ATM Dependent DUSP6 Modulation of p53 Involved in Synergistic Targeting of MAPK and p53 Pathways with Trametinib and MDM2 Inhibitors in Cutaneous Melanoma

 , ,
, ,
Abstract
:1. Introduction
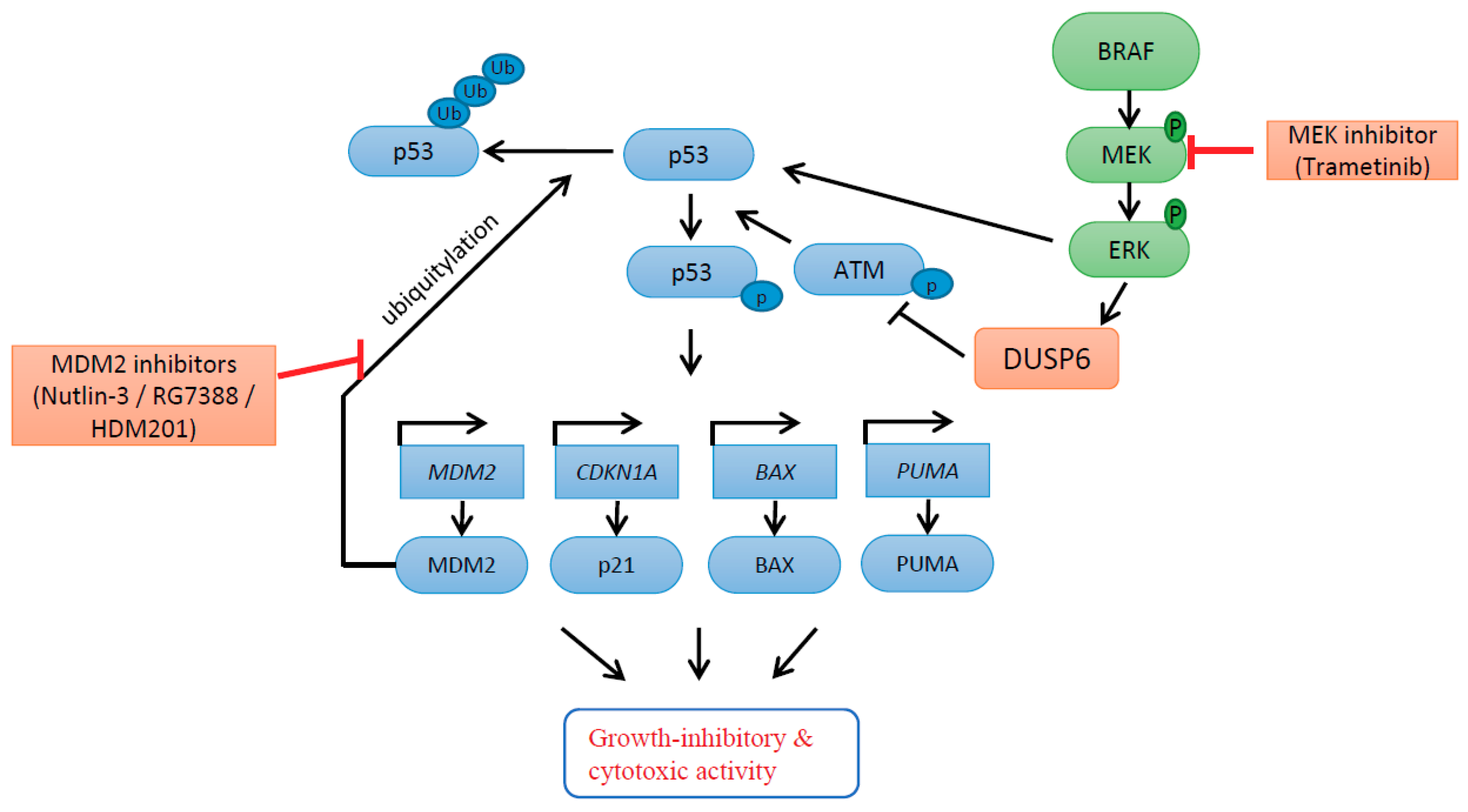
2. Results
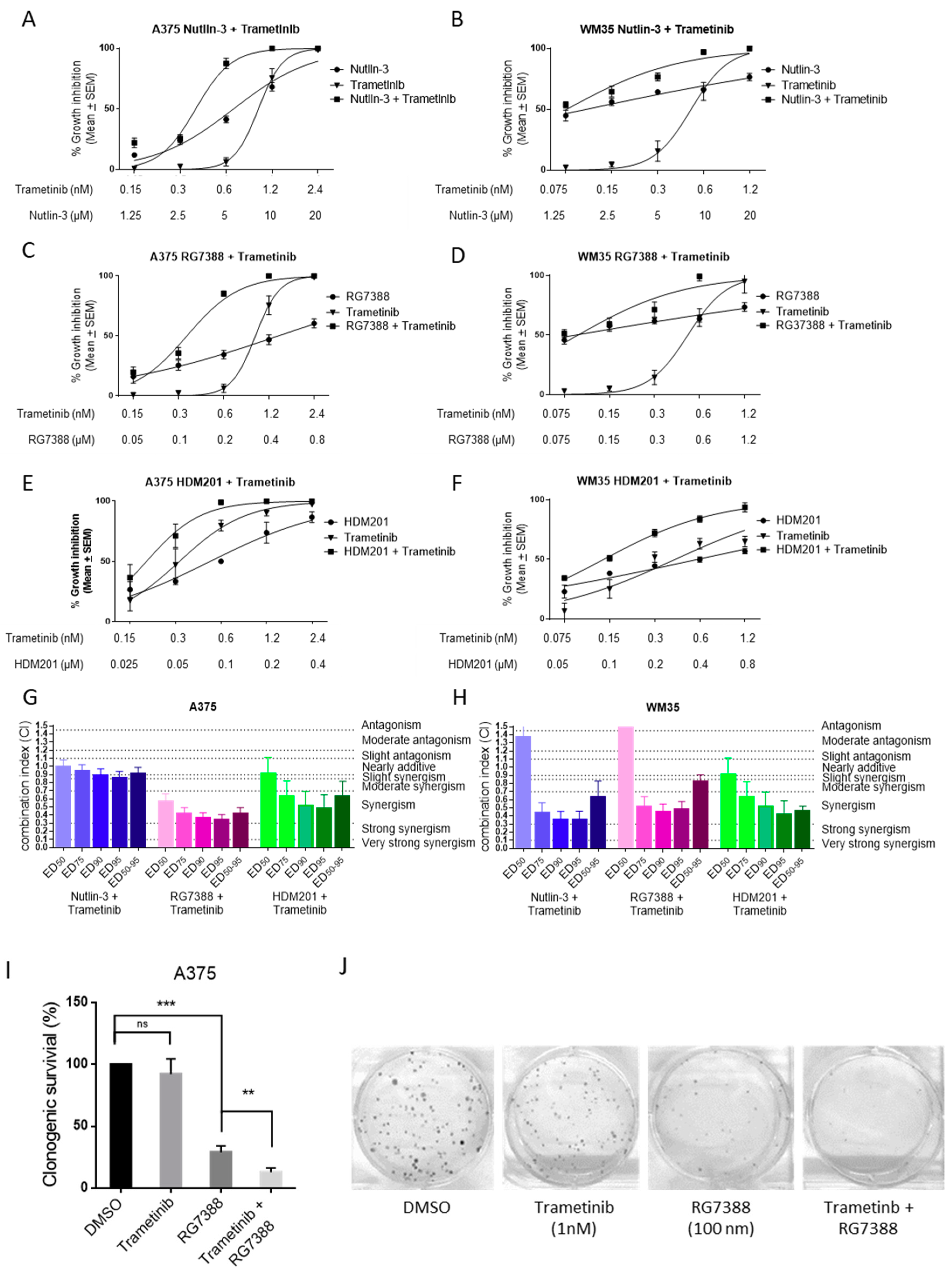
2.1. Combination Treatment of MDM2 Inhibitors and Trametinib in BRAFV600E and p53WT Melanoma Cells
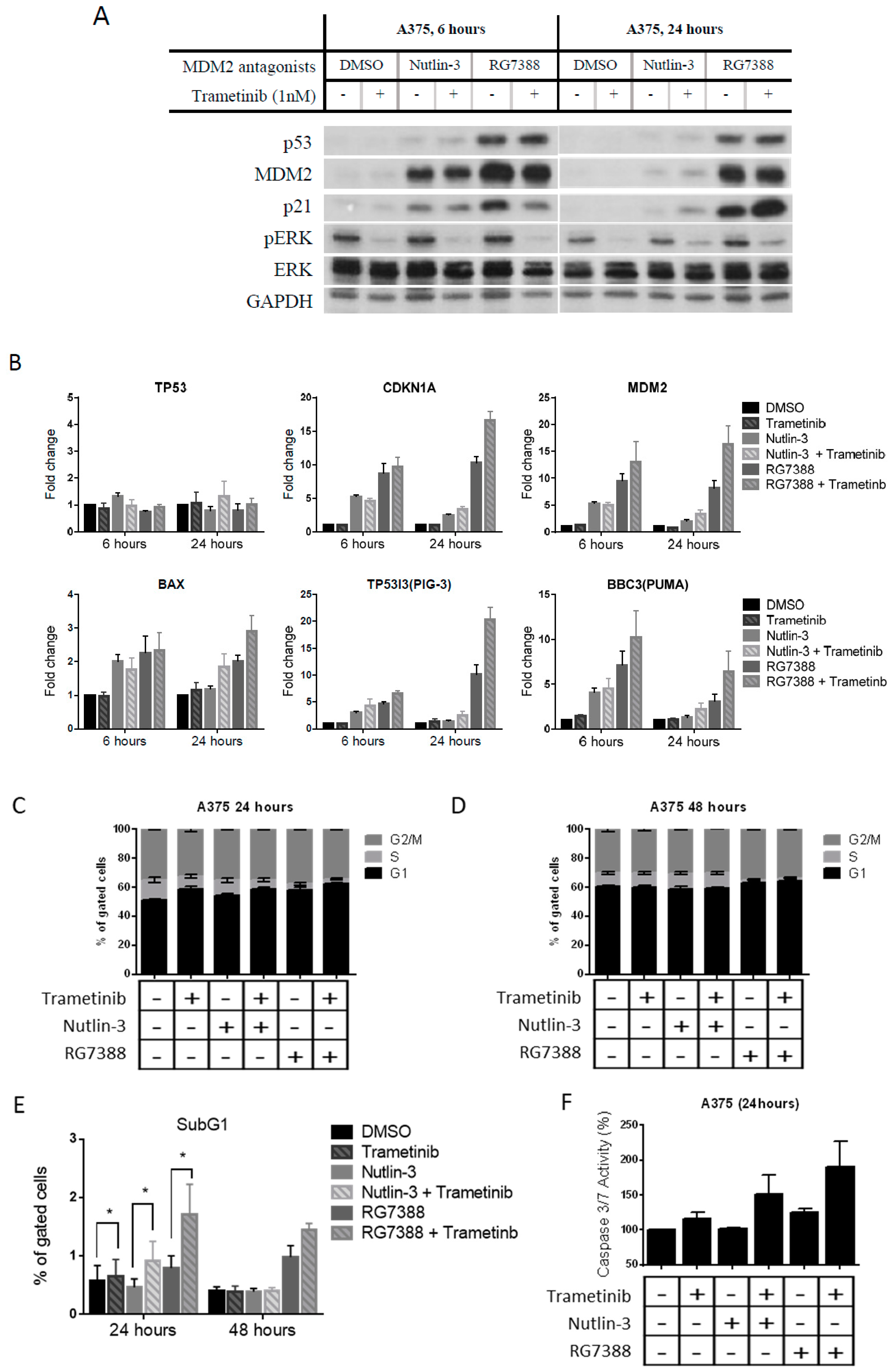
2.2. Combination of Trametinib and MDM2 Inhibitors Increased the Induction of p53 Transcriptional Targets, Resulting in Further Cell Cycle Arrest and Apoptosis
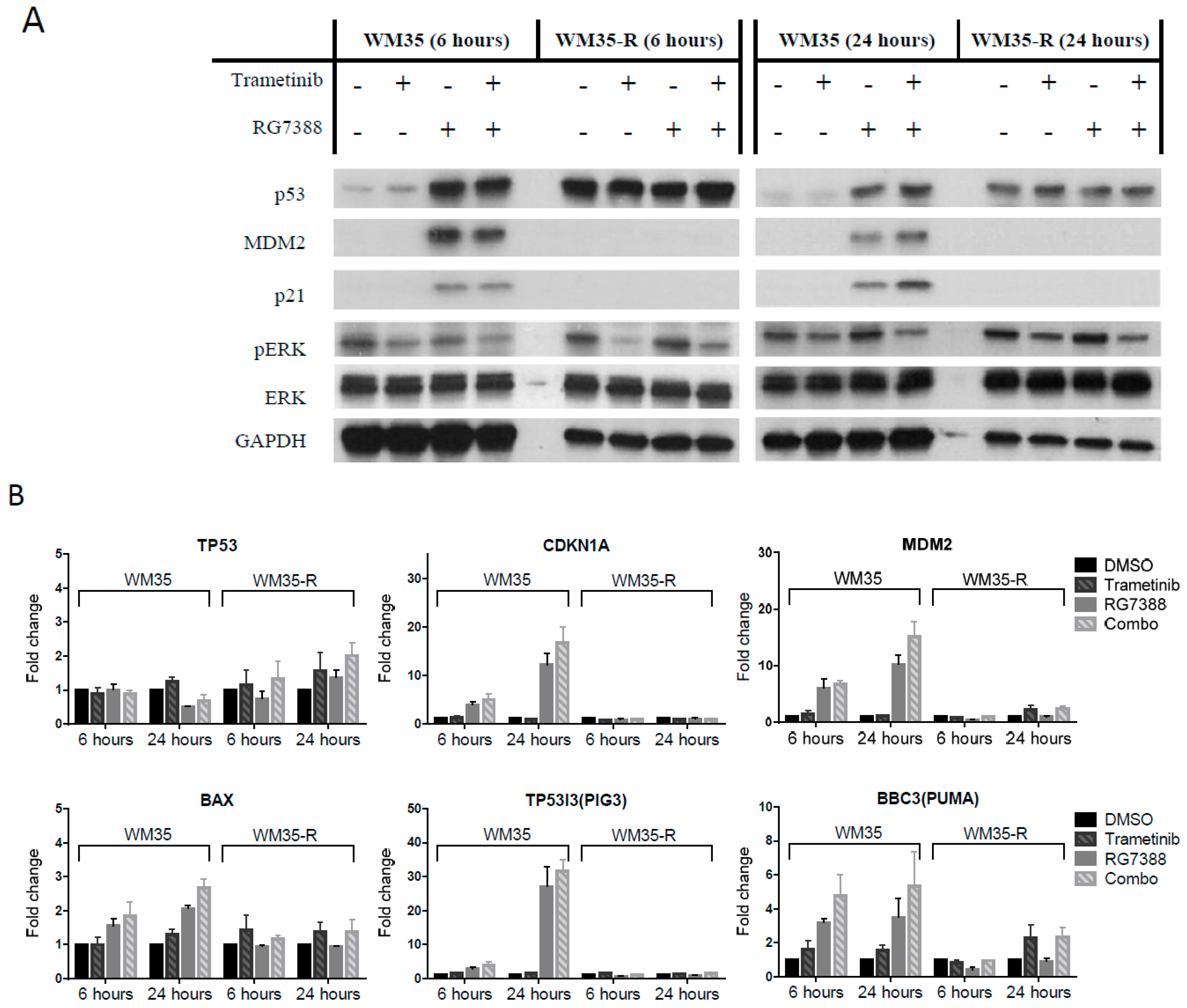
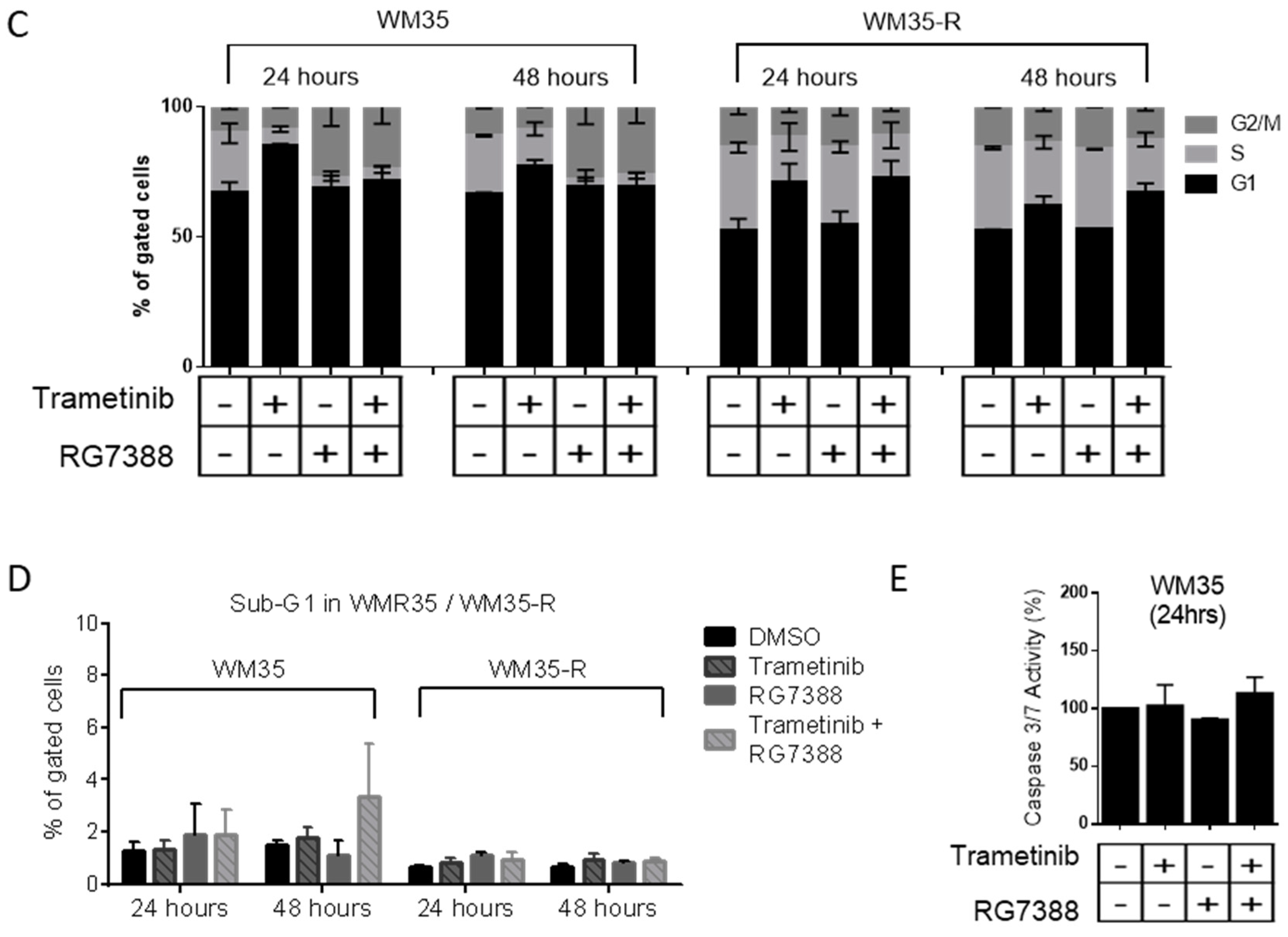
2.3. The Combination Effects for Trametinib and MDM2 Inhibitors in Paired WM35/WM35-R Cell Lines
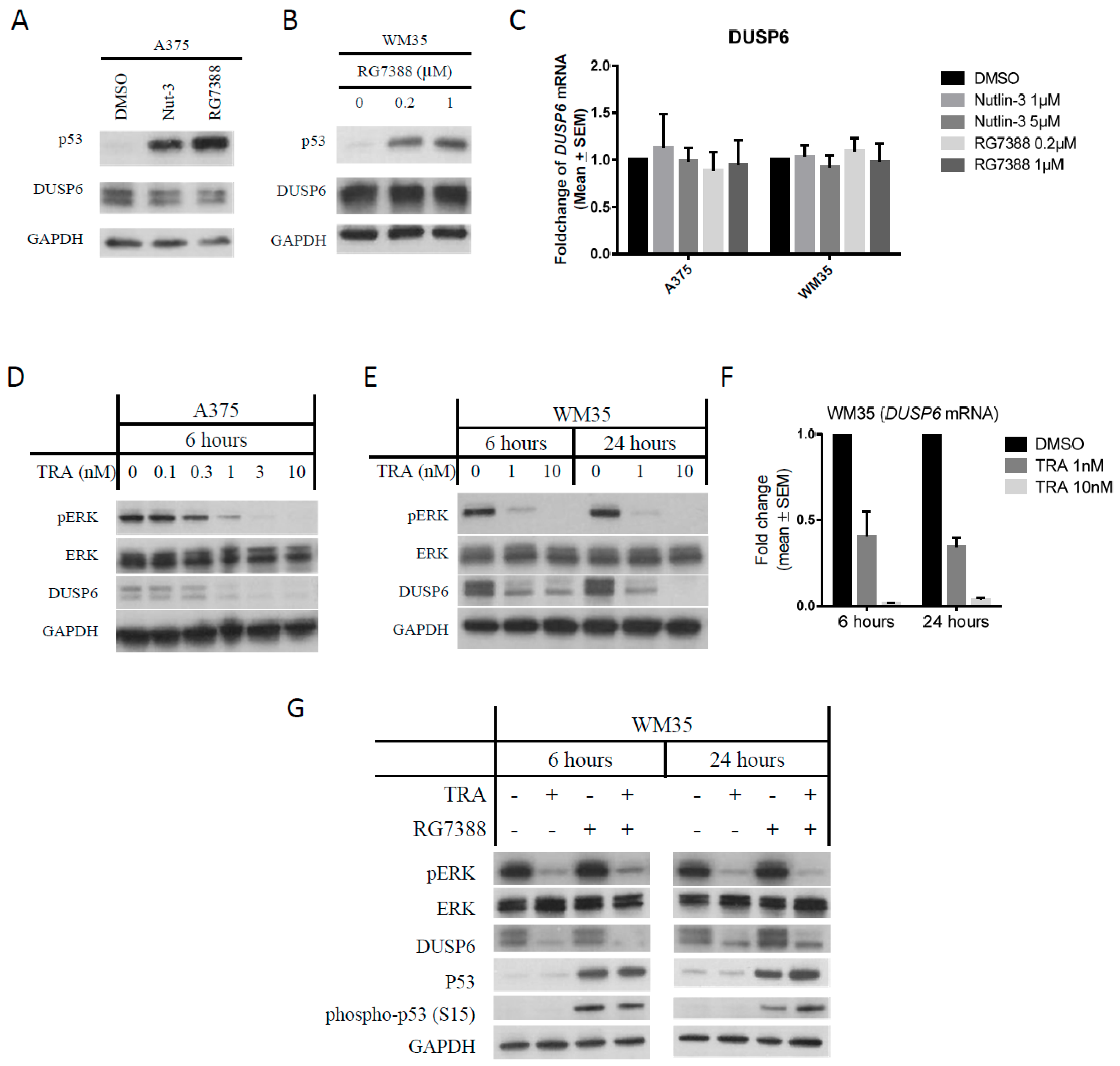
2.4. DUSP6 Regulation by pERK Rather Than p53
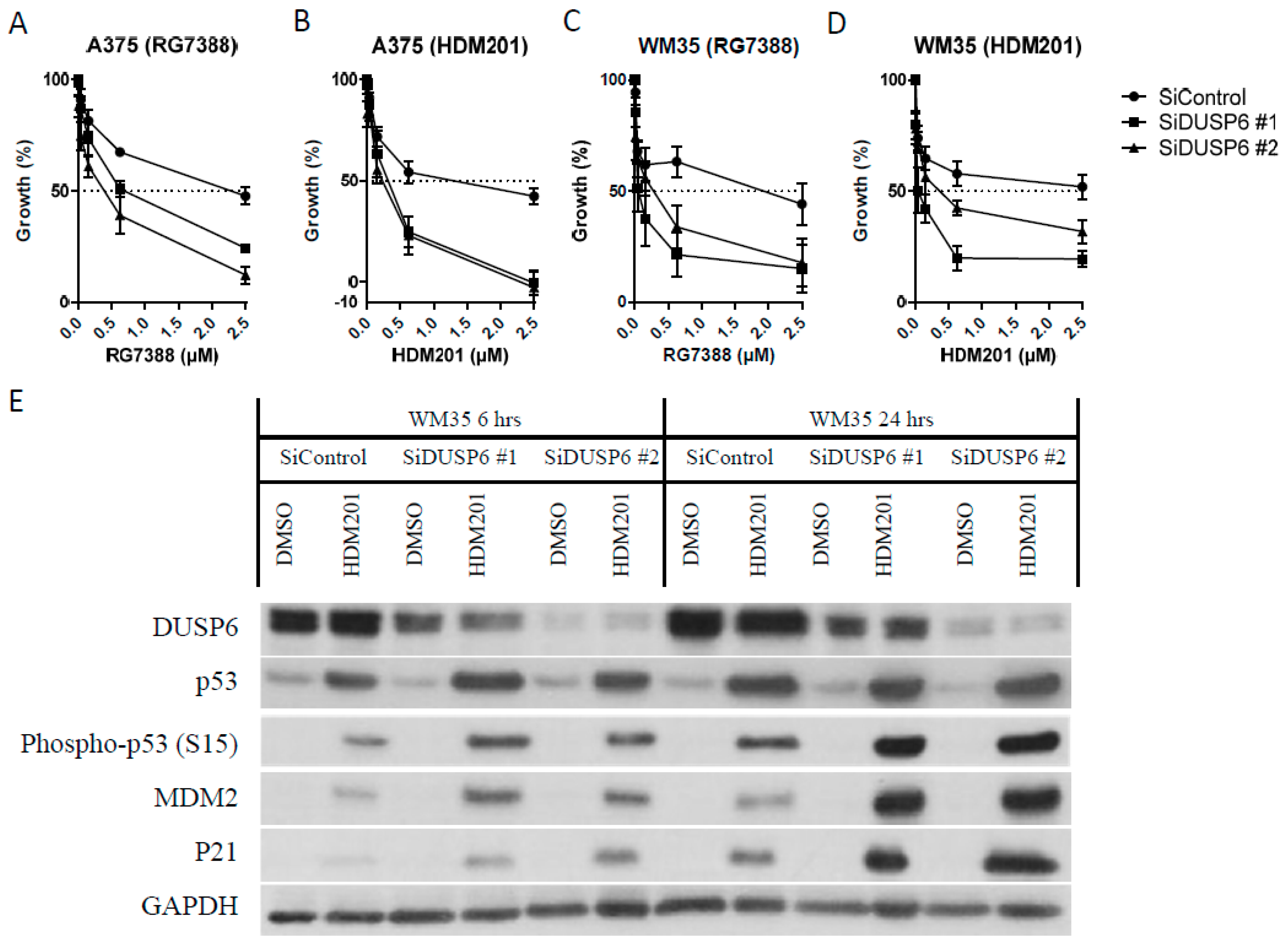
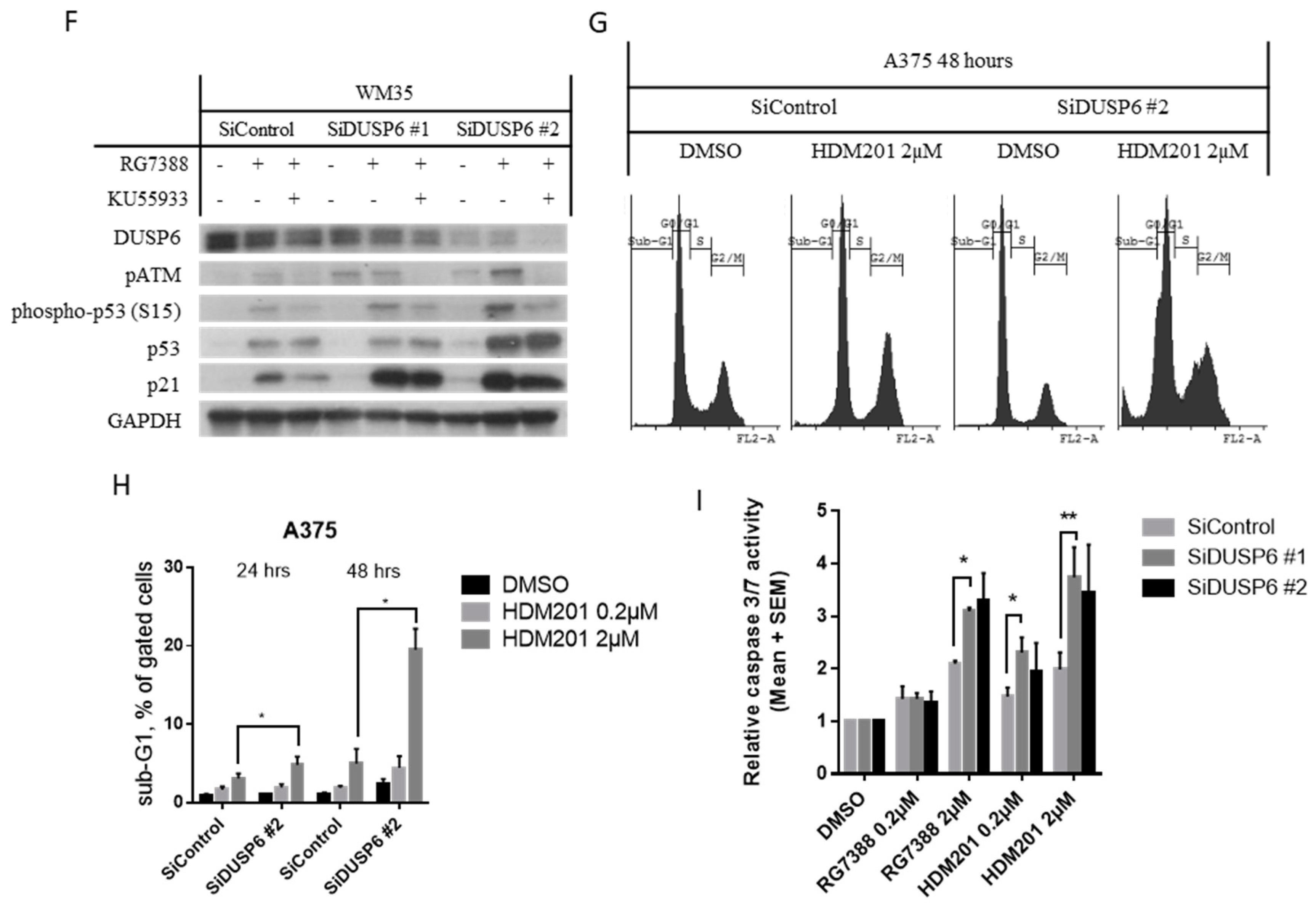
2.5. DUSP6 Suppression by siRNA Promotes the Effect of MDM2 Inhibitors through ATM-Mediated p53 Phosphorylation
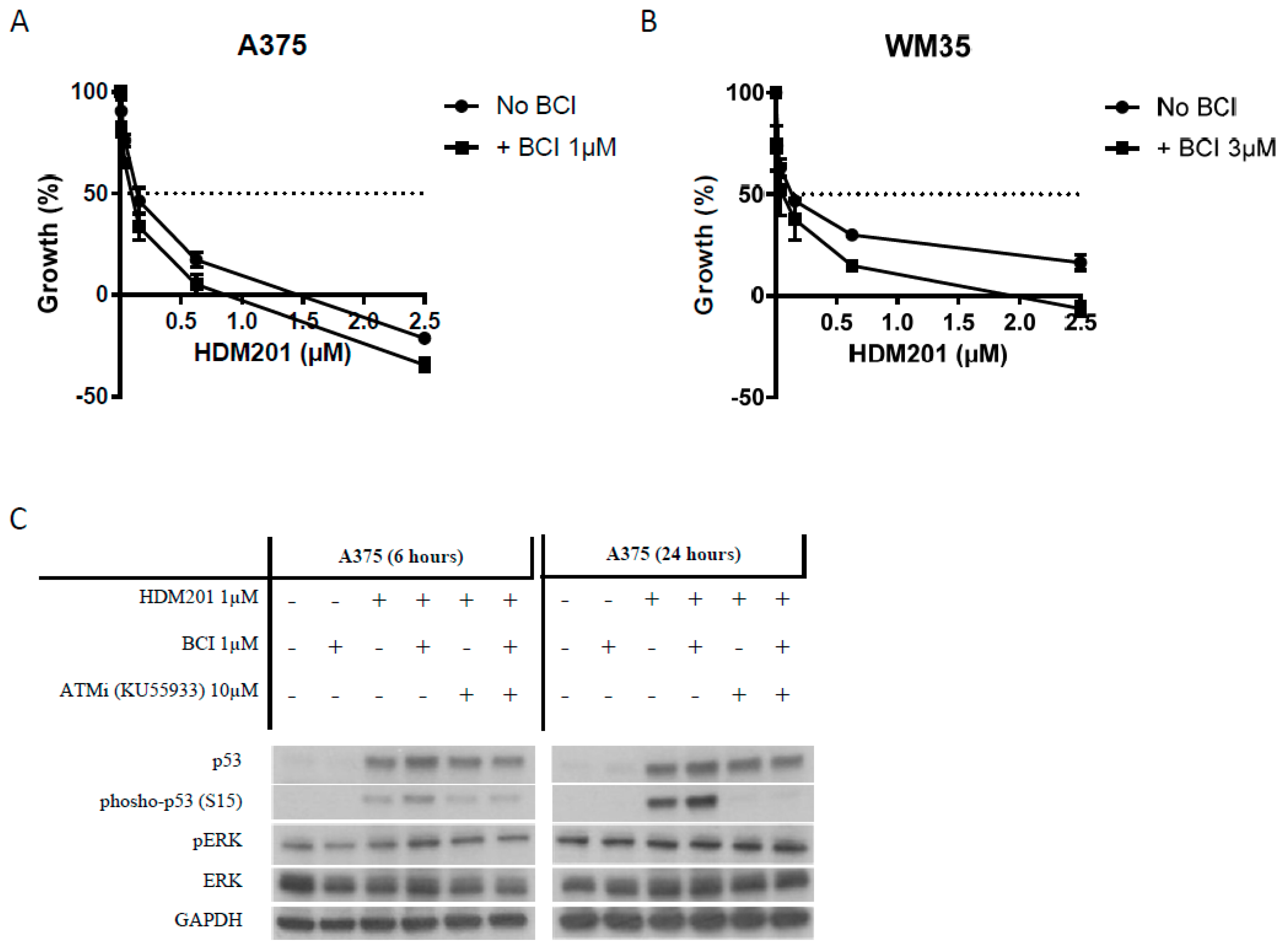
2.6. DUSP6 Enzymatic Inhibitor (BCI) at a Dose without Cytotoxicity Promotes the Anticancer Effect of MDM2 Inhibitors through p53 Phosphorylation
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Growth Inhibition and Clonogenic Survival Assays
4.3. Combination Treatment
4.4. Immunoblotting
4.5. RNA Extraction and qRT-PCR
4.6. Fluorescence-Activated Cell Sorting (FACS)
4.7. Caspase 3/7 Activity Assay
4.8. SiRNAs and Transfection
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demunter, A.; Stas, M.; Degreef, H.; De Wolf-Peeters, C.; van den Oord, J.J. Analysis of N- and K-ras mutations in the distinctive tumor progression phases of melanoma. J. Investig. Dermatol. 2001, 117, 1483–1489. [Google Scholar] [PubMed]
- Chin, L. The genetics of malignant melanoma: Lessons from mouse and man. Nat. Rev. Cancer 2003, 3, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [Green Version]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Miller, P.; Lu, X. Restoring the tumour suppressive function of p53 as a parallel strategy in melanoma therapy. FEBS Lett. 2014, 588, 2616–2621. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.E.; Esfandiari, A.; Ho, Y.H.; Wang, N.; Mahdi, A.K.; Aptullahoglu, E.; Lovat, P.; Lunec, J. Targeting negative regulation of p53 by MDM2 and WIP1 as a therapeutic strategy in cutaneous melanoma. Br. J. Cancer 2018, 118, 495–508. [Google Scholar] [CrossRef]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The genomic landscape of cutaneous melanoma. Pigm. Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Chen, G.; Jukofsky, L.; Geho, D.; Han, S.W.; Birzele, F.; Bader, S.; Himmelein, L.; Cai, J.; Albertyn, Z.; et al. MDM2 antagonist clinical response association with a gene expression signature in acute myeloid leukaemia. Br. J. Haematol. 2015, 171, 432–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Sachweh, M.C.; Drummond, C.J.; Higgins, M.; Campbell, J.; Lain, S. Incompatible effects of p53 and HDAC inhibition on p21 expression and cell cycle progression. Cell Death Dis. 2013, 4, e533. [Google Scholar] [CrossRef]
- Polanski, R.; Noon, A.P.; Blaydes, J.; Phillips, A.; Rubbi, C.P.; Parsons, K.; Vlatković, N.; Boyd, M.T. Senescence induction in renal carcinoma cells by Nutlin-3: A potential therapeutic strategy based on MDM2 antagonism. Cancer Lett. 2014, 353, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.-J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.; Chatterjee, M.; Langenberg, M.H.G.; Lin, C.C.; Suárez, C.; Tai, D.; Cassier, P.; Yamamoto, N.; De Weger, V.A.; Jeay, S.; et al. Dose- and regimen-finding phase I study of NVP-HDM201 in patients (pts) with TP53 wild-type (wt) advanced tumors. Eur. J. Cancer 2016, 69, S128–S129. [Google Scholar] [CrossRef]
- Piya, S.; Kim, J.Y.; Bae, J.; Seol, D.W.; Moon, A.R.; Kim, T.H. DUSP6 is a novel transcriptional target of p53 and regulates p53-mediated apoptosis by modulating expression levels of Bcl-2 family proteins. FEBS Lett. 2012, 586, 4233–4240. [Google Scholar] [CrossRef] [Green Version]
- Bermudez, O.; Jouandin, P.; Rottier, J.; Bourcier, C.; Pagès, G.; Gimond, C. Post-transcriptional regulation of the DUSP6/MKP-3 phosphatase by MEK/ERK signaling and hypoxia. J. Cell. Physiol. 2011, 226, 276–284. [Google Scholar] [CrossRef]
- Marchetti, S.; Gimond, C.; Chambard, J.C.; Touboul, T.; Roux, D.; Pouysségur, J.; Pagès, G. Extracellular signal-regulated kinases phosphorylate mitogen-activated protein kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites critical for its proteasomal degradation. Mol. Cell. Biol. 2005, 25, 854–864. [Google Scholar] [CrossRef]
- Cancer Cell Line Encyclopedia. Available online: https://portals.broadinstitute.org/ccle/page?gene=DUSP6 (accessed on 31 October 2017).
- cBioPortal. Available online: http://www.cbioportal.org/ (accessed on 31 October 2017).
- Bloethner, S.; Chen, B.; Hemminki, K.; Müller-Berghaus, J.; Ugurel, S.; Schadendorf, D.; Kumar, R. Effect of common B-RAF and N-RAS mutations on global gene expression in melanoma cell lines. Carcinogenesis 2005, 26, 1224–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, J.; Greshock, J.; Holbrook, J.D.; Gilmartin, A.; Zhang, X.; McNeil, E.; Conway, T.; Moy, C.; Laquerre, S.; Bachman, K.; et al. Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol. Cancer Ther. 2012, 11, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Regulation of the p53 response and its relationship to cancer. Biochem. J. 2015, 469, 325–346. [Google Scholar] [CrossRef] [PubMed]
- Bagnyukova, T.V.; Restifo, D.; Beeharry, N.; Gabitova, L.; Li, T.; Serebriiskii, I.G.; Golemis, E.A.; Astsaturov, I. DUSP6 regulates drug sensitivity by modulating DNA damage response. Br. J. Cancer 2013, 109, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, G.; Vogt, A.; Bakan, A.; Dai, W.; De Oliveira, P.Q.; Znosko, W.; Smithgall, T.E.; Bahar, I.; Lazo, J.S.; Day, B.W.; et al. Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat. Chem. Biol. 2009, 5, 680–687. [Google Scholar] [CrossRef]
- Shojaee, S.; Caeser, R.; Buchner, M.; Park, E.; Swaminathan, S.; Hurtz, C.; Geng, H.; Chan, L.N.; Klemm, L.; Hofmann, W.-K.; et al. Erk Negative Feedback Control Enables Pre-B Cell Transformation and Represents a Therapeutic Target in Acute Lymphoblastic Leukemia. Cancer Cell 2015, 28, 114–128. [Google Scholar] [CrossRef] [Green Version]
- Korotchenko, V.N.; Saydmohammed, M.; Vollmer, L.L.; Bakan, A.; Sheetz, K.; Debiec, K.T.; Greene, K.A.; Agliori, C.S.; Bahar, I.; Day, B.W.; et al. In vivo structure-activity relationship studies support allosteric targeting of a dual specificity phosphatase. ChemBiochem 2014, 15, 1436–1445. [Google Scholar] [CrossRef]
- She, Q.B.; Chen, N.; Dong, Z. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J. Biol. Chem. 2000, 275, 20444–20449. [Google Scholar] [CrossRef]
- Ji, Z.; Njauw, C.N.; Taylor, M.; Neel, V.; Flaherty, K.T.; Tsao, H. p53 rescue through HDM2 antagonism suppresses melanoma growth and potentiates MEK inhibition. J. Investig. Dermatol. 2012, 132, 356–364. [Google Scholar] [CrossRef]
- Lee, J.T.; Herlyn, M. MEK’ing the most of p53 reactivation therapy in melanoma. J. Investig. Dermatol. 2012, 132, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Phelps, M.; Phillips, A.; Darley, M.; Blaydes, J.P. MEK-ERK signaling controls Hdm2 oncoprotein expression by regulating hdm2 mRNA export to the cytoplasm. J. Biol. Chem. 2005, 280, 16651–16658. [Google Scholar] [CrossRef] [PubMed]
- Siu, L.L.; Italiano, A.; Miller, W.H.; Blay, J.Y.; Gietema, J.A.; Bang, Y.J.; Mileshkin, L.R.; Hirte, H.W.; Reckner, M.; Higgins, B.; et al. Phase 1 dose escalation, food effect, and biomarker study of RG7388, a more potent second-generation MDM2 antagonist, in patients (pts) with solid tumors. J. Clin. Oncol. 2014, 32, 2535. [Google Scholar]
- Infante, J.R.; Fecher, L.A.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef]
- Wei, C.L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.; Weng, Z.; et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Esfandiari, A.; Hawthorne, T.A.; Nakjang, S.; Lunec, J. Chemical Inhibition of Wild-Type p53-Induced Phosphatase 1 (WIP1/PPM1D) by GSK2830371 Potentiates the Sensitivity to MDM2 Inhibitors in a p53-Dependent Manner. Mol. Cancer Ther. 2016, 15, 379–391. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | ED50 | ED75 | ED90 | |||
|---|---|---|---|---|---|---|
| Nutlin-3 | Trametinib | Nutlin-3 | Trametinib | Nutlin-3 | Trametinib | |
| A375 | 1.52 ± 0.05 | 3.43 ± 0.86 | 1.63 ± 0.05 | 3.49 ± 0.79 | 1.75 ± 0.07 | 3.58 ± 0.74 |
| WM35 | 1.66 ± 0.73 | 4.15 ± 1.19 | 7.34 ± 1.45 | 3.93 ± 1.12 | 62.1 ± 14.3 | 3.72 ± 1.05 |
| RG7388 | Trametinib | RG7388 | Trametinib | RG7388 | Trametinib | |
| A375 | 4.84 ± 0.75 | 3.30 ± 0.77 | 18.3 ± 5.94 | 3.31 ± 0.71 | 42.6 ± 20.6 | 3.33 ± 0.66 |
| WM35 | 0.87 ± 0.19 | 2.85 ± 1.12 | 9.24 ± 4.57 | 3.70 ± 0.82 | 61.7 ± 2.37 | 9.93 ± 6.71 |
| HDM201 | Trametinib | HDM201 | Trametinib | HDM201 | Trametinib | |
| A375 | 2.44 ± 0.57 | 2.63 ± 0.48 | 2.97 ± 0.51 | 7.17 ± 2.27 | 3.84 ± 0.76 | 22.2 ± 10.1 |
| WM35 | 3.23 ± 0.58 | 4.48 ± 0.86 | 2.30 ± 0.58 | 9.41 ± 2.06 | 2.41 ± 0.97 | 25.9 ± 9.57 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-E.; Koay, T.S.; Esfandiari, A.; Ho, Y.-H.; Lovat, P.; Lunec, J. ATM Dependent DUSP6 Modulation of p53 Involved in Synergistic Targeting of MAPK and p53 Pathways with Trametinib and MDM2 Inhibitors in Cutaneous Melanoma. Cancers 2019, 11, 3. https://doi.org/10.3390/cancers11010003
Wu C-E, Koay TS, Esfandiari A, Ho Y-H, Lovat P, Lunec J. ATM Dependent DUSP6 Modulation of p53 Involved in Synergistic Targeting of MAPK and p53 Pathways with Trametinib and MDM2 Inhibitors in Cutaneous Melanoma. Cancers. 2019; 11(1):3. https://doi.org/10.3390/cancers11010003
Chicago/Turabian StyleWu, Chiao-En, Tsin Shue Koay, Arman Esfandiari, Yi-Hsuan Ho, Penny Lovat, and John Lunec. 2019. "ATM Dependent DUSP6 Modulation of p53 Involved in Synergistic Targeting of MAPK and p53 Pathways with Trametinib and MDM2 Inhibitors in Cutaneous Melanoma" Cancers 11, no. 1: 3. https://doi.org/10.3390/cancers11010003
APA StyleWu, C.-E., Koay, T. S., Esfandiari, A., Ho, Y.-H., Lovat, P., & Lunec, J. (2019). ATM Dependent DUSP6 Modulation of p53 Involved in Synergistic Targeting of MAPK and p53 Pathways with Trametinib and MDM2 Inhibitors in Cutaneous Melanoma. Cancers, 11(1), 3. https://doi.org/10.3390/cancers11010003