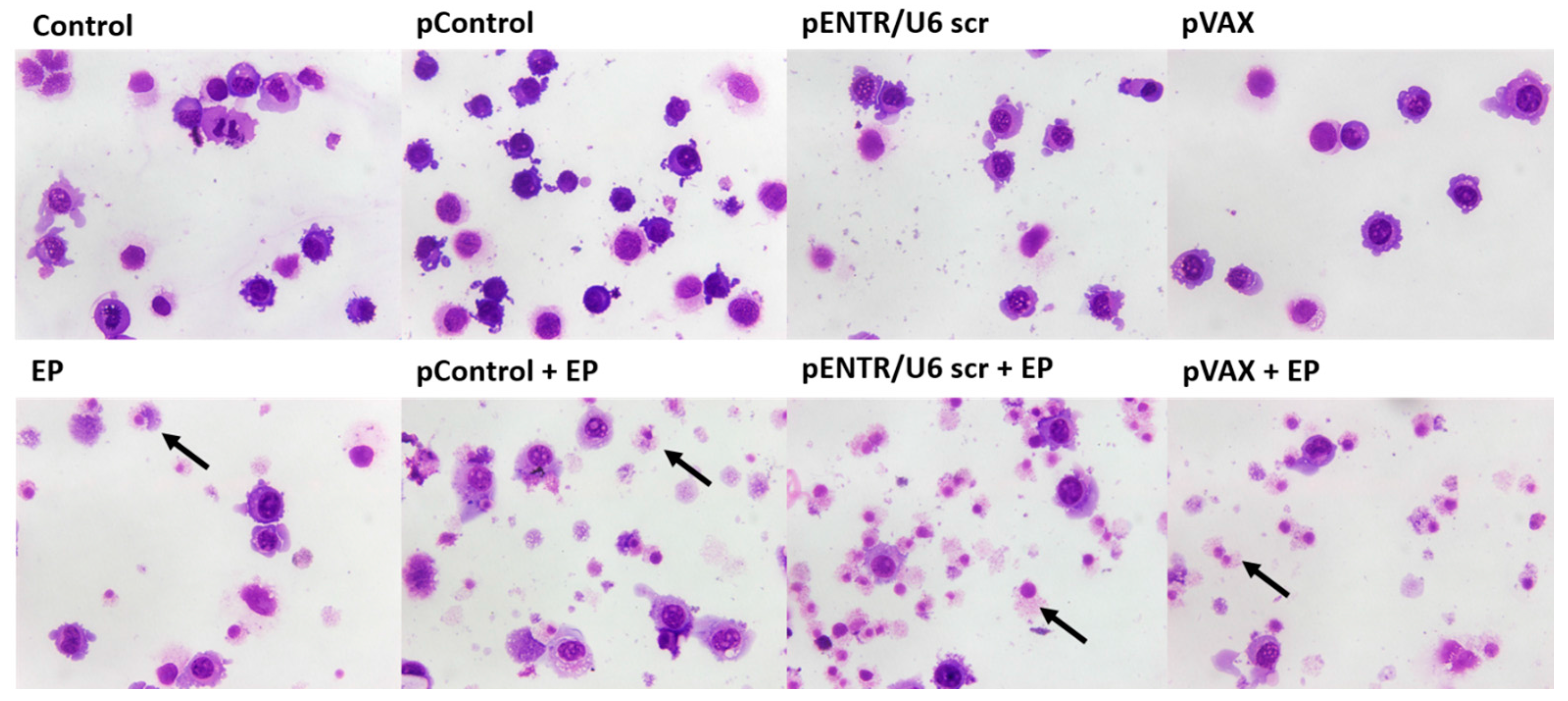
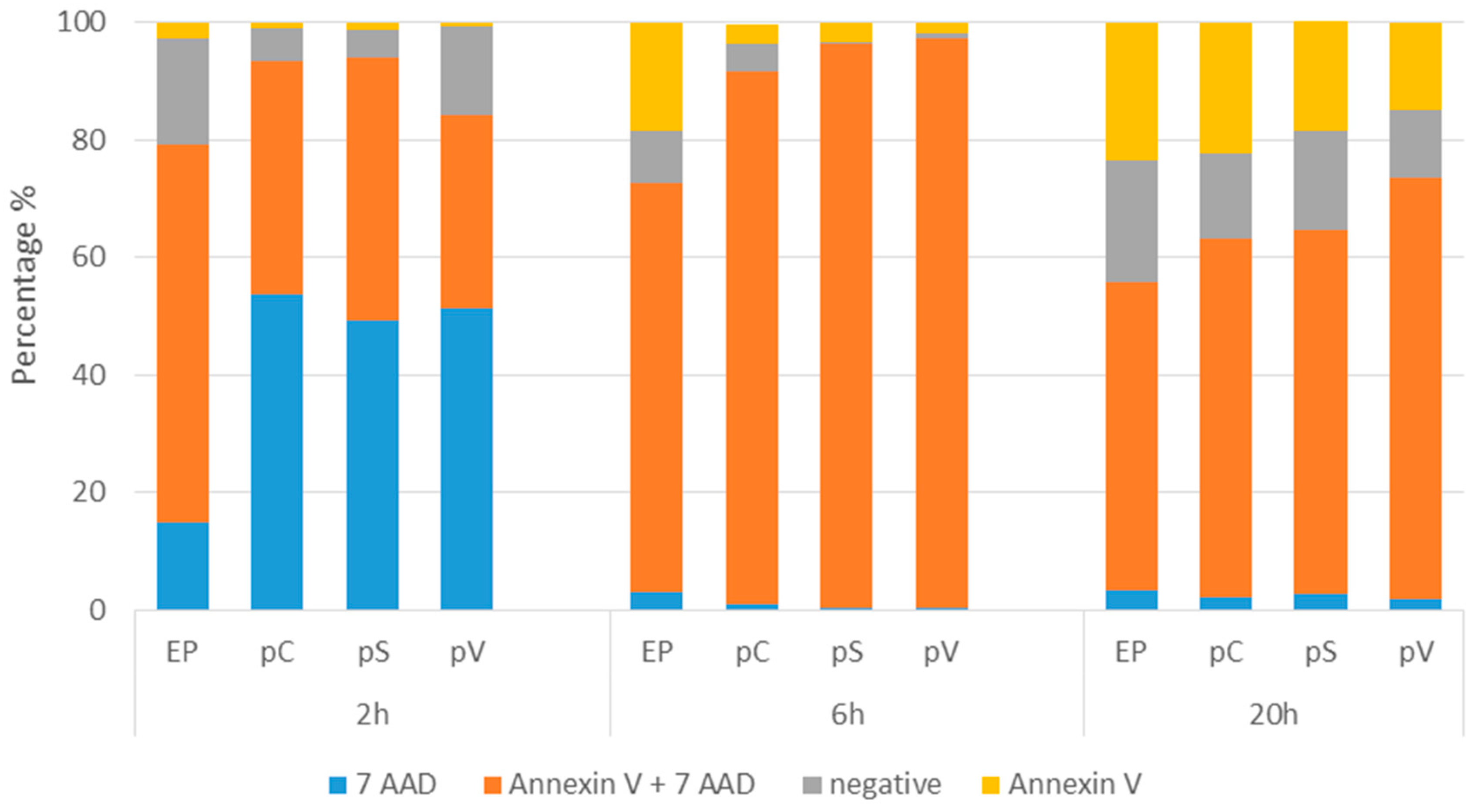
2.2. In Vitro Gene Electrotransfer of Control Plasmids Caused Necrotic Cell Death and Expression of DNA Sensor and Signaling Pathway mRNAs
3. Discussion
4. Materials and Methods
4.1. Plasmids
4.2. Cells
4.3. Animals and Tumors
4.4. GET in Vitro
4.5. GET in Vivo
4.6. Tumor Growth
4.7. Histology
4.8. Extraction of RNA from Cells or Tumors and Determination of DNA Sensors mRNAs by Quantitative Real-Time PCR (qPCR)
4.9. Cytotoxicity Assay
4.10. Giemsa Staining
4.11. Determination of Cell Death Mechanism
4.12. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mir, L.M.; Glass, L.F.; Sersa, G.; Teissié, J.; Domenge, C.; Miklavcic, D.; Jaroszeski, M.J.; Orlowski, S.; Reintgen, D.S.; Rudolf, Z.; et al. Effective treatment of cutaneous and subcutaneous malignant tumours by electrochemotherapy. Br. J. Cancer 1998, 77, 2336–2342. [Google Scholar] [CrossRef] [PubMed]
- Dolinsek, T.; Sersa, G.; Prosen, L.; Bosnjak, M.; Stimac, M.; Razborsek, U.; Cemazar, M. Electrotransfer of plasmid DNA encoding an anti-mouse endoglin (CD105) shRNA to B16 melanoma tumors with low and high metastatic potential results in pronounced anti-tumor effects. Cancers 2015, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Golzio, M.; Escoffre, J.-M.; Portet, T.; Mauroy, C.; Teissié, J.; Dean, D.S.; Rols, M.-P. Observations of the mechanisms of electromediated DNA uptake—From vesicles to tissues. Curr. Gene Ther. 2010, 10, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Neumann, E.; Rosenheck, K. Permeability changes induced by electric impulses in vesicular membranes. J. Membr. Biol. 1972, 10, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Mir, L.M.; Banoun, H.; Paoletti, C. Introduction of definite amounts of nonpermeant molecules into living cells after electropermeabilization: Direct access to the cytosol. Exp. Cell Res. 1988, 175, 15–25. [Google Scholar] [CrossRef]
- Bosnjak, M.; Dolinsek, T.; Cemazar, M.; Kranjc, S.; Blagus, T.; Markelc, B.; Stimac, M.; Zavrsnik, J.; Kamensek, U.; Heller, L.; et al. Gene electrotransfer of plasmid AMEP, an integrin-targeted therapy, has antitumor and antiangiogenic action in murine B16 melanoma. Gene Ther. 2015, 22. [Google Scholar] [CrossRef] [PubMed]
- Prosen, L.; Markelc, B.; Dolinsek, T.; Music, B.; Cemazar, M.; Sersa, G. Mcam Silencing With RNA Interference Using Magnetofection has Antitumor Effect in Murine Melanoma. Mol. Ther. Nucleic Acids 2014, 3, e205. [Google Scholar] [CrossRef] [PubMed]
- Dolinsek, T.; Markelc, B.; Bosnjak, M.; Blagus, T.; Prosen, L.; Kranjc, S.; Stimac, M.; Lampreht, U.; Sersa, G.; Cemazar, M. Endoglin silencing has significant antitumor effect on murine mammary adenocarcinoma mediated by vascular targeted effect. Curr. Gene Ther. 2015, 15, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Spanggaard, I.; Snoj, M.; Cavalcanti, A.; Bouquet, C.; Sersa, G.; Robert, C.; Cemazar, M.; Dam, E.; Vasseur, B.; Attali, P.; et al. Gene electrotransfer of plasmid antiangiogenic metargidin peptide (AMEP) in disseminated melanoma: Safety and efficacy results of a phase I first-in-man study. Hum. Gene Ther. Clin. Dev. 2013, 24, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Cha, E.; Daud, A. Plasmid IL-12 electroporation in melanoma. Hum. Vaccin. Immunother. 2012, 8, 1734–1738. [Google Scholar] [CrossRef] [PubMed]
- Dean, D.A.; Machado-Aranda, D.; Blair-Parks, K.; Yeldandi, A.V.; Young, J.L. Electroporation as a method for high-level nonviral gene transfer to the lung. Gene Ther. 2003, 10, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Sardesai, N.Y.; Weiner, D.B. Electroporation delivery of DNA vaccines: Prospects for success. Curr. Opin. Immunol. 2011, 23, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Wells, D.J. Gene therapy progress and prospects: Electroporation and other physical methods. Gene Ther. 2004, 11, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Gothelf, A.; Gehl, J. Gene electrotransfer to skin; review of existing literature and clinical perspectives. Curr. Gene Ther. 2010, 10, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Calvet, C.Y.; Thalmensi, J.; Liard, C.; Pliquet, E.; Bestetti, T.; Huet, T.; Langlade-Demoyen, P.; Mir, L.M. Optimization of a gene electrotransfer procedure for efficient intradermal immunization with an hTERT-based DNA vaccine in mice. Mol. Ther. 2014, 1, 14045. [Google Scholar] [CrossRef] [PubMed]
- Bosnjak, M.; Kamensek, U.; Sersa, G.; Stolfa, D.; Lavrencak, J.; Cemazar, M. Inhibition of the Innate Immune Receptors for Foreign DNA Sensing Improves Transfection Efficiency of Gene Electrotransfer in Melanoma B16F10 Cells. J. Membr. Biol. 2017. [Google Scholar] [CrossRef]
- Znidar, K.; Bosnjak, M.; Cemazar, M.; Heller, L.C. Cytosolic DNA Sensor Upregulation Accompanies DNA Electrotransfer in B16.F10 Melanoma Cells. Mol. Ther. Acids 2016, 5, e322. [Google Scholar] [CrossRef] [PubMed]
- Desmet, C.J.; Ishii, K.J. Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nat. Rev. Immunol. 2012, 12, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Heller, L.; Todorovic, V.; Cemazar, M. Electrotransfer of single-stranded or double-stranded DNA induces complete regression of palpable B16.F10 mouse melanomas. Cancer Gene Ther. 2013, 20, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Heller, L.C.; Coppola, D. Electrically mediated delivery of vector plasmid DNA elicits an antitumor effect. Gene Ther. 2002, 9, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Marrero, B.; Shirley, S.; Heller, R. Delivery of interleukin-15 to B16 melanoma by electroporation leads to tumor regression and long-term survival. Technol. Cancer Res. Treat. 2014, 13, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Shirley, S.A.; Lundberg, C.G.; Li, F.; Burcus, N.; Heller, R. Controlled gene delivery can enhance therapeutic outcome for cancer immune therapy for melanoma. Curr. Gene Ther. 2015, 15, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Savarin, M.; Kamensek, U.; Cemazar, M.; Heller, R.; Sersa, G. Electrotransfer of plasmid DNA radiosensitizes B16F10 tumors through activation of immune response. Radiol. Oncol. 2017, 51, 30–39. [Google Scholar] [CrossRef] [PubMed]
- McCray, A.N.; Ugen, K.E.; Muthumani, K.; Kim, J.J.; Weiner, D.B.; Heller, R. Complete Regression of Established Subcutaneous B16 Murine Melanoma Tumors after Delivery of an HIV-1 Vpr-Expressing Plasmid by in Vivo Electroporation. Mol. Ther. 2006, 14, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Ugen, K.E.; Kutzler, M.A.; Marrero, B.; Westover, J.; Coppola, D.; Weiner, D.B.; Heller, R. Regression of subcutaneous B16 melanoma tumors after intratumoral delivery of an IL-15-expressing plasmid followed by in vivo electroporation. Cancer Gene Ther. 2006, 13, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Rosato, A.; Dalla Santa, S.; Zoso, A.; Giacomelli, S.; Milan, G.; Macino, B.; Tosello, V.; Dellabona, P.; Lollini, P.L.; De Giovanni, C.; et al. The cytotoxic T-lymphocyte response against a poorly immunogenic mammary adenocarcinoma is focused on a single immunodominant class I epitope derived from the gp70 Env product of an endogenous retrovirus. Cancer Res. 2003, 63, 2158–2163. [Google Scholar] [PubMed]
- Nanni, P.; De Giovanni, C.; Lollini, P.L.; Nicoletti, G.; Prodi, G. TS/A: A new metastasizing cell line from a BALB/c spontaneous mammary adenocarcinoma. Clin. Exp. Metastasis 1983, 1, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014, 41, 543–852. [Google Scholar] [CrossRef] [PubMed]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Grosso, J.F.; Jure-Kunkel, M.N. CTLA-4 blockade in tumor models: An overview of preclinical and translational research. Cancer Immun. 2013, 13, 5. [Google Scholar] [PubMed]
- Vanpouille-Box, C.; Formenti, S.C.; Demaria, S. TREX1 dictates the immune fate of irradiated cancer cells. Oncoimmunology 2017, 6, e1339857. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Maelfait, J.; Liverpool, L.; Bridgeman, A.; Ragan, K.B.; Upton, J.W.; Rehwinkel, J. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 2017, 36, 2529–2543. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Miyashita, M.; Okamoto, M.; Morioka, Y.; Okabe, M.; Matsumoto, M.; Seya, T. DDX60 Is Involved in RIG-I-Dependent and Independent Antiviral Responses, and Its Function Is Attenuated by Virus-Induced EGFR Activation. Cell Rep. 2015, 11, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Morrone, S.R.; Wang, T.; Constantoulakis, L.M.; Hooy, R.M.; Delannoy, M.J.; Sohn, J. Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc. Natl. Acad. Sci. 2014, 111, E62–E71. [Google Scholar] [CrossRef] [PubMed]
- Kenworthy, R.; Lambert, D.; Yang, F.; Wang, N.; Chen, Z.; Zhu, H.; Zhu, F.; Liu, C.; Li, K.; Tang, H. Short-hairpin RNAs delivered by lentiviral vector transduction trigger RIG-I-mediated IFN activation. Nucleic Acids Res. 2009, 37, 6587–6599. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.E.; Baran, M.; Bowie, A.G. Cytosolic DNA sensors regulating type I interferon induction. Trends Immunol. 2011, 32, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Latz, E. Intracellular DNA recognition. Nat. Rev. Immunol. 2010, 10, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Kandimalla, E.R.; Zhao, Q.; Bhagat, L.; Cong, Y.; Agrawal, S. Requirement of nucleobase proximal to CpG dinucleotide for immunostimulatory activity of synthetic CpG DNA. Bioorganic Med. Chem. 2003, 11, 459–464. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | N | Growth Delay (to 40 mm3) (days) (AM ± SEM) | Complete Response |
|---|---|---|---|
| Control | 12 | 0.0 ± 0.4 | 0 |
| EP | 12 | 0.7 ± 0.1 | 0 |
| pControl + EP | 12 | 4.6 ± 1.5 * | 1/12 (8.3%) |
| pENTR/U6 scr + EP | 12 | 7.4 ± 0.6 * | 3/12 (25%) |
| pVAX + EP | 6 | 10.5 ± 2.4 *,** | 2/6 (33%) |
| Group | N | DNA Sensors | Cytokines | |||
|---|---|---|---|---|---|---|
| DDX60 (AM ± SEM) | DAI/ZBP1 (AM ± SEM) | p204 (AM ± SEM) | IFN β (AM ± SEM) | TNF α (AM ± SEM) | ||
| Control | 3 | 1.09 ± 0.31 | 1.06 ± 0.26 | 1.22 ± 0.57 | 1.13 ± 0.42 | 1.06 ± 0.26 |
| EP | 3 | 1.05 ± 0.15 | 0.98 ± 0.18 | 1.63 ± 0.19 | 13.27 ± 3.36 | 0.61 ± 0.12 |
| pContol + EP | 6 | 2.41 ± 0.23 | 2.43 ± 0.33 | 3.57 ± 0.61 | 126.22 ± 20.03 * | 9.98 ± 1.63 |
| pENTR/U6 scr + EP | 6 | 39.42 ± 20.86 | 14.16 ± 5.25 | 15.62 ± 4.49 ** | 967.45 ± 184.35 * | 5.08 ± 2.47 |
| pVAX + EP | 6 | 5.21 ± 1.45 | 5.88 ± 2.55 | 11.39 ± 2.37 | 743.48 ± 174.34 * | 12.08 ± 2.60 * |
| Group | N | DNA Sensors | Cytokines | |||
|---|---|---|---|---|---|---|
| DDX60 (AM ± SEM) | DAI/ZBP1 (AM ± SEM) | p204 (AM ± SEM) | IFN β (AM ± SEM) | TNF α (AM ± SEM) | ||
| Control | 8 | 1.00 ± 0.04 | 1.01 ± 0.06 | 1.01 ± 0.04 | 1.01 ± 0.05 | 1.28 ± 0.29 |
| pContol + EP | 8 | 39.30 ± 5.52 * | 109.85 ± 24.74 | 64.83 ± 16.46 * | 74.43 ± 27.54 | 1.69 ± 0.43 |
| pENTR/U6 scr + EP | 8 | 52.74 ± 14.01 * | 161.78 ± 54.23 * | 68.76 ± 21.38 * | 78.90 ± 19.86 | 2.44 ± 0.98 |
| pVAX + EP | 8 | 62.56 ± 13.29 * | 153.85 ± 60.12 * | 82.25 ± 28.97 * | 102.88 ± 27.46 * | 4.35 ± 1.81 |
| Plasmid | Base Pairs | Molecular Weight * | Copies/100 ug | Motifs **/Copy | Total Motifs ** Injected | Promotor | Antibiotic Resistance Gene |
|---|---|---|---|---|---|---|---|
| pVAX1 | 2999 | 1.85 × 106 | 3.25 × 1013 | 7 | 2.27 × 1014 | CMV | kanamycin |
| pControl | 3390 | 2.09 × 106 | 2.87 × 1013 | 10 | 2.87 × 1014 | CMV | ampicillin |
| pENTR/U6 scr | 2909 | 1.79 × 106 | 3.35 × 1013 | 13 | 4.36 × 1014 | U6 | kanamycin |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosnjak, M.; Jesenko, T.; Kamensek, U.; Sersa, G.; Lavrencak, J.; Heller, L.; Cemazar, M. Electrotransfer of Different Control Plasmids Elicits Different Antitumor Effectiveness in B16.F10 Melanoma. Cancers 2018, 10, 37. https://doi.org/10.3390/cancers10020037
Bosnjak M, Jesenko T, Kamensek U, Sersa G, Lavrencak J, Heller L, Cemazar M. Electrotransfer of Different Control Plasmids Elicits Different Antitumor Effectiveness in B16.F10 Melanoma. Cancers. 2018; 10(2):37. https://doi.org/10.3390/cancers10020037
Chicago/Turabian StyleBosnjak, Masa, Tanja Jesenko, Urska Kamensek, Gregor Sersa, Jaka Lavrencak, Loree Heller, and Maja Cemazar. 2018. "Electrotransfer of Different Control Plasmids Elicits Different Antitumor Effectiveness in B16.F10 Melanoma" Cancers 10, no. 2: 37. https://doi.org/10.3390/cancers10020037