Understanding Intratumor Heterogeneity and Evolution in NSCLC and Potential New Therapeutic Approach


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
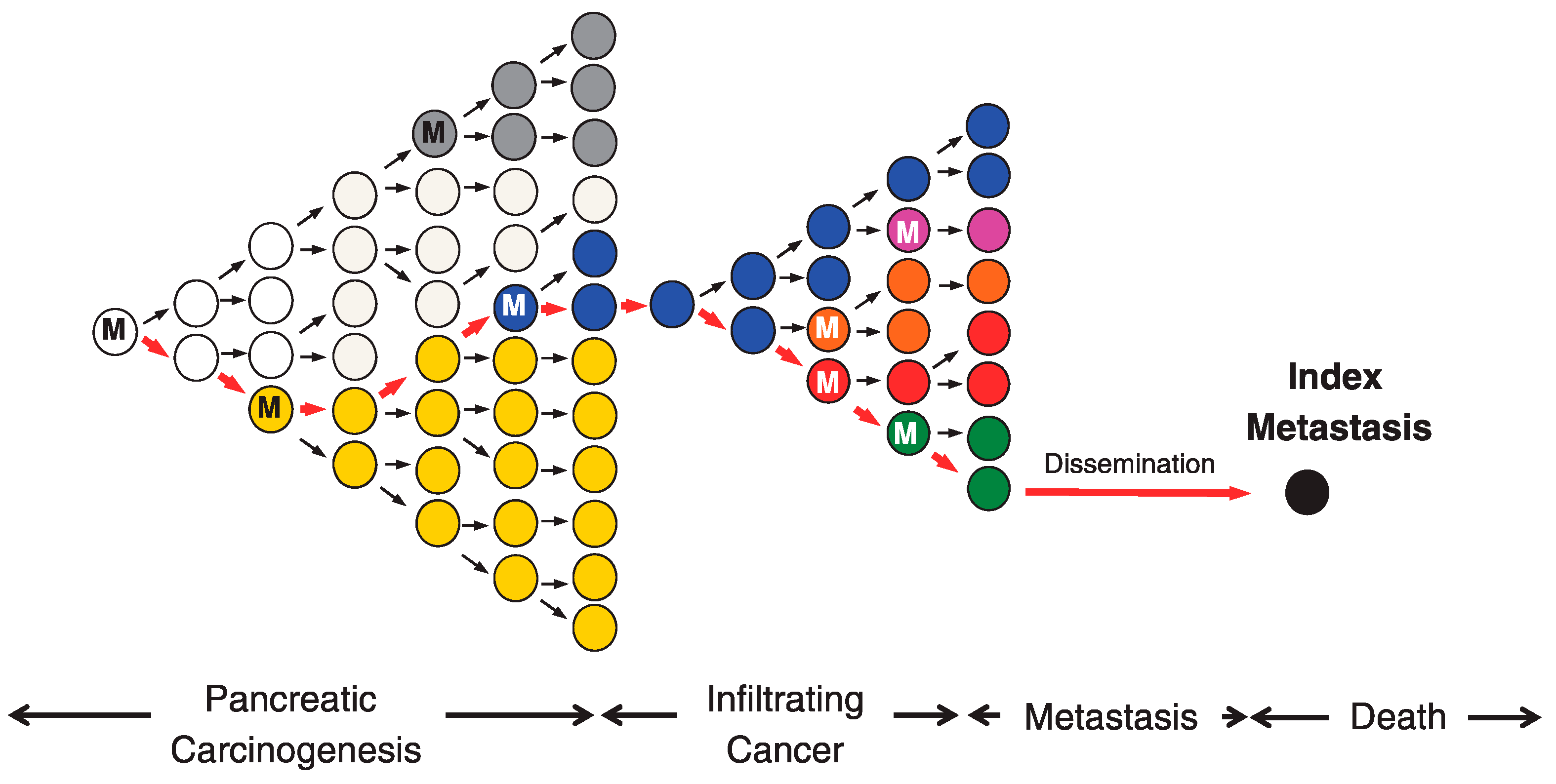
2. Intratumor Heterogeneity and Pattern of Tumor Development
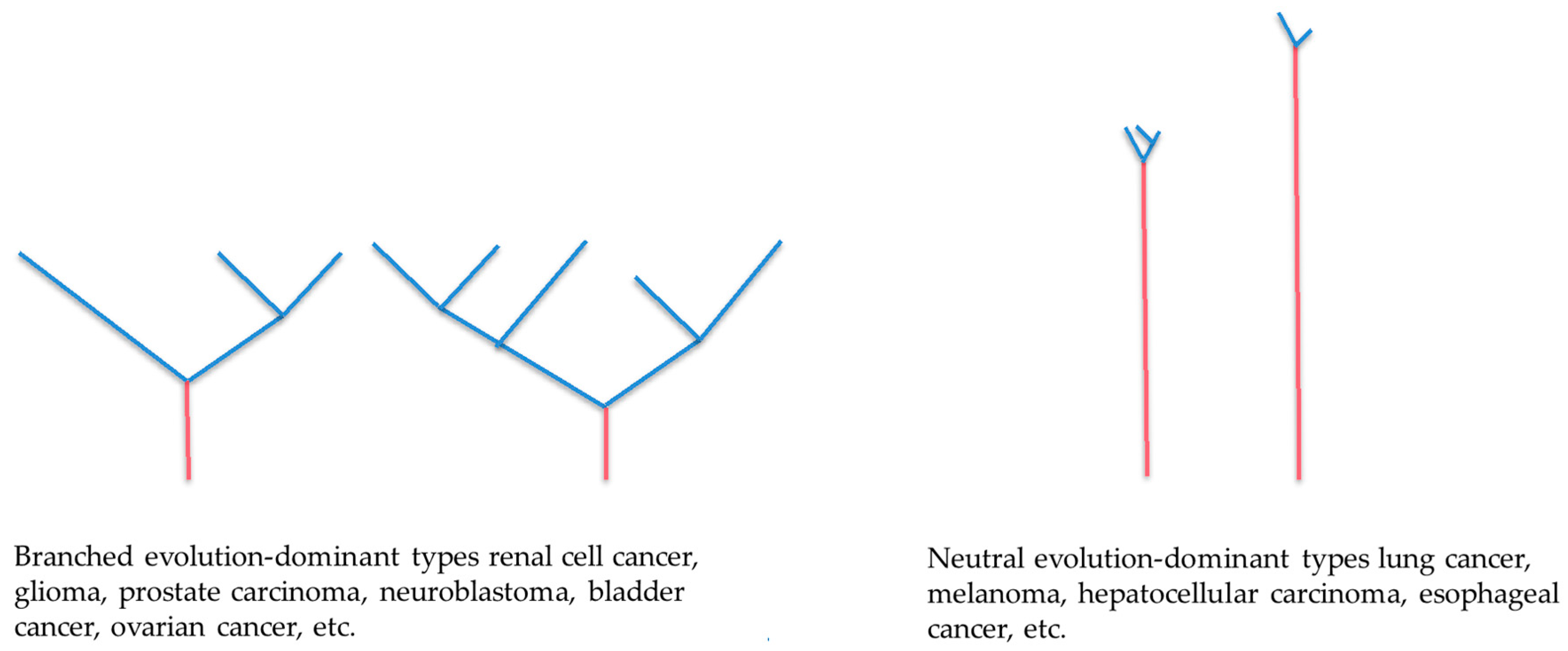
2.1. Branched Evolution
2.2. Neutral Evolution
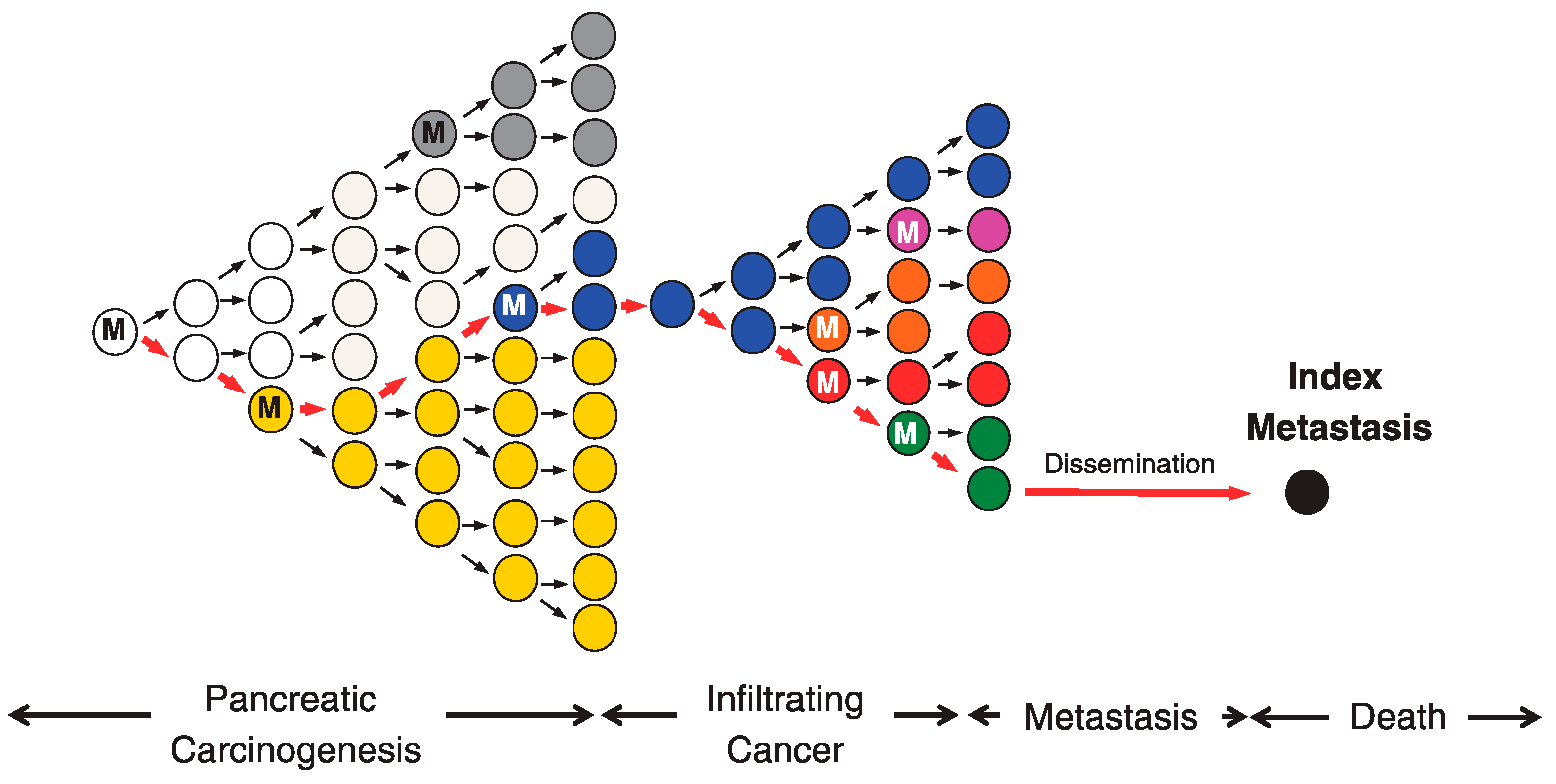
3. Temporal Diversity in the Cancer Genome Evolutionary Processes
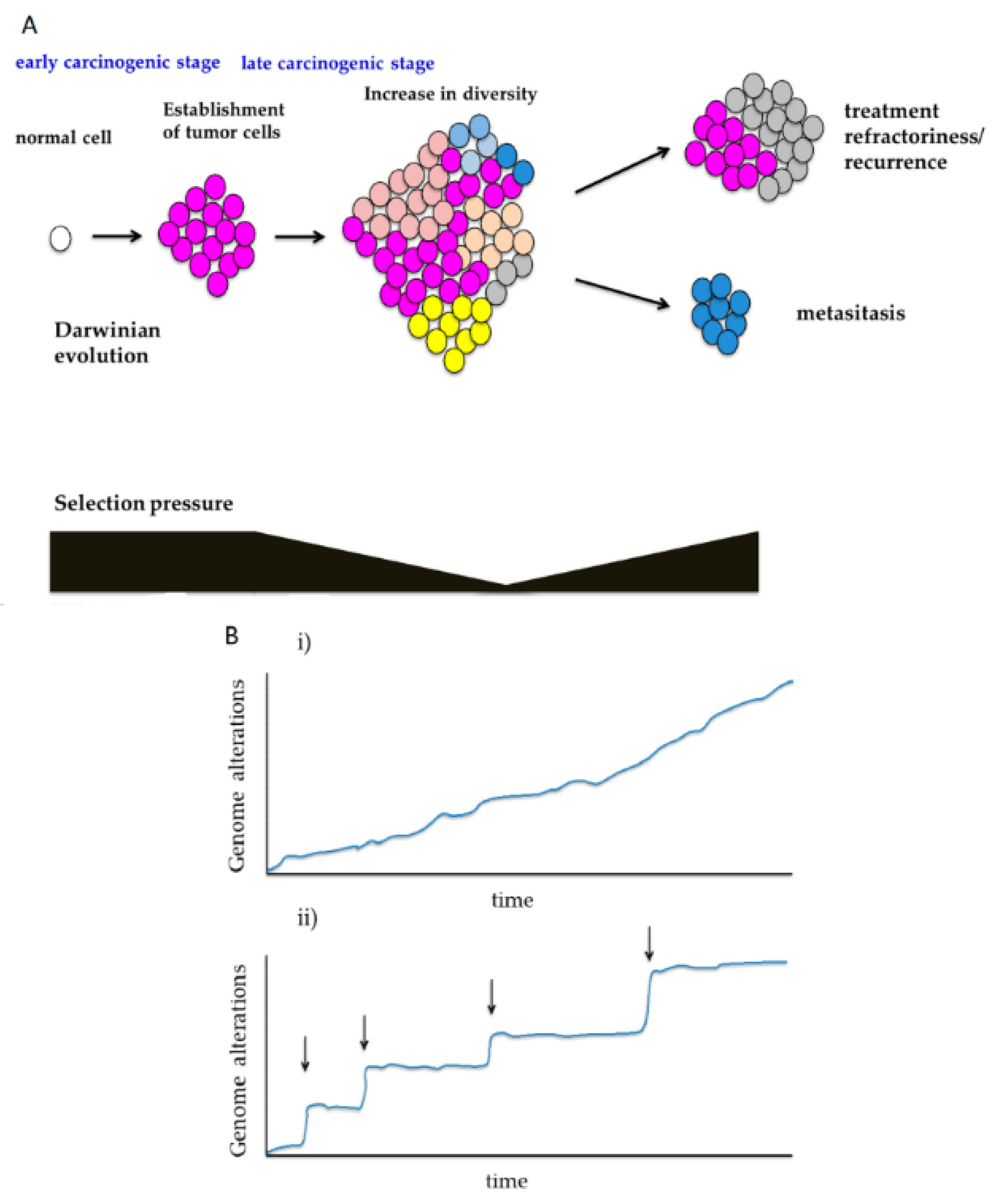
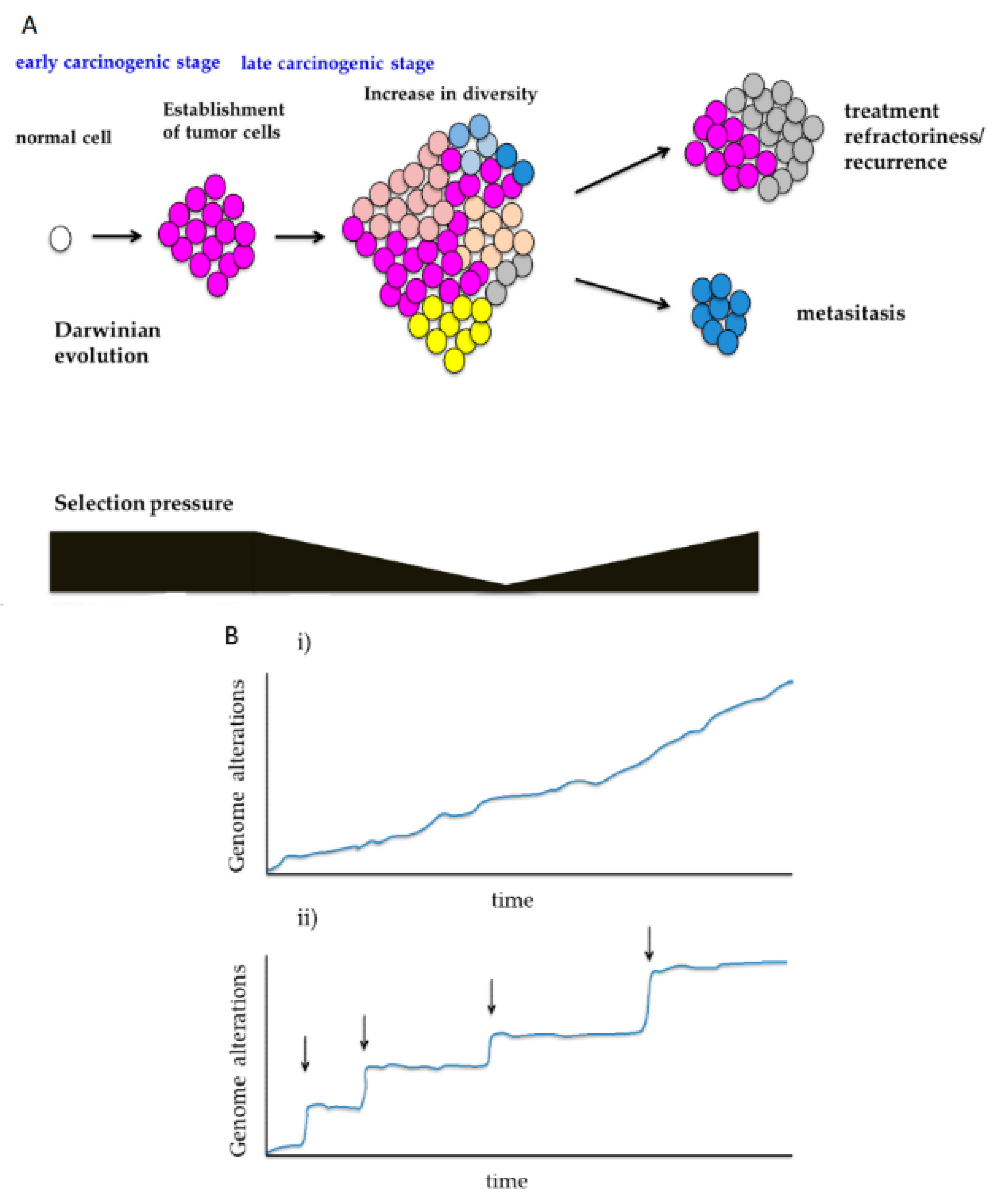
3.1. Shifts in Selection Pressure During Carcinogenesis
3.2. Sequential Evolution and Punctuated Evolution
4. Intercellular Interaction in Cancer Genome Evolution
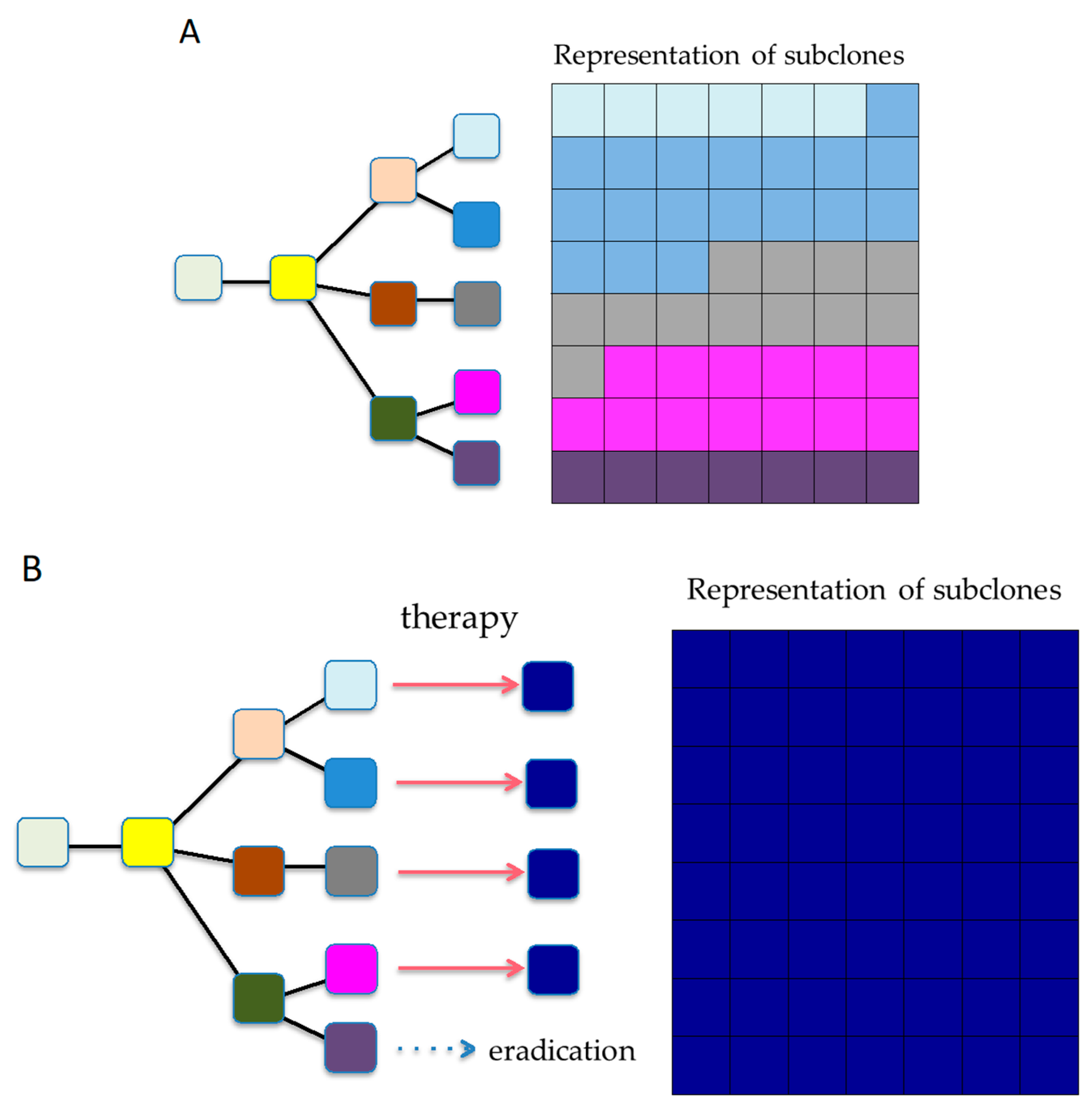
5. Degree of Heterogeneity and Limiting Points
6. Significance of ITH in Treatment Refractoriness, Recurrence, and Metastasis
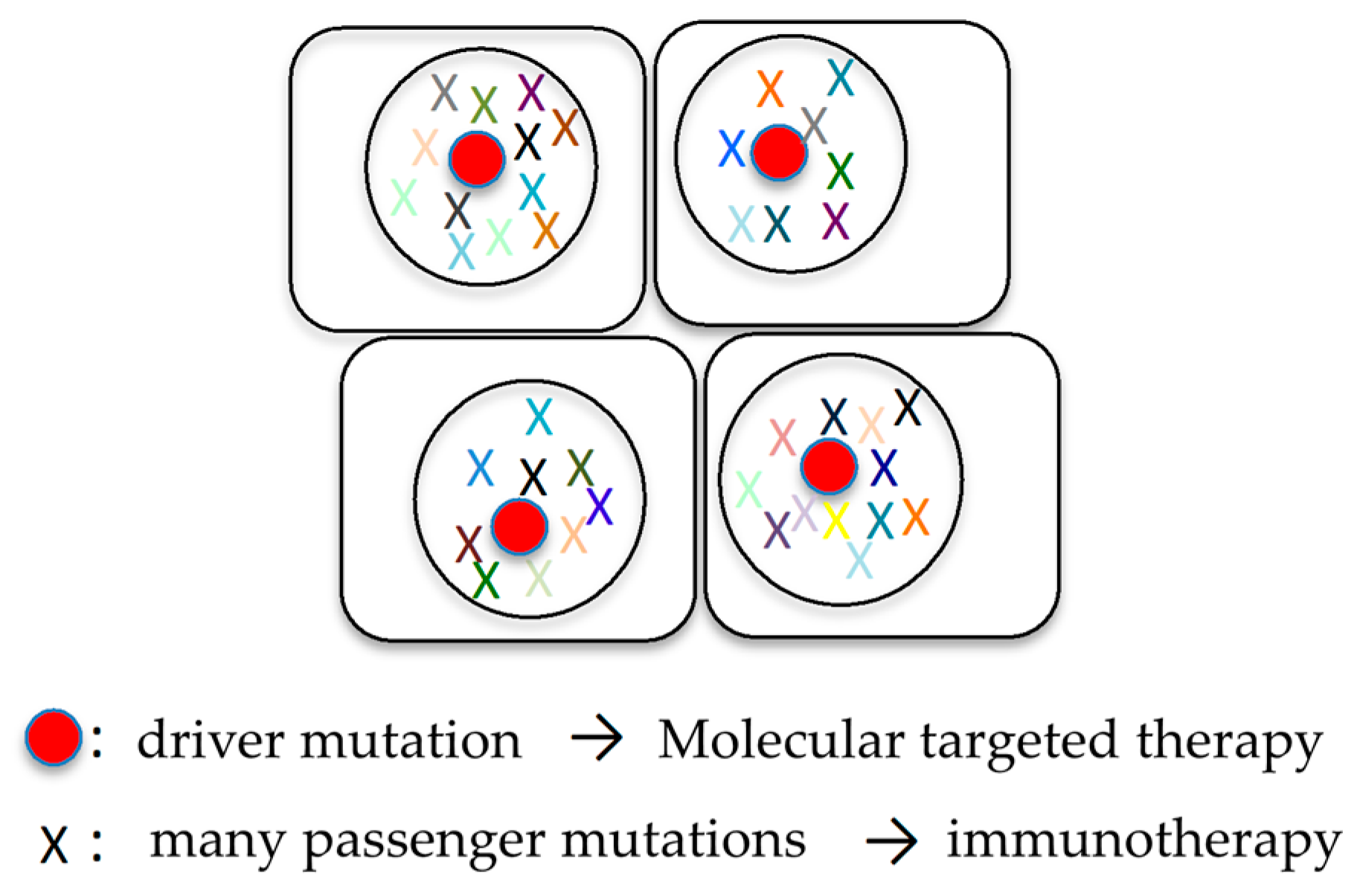
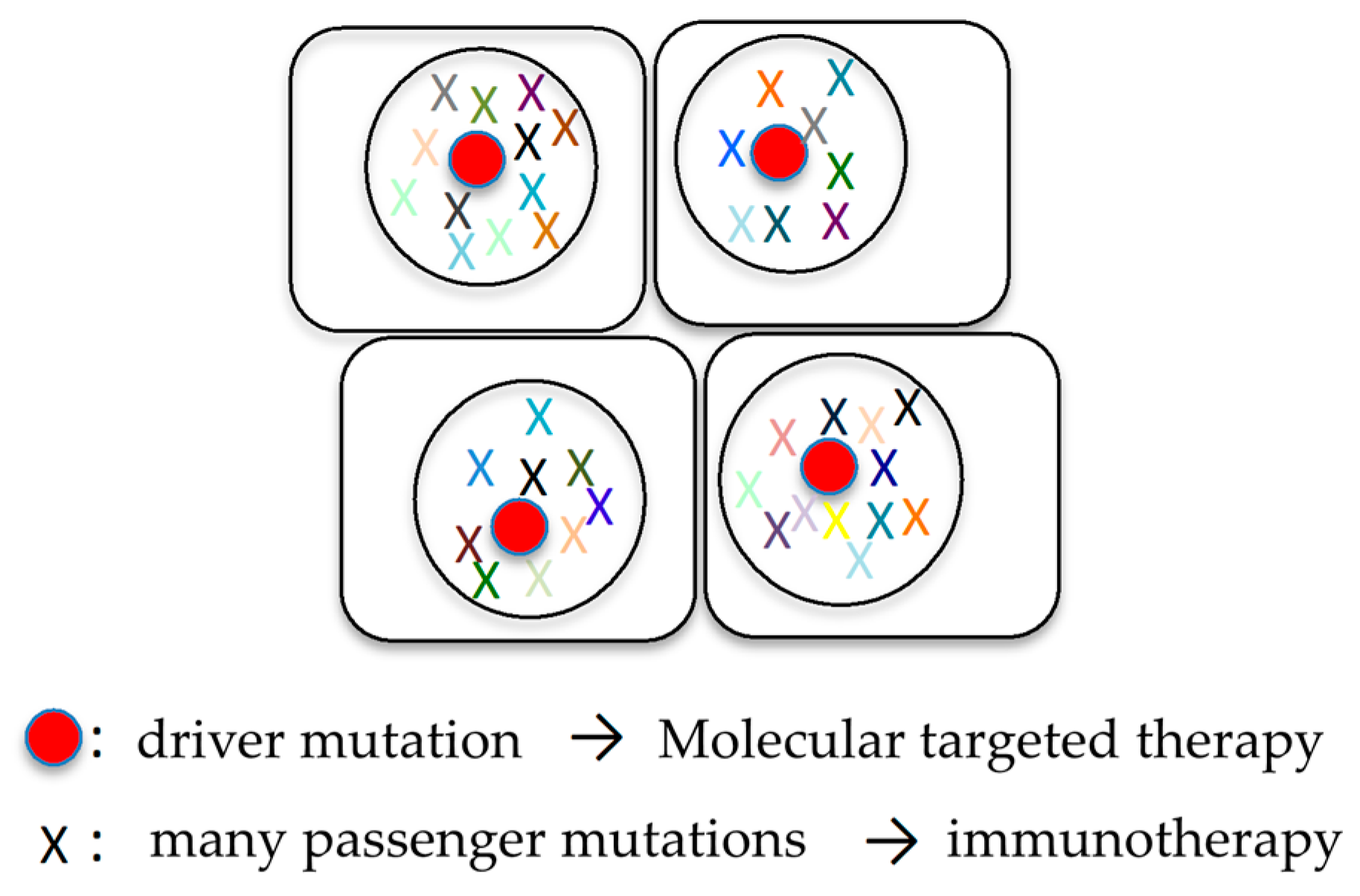
6.1. Combination of Molecular Targeted Therapy and Immunotherapy
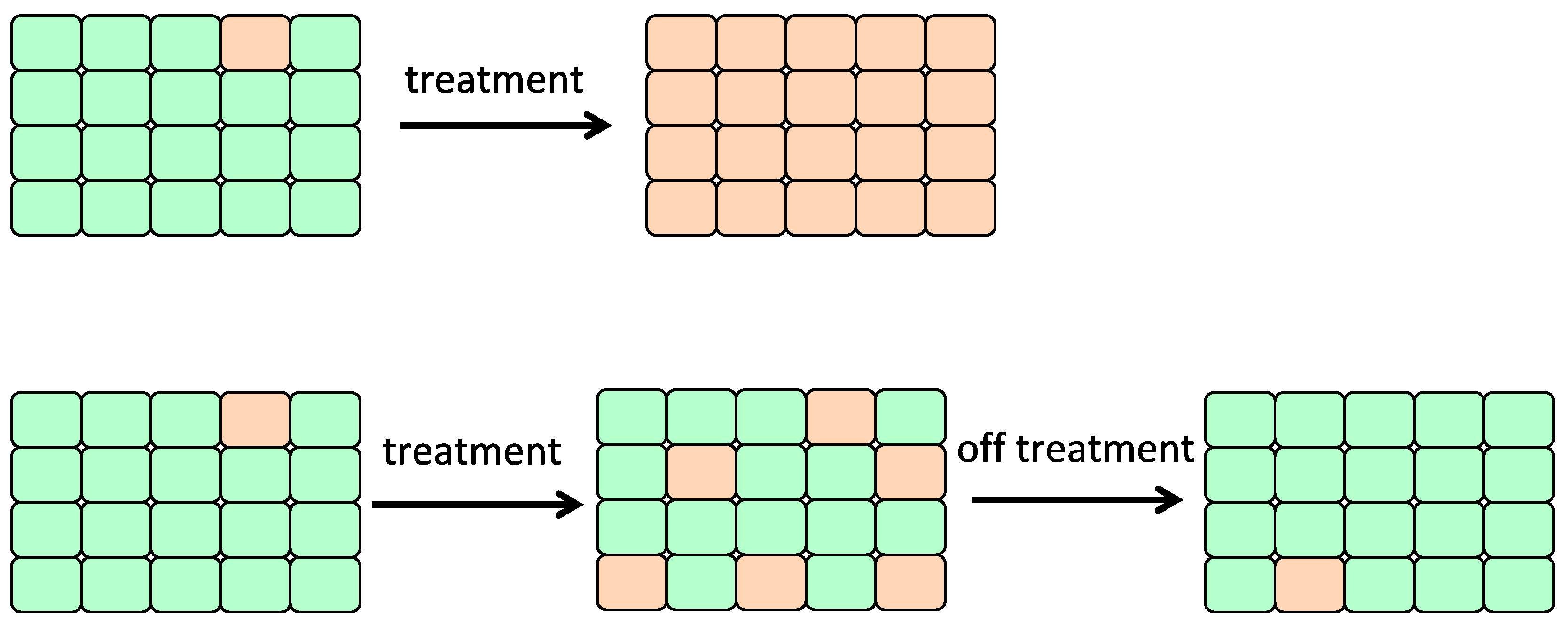
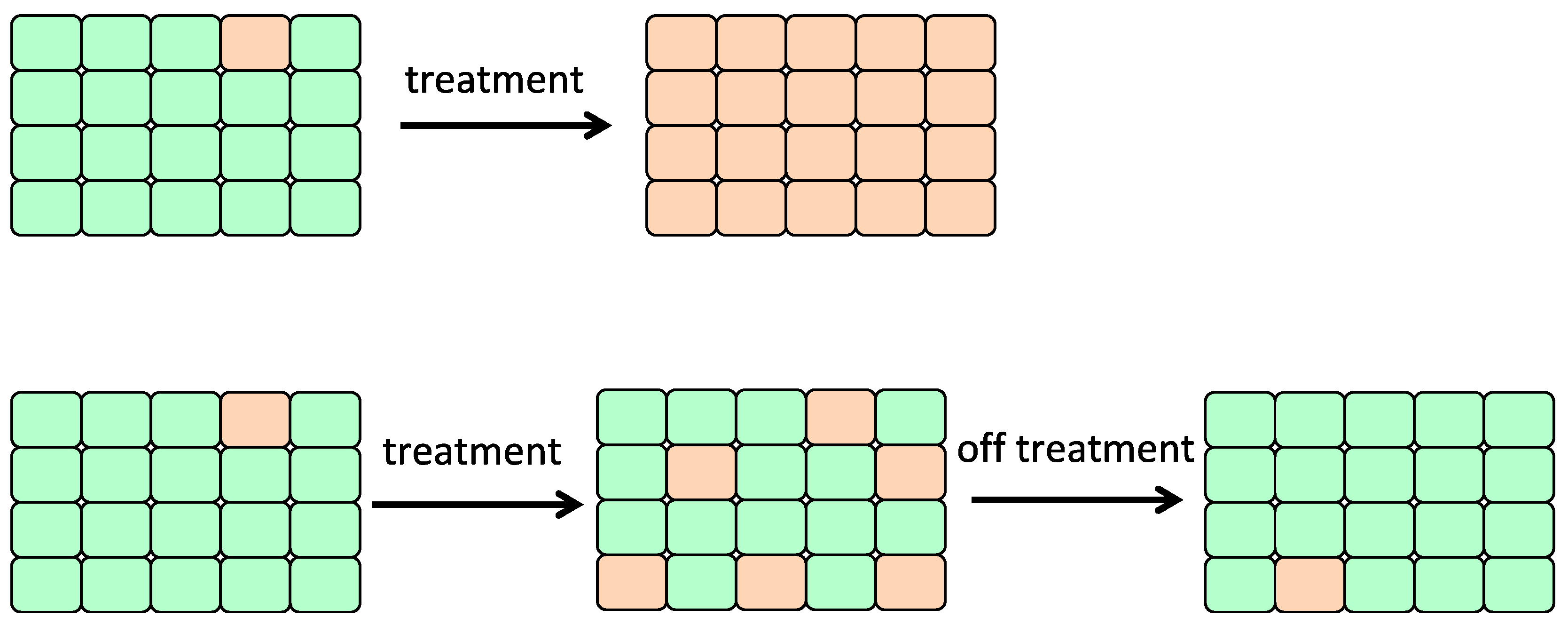
6.2. Adaptive Therapy
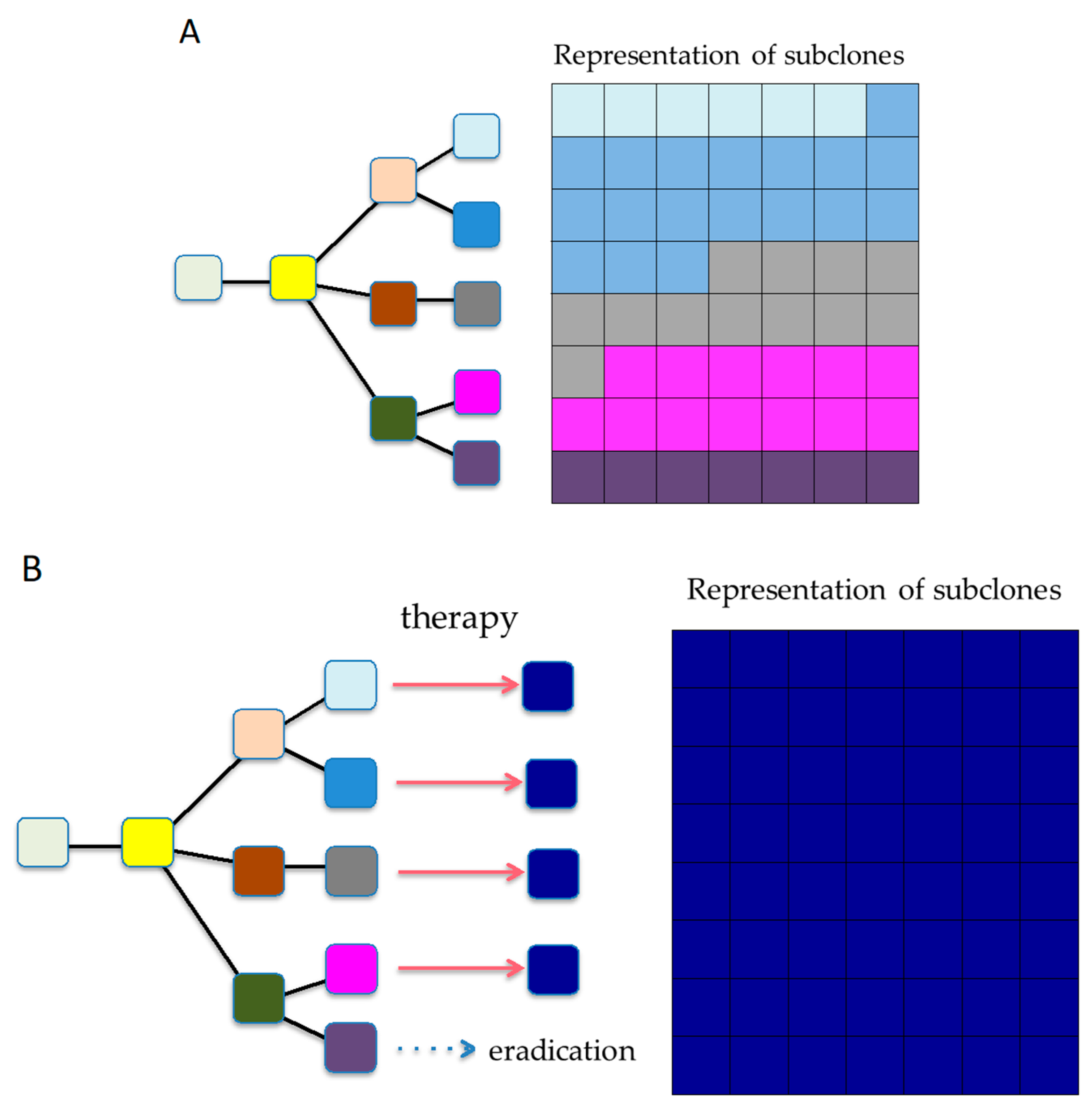
6.3. Reshaping Tumor Evolution
7. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindan, R.; Ding, L.; Griffith, M.; Subramanian, J.; Dees, N.D.; Kanchi, K.L.; Maher, C.A.; Fulton, R.; Fulton, L.; Wallis, J.; et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012, 150, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fujimoto, J.; Zhang, J.; Wedge, D.C.; Song, X.; Zhang, J.; Seth, S.; Chow, C.W.; Cao, Y.; Gumbs, C.; et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.; Lutz, C.; van Delft, F.W.; Bateman, C.M.; Guo, Y.; Colman, S.M.; Kempski, H.; Moorman, A.V.; Titley, I.; Swansbury, J.; et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011, 469, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Van Loo, P.; Wedge, D.C.; Alexandrov, L.B.; Greenman, C.D.; Lau, K.W.; Raine, K.; Jones, D.; Marshall, J.; Ramakrishna, M.; et al. The life history of 21 breast cancers. Cell 2012, 149, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Flannery, M.A.; Dembski, W.A.; Wallace, A.R. Alfred Russel Wallace’s Theory of Intelligent Evolution: How Wallace’s World of Life Challenged Darwinism: With an Abridged Version of the World of Life; Erasmus Press: Riesel, TX, USA, 2008; p. 225. [Google Scholar]
- Gillies, R.J.; Brown, J.S.; Anderson, A.R.A.; Gatenby, R.A. Eco-evolutionary causes and consequences of temporal changes in intratumoural blood flow. Nat. Rev. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Hirotsu, Y.; Mochizuki, H.; Nakagomi, T.; Oyama, T.; Amemiya, K.; Omata, M. Stepwise addition of genetic changes correlated with histological change from “well-differentiated” to “sarcomatoid” phenotypes: A case report. BMC Cancer 2017, 17, 65. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, T.; Goto, T.; Hirotsu, Y.; Shikata, D.; Yokoyama, Y.; Higuchi, R.; Amemiya, K.; Okimoto, K.; Oyama, T.; Mochizuki, H.; et al. New therapeutic targets for pulmonary sarcomatoid carcinomas based on their genomic and phylogenetic profiles. Oncotarget 2018, 9, 10635–10649. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Murugaesu, N.; Wilson, G.A.; Birkbak, N.J.; Watkins, T.; McGranahan, N.; Kumar, S.; Abbassi-Ghadi, N.; Salm, M.; Mitter, R.; Horswell, S.; et al. Tracking the genomic evolution of esophageal adenocarcinoma through neoadjuvant chemotherapy. Cancer Discov. 2015, 5, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.M.; Zhang, C.Z.; Maire, C.L.; Jung, J.; Manzo, V.E.; Adalsteinsson, V.A.; Homer, H.; Haidar, S.; Blumenstiel, B.; Pedamallu, C.S.; et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 2014, 4, 956–971. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during braf inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A big bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Werner, B.; Barnes, C.P.; Graham, T.A.; Sottoriva, A. Identification of neutral tumor evolution across cancer types. Nat. Genet. 2016, 48, 238–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yachida, S.; Iacobuzio-Donahue, C.A. Evolution and dynamics of pancreatic cancer progression. Oncogene 2013, 32, 5253–5260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Xing, K.; He, X. The DJ/DS ratio test reveals hundreds of novel putative cancer drivers. Mol. Biol. Evol. 2015, 32, 2181–2185. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.D. The ka/ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486. [Google Scholar] [CrossRef]
- Mathews, S. Analytical methods for studying the evolution of paralogs using duplicate gene datasets. Methods Enzymol. 2005, 395, 724–745. [Google Scholar] [PubMed]
- Nekrutenko, A.; Makova, K.D.; Li, W.H. The K(A)/K(S) ratio test for assessing the protein-coding potential of genomic regions: An empirical and simulation study. Genome Res. 2002, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.I.; Wang, H.Y.; Ling, S.; Lu, X. The ecology and evolution of cancer: The ultra-microevolutionary process. Annu. Rev. Genet. 2016, 50, 347–369. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, J.; Li, L.; Yin, G.; Zhang, J.; Zheng, S.; Cheung, H.; Wu, N.; Lu, N.; Mao, X.; et al. Genomic heterogeneity of multiple synchronous lung cancer. Nat. Commun. 2016, 7, 13200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, T.; Hirotsu, Y.; Mochizuki, H.; Nakagomi, T.; Shikata, D.; Yokoyama, Y.; Oyama, T.; Amemiya, K.; Okimoto, K.; Omata, M. Mutational analysis of multiple lung cancers: Discrimination between primary and metastatic lung cancers by genomic profile. Oncotarget 2017, 8, 31133–31143. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Galanos, P.; Pappas, G.; Polyzos, A.; Kotsinas, A.; Svolaki, I.; Giakoumakis, N.N.; Glytsou, C.; Pateras, I.S.; Swain, U.; Souliotis, V.L.; et al. Mutational signatures reveal the role of RAD52 in p53-independent p21-driven genomic instability. Genome Biol. 2018, 19, 37. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.; Hu, Z.; Sottoriva, A.; Graham, T.A.; Harpak, A.; Ma, Z.; Fischer, J.M.; Shibata, D.; Curtis, C. Between-region genetic divergence reflects the mode and tempo of tumor evolution. Nat. Genet. 2017, 49, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A.; Roth, A.; Laks, E.; Masud, T.; Bashashati, A.; Zhang, A.W.; Ha, G.; Biele, J.; Yap, D.; Wan, A.; et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet. 2016, 48, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, A.; Lindberg, M.; Faust, G.G.; Leibowitz, M.L.; Clark, R.A.; Layer, R.M.; Quinlan, A.R.; Hall, I.M. Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome Res. 2013, 23, 762–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, C.D.; Liu, P.; Woloszynska-Read, A.; Zhang, J.; Luo, W.; Qin, M.; Bshara, W.; Conroy, J.M.; Sabatini, L.; Vedell, P.; et al. Whole-genome sequencing identifies genomic heterogeneity at a nucleotide and chromosomal level in bladder cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E672–681. [Google Scholar] [CrossRef] [PubMed]
- Nones, K.; Waddell, N.; Wayte, N.; Patch, A.M.; Bailey, P.; Newell, F.; Holmes, O.; Fink, J.L.; Quinn, M.C.J.; Tang, Y.H.; et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat. Commun. 2014, 5, 5224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Eklund, A.C.; Li, Q.; McClelland, S.E.; Endesfelder, D.; Tan, P.; Tan, I.B.; Richardson, A.L.; Szallasi, Z.; Swanton, C. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 2011, 71, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.H.; Gao, J.L.; Liu, Q.; Siwicki, M.; Martens, C.; Jacobs, P.; Velez, D.; Yim, E.; Bryke, C.R.; Hsu, N.; et al. Chromothriptic cure of whim syndrome. Cell 2015, 160, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Bouge, A.L.; Ruffle, F.; Riquier, S.; Guibert, B.; Audoux, J.; Commes, T. RNA-seq analysis to detect abnormal fusion transcripts linked to chromothripsis. Methods Mol. Biol. 2018, 1769, 133–156. [Google Scholar] [PubMed]
- Kloosterman, W.P.; Coebergh van den Braak, R.R.J.; Pieterse, M.; van Roosmalen, M.J.; Sieuwerts, A.M.; Stangl, C.; Brunekreef, R.; Lalmahomed, Z.S.; Ooft, S.; van Galen, A.; et al. A systematic analysis of oncogenic gene fusions in primary colon cancer. Cancer Res. 2017, 77, 3814–3822. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Cleveland, D.W. Rebuilding chromosomes after catastrophe: Emerging mechanisms of chromothripsis. Trends Cell Biol. 2017, 27, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Cleary, A.S.; Leonard, T.L.; Gestl, S.A.; Gunther, E.J. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature 2014, 508, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marusyk, A.; Tabassum, D.P.; Altrock, P.M.; Almendro, V.; Michor, F.; Polyak, K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 2014, 514, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efremova, M.; Finotello, F.; Rieder, D.; Trajanoski, Z. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front. Immunol. 2017, 8, 1679. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Fang, W.; Zhan, J.; Hong, S.; Tang, Y.; Kang, S.; Zhang, Y.; He, X.; Zhou, T.; Qin, T.; et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: Implication for optional immune targeted therapy for nsclc patients with EGFR mutation. J. Thorac. Oncol. 2015, 10, 910–923. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Chen, X.; Li, M.; Liu, J.; Qi, X.; Yang, W.; Zhang, H.; Cai, Z.; Dai, Y.; Ouyang, X. Programmed death-ligand 1 expression predicts tyrosine kinase inhibitor response and better prognosis in a cohort of patients with epidermal growth factor receptor mutation-positive lung adenocarcinoma. Clin. Lung Cancer 2015, 16, e25–e35. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 2017, 171, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 1212–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agaram, N.P.; Shia, J.; Tang, L.H.; Klimstra, D.S. DNA mismatch repair deficiency in ampullary carcinoma: A morphologic and immunohistochemical study of 54 cases. Am. J. Clin. Pathol. 2010, 133, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [Green Version]
- Cancer Genome Atlas Research, N.; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Poulogiannis, G.; Frayling, I.M.; Arends, M.J. DNA mismatch repair deficiency in sporadic colorectal cancer and lynch syndrome. Histopathology 2010, 56, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.N.; Hile, S.E.; Eckert, K.A. Defective mismatch repair, microsatellite mutation bias, and variability in clinical cancer phenotypes. Cancer Res. 2010, 70, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Stoehr, C.; Burger, M.; Stoehr, R.; Bertz, S.; Ruemmele, P.; Hofstaedter, F.; Denzinger, S.; Wieland, W.F.; Hartmann, A.; Walter, B. Mismatch repair proteins HMLH1 and HMSH2 are differently expressed in the three main subtypes of sporadic renal cell carcinoma. Pathobiology 2012, 79, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Timmermann, B.; Kerick, M.; Roehr, C.; Fischer, A.; Isau, M.; Boerno, S.T.; Wunderlich, A.; Barmeyer, C.; Seemann, P.; Koenig, J.; et al. Somatic mutation profiles of MSI and mss colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS ONE 2010, 5, e15661. [Google Scholar] [CrossRef] [PubMed]
- Warth, A.; Korner, S.; Penzel, R.; Muley, T.; Dienemann, H.; Schirmacher, P.; von Knebel-Doeberitz, M.; Weichert, W.; Kloor, M. Microsatellite instability in pulmonary adenocarcinomas: A comprehensive study of 480 cases. Virchows Arch. 2016, 468, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.S.; Huang, W.Y. The analysis of microsatellite instability in extracolonic gastrointestinal malignancy. Pathology 2013, 45, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Shlien, A.; Campbell, B.B.; de Borja, R.; Alexandrov, L.B.; Merico, D.; Wedge, D.; Van Loo, P.; Tarpey, P.S.; Coupland, P.; Behjati, S.; et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 2015, 47, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Loewe, L. Quantifying the genomic decay paradox due to muller’s ratchet in human mitochondrial DNA. Genet. Res. 2006, 87, 133–159. [Google Scholar] [CrossRef] [PubMed]
- Savas, P.; Teo, Z.L.; Lefevre, C.; Flensburg, C.; Caramia, F.; Alsop, K.; Mansour, M.; Francis, P.A.; Thorne, H.A.; Silva, M.J.; et al. The subclonal architecture of metastatic breast cancer: Results from a prospective community-based rapid autopsy program “cascade”. PLoS Med. 2016, 13, e1002204. [Google Scholar] [CrossRef] [PubMed]
- Faltas, B.M.; Prandi, D.; Tagawa, S.T.; Molina, A.M.; Nanus, D.M.; Sternberg, C.; Rosenberg, J.; Mosquera, J.M.; Robinson, B.; Elemento, O.; et al. Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nat. Genet. 2016, 48, 1490–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [PubMed]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctdna analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Hirotsu, Y.; Amemiya, K.; Nakagomi, T.; Shikata, D.; Yokoyama, Y.; Okimoto, K.; Oyama, T.; Mochizuki, H.; Omata, M. Distribution of circulating tumor DNA in lung cancer: Analysis of the primary lung and bone marrow along with the pulmonary venous and peripheral blood. Oncotarget 2017, 8, 59268–59281. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating tumor DNA as a liquid biopsy for cancer. Clin. Chem. 2015, 61, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Hench, I.B.; Hench, J.; Tolnay, M. Liquid biopsy in clinical management of breast, lung, and colorectal cancer. Front. Med. (Lausanne) 2018, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.A.; Chabon, J.J.; Lovejoy, A.F.; Newman, A.M.; Stehr, H.; Azad, T.D.; Khodadoust, M.S.; Esfahani, M.S.; Liu, C.L.; Zhou, L.; et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 2017, 7, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Rosenfeld, N.; Caldas, C. Circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 369, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage ii colon cancer. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.A.; Rooney, M.S.; Rajasagi, M.; Tiao, G.; Dixon, P.M.; Lawrence, M.S.; Stevens, J.; Lane, W.J.; Dellagatta, J.L.; Steelman, S.; et al. Comprehensive analysis of cancer-associated somatic mutations in class i hla genes. Nat. Biotechnol. 2015, 33, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Thanopoulou, E.; Peggs, K.S.; Quezada, S.A.; Swanton, C. Tumour heterogeneity and immune-modulation. Curr. Opin. Pharmacol. 2013, 13, 497–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallaher, J.A.; Enriquez-Navas, P.M.; Luddy, K.A.; Gatenby, R.A.; Anderson, A.R.A. Spatial heterogeneity and evolutionary dynamics modulate time to recurrence in continuous and adaptive cancer therapies. Cancer Res. 2018, 78, 2127–2139. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Silva, A.S.; Gillies, R.J.; Frieden, B.R. Adaptive therapy. Cancer Res. 2009, 69, 4894–4903. [Google Scholar] [CrossRef] [PubMed]
- Enriquez-Navas, P.M.; Kam, Y.; Das, T.; Hassan, S.; Silva, A.; Foroutan, P.; Ruiz, E.; Martinez, G.; Minton, S.; Gillies, R.J.; et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci. Transl. Med. 2016, 8, 327ra324. [Google Scholar] [CrossRef] [PubMed]
- Inukai, M.; Toyooka, S.; Ito, S.; Asano, H.; Ichihara, S.; Soh, J.; Suehisa, H.; Ouchida, M.; Aoe, K.; Aoe, M.; et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res. 2006, 66, 7854–7858. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Martins, R.G.; Spigel, D.; Grunberg, S.M.; Spira, A.; Janne, P.A.; Joshi, V.A.; McCollum, D.; Evans, T.L.; Muzikansky, A.; et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J. Clin. Oncol. 2008, 26, 2442–2449. [Google Scholar] [CrossRef] [PubMed]
- Soh, J.; Toyooka, S.; Ichihara, S.; Suehisa, H.; Kobayashi, N.; Ito, S.; Yamane, M.; Aoe, M.; Sano, Y.; Kiura, K.; et al. EGFR mutation status in pleural fluid predicts tumor responsiveness and resistance to gefitinib. Lung Cancer 2007, 56, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Friboulet, L.; Leshchiner, I.; Gainor, J.F.; Bergqvist, S.; Brooun, A.; Burke, B.J.; Deng, Y.L.; Liu, W.; Dardaei, L.; et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation l1198f. N. Engl. J. Med. 2016, 374, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The allelic context of the c797s mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef] [PubMed]
- Curioni-Fontecedro, A. A new era of oncology through artificial intelligence. ESMO Open 2017, 2, e000198. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goto, T.; Hirotsu, Y.; Amemiya, K.; Mochizuki, H.; Omata, M. Understanding Intratumor Heterogeneity and Evolution in NSCLC and Potential New Therapeutic Approach. Cancers 2018, 10, 212. https://doi.org/10.3390/cancers10070212
Goto T, Hirotsu Y, Amemiya K, Mochizuki H, Omata M. Understanding Intratumor Heterogeneity and Evolution in NSCLC and Potential New Therapeutic Approach. Cancers. 2018; 10(7):212. https://doi.org/10.3390/cancers10070212
Chicago/Turabian StyleGoto, Taichiro, Yosuke Hirotsu, Kenji Amemiya, Hitoshi Mochizuki, and Masao Omata. 2018. "Understanding Intratumor Heterogeneity and Evolution in NSCLC and Potential New Therapeutic Approach" Cancers 10, no. 7: 212. https://doi.org/10.3390/cancers10070212
APA StyleGoto, T., Hirotsu, Y., Amemiya, K., Mochizuki, H., & Omata, M. (2018). Understanding Intratumor Heterogeneity and Evolution in NSCLC and Potential New Therapeutic Approach. Cancers, 10(7), 212. https://doi.org/10.3390/cancers10070212