High Frequency of ERBB2 Activating Mutations in Invasive Lobular Breast Carcinoma with Pleomorphic Features


Abstract
:1. Introduction
2. Materials and Methods
2.1. Case Selection
2.2. Immunohistochemistry
2.3. Fluorescent In-Situ Hybridisation (FISH)
2.4. Massive Parallel Sequencing
2.5. Estimation of LOH Status in CDH1
3. Results
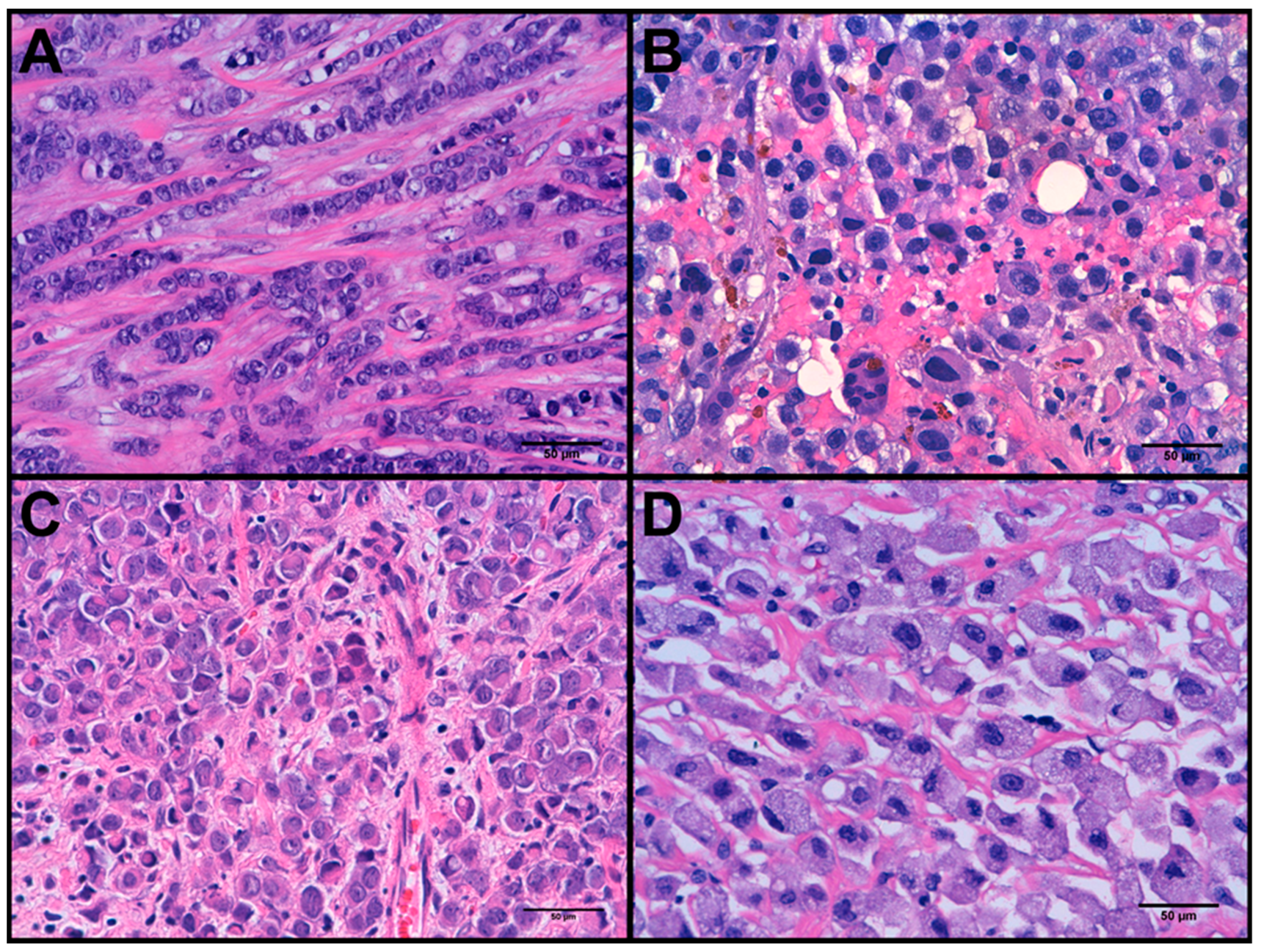
3.1. Clinicopathological Features
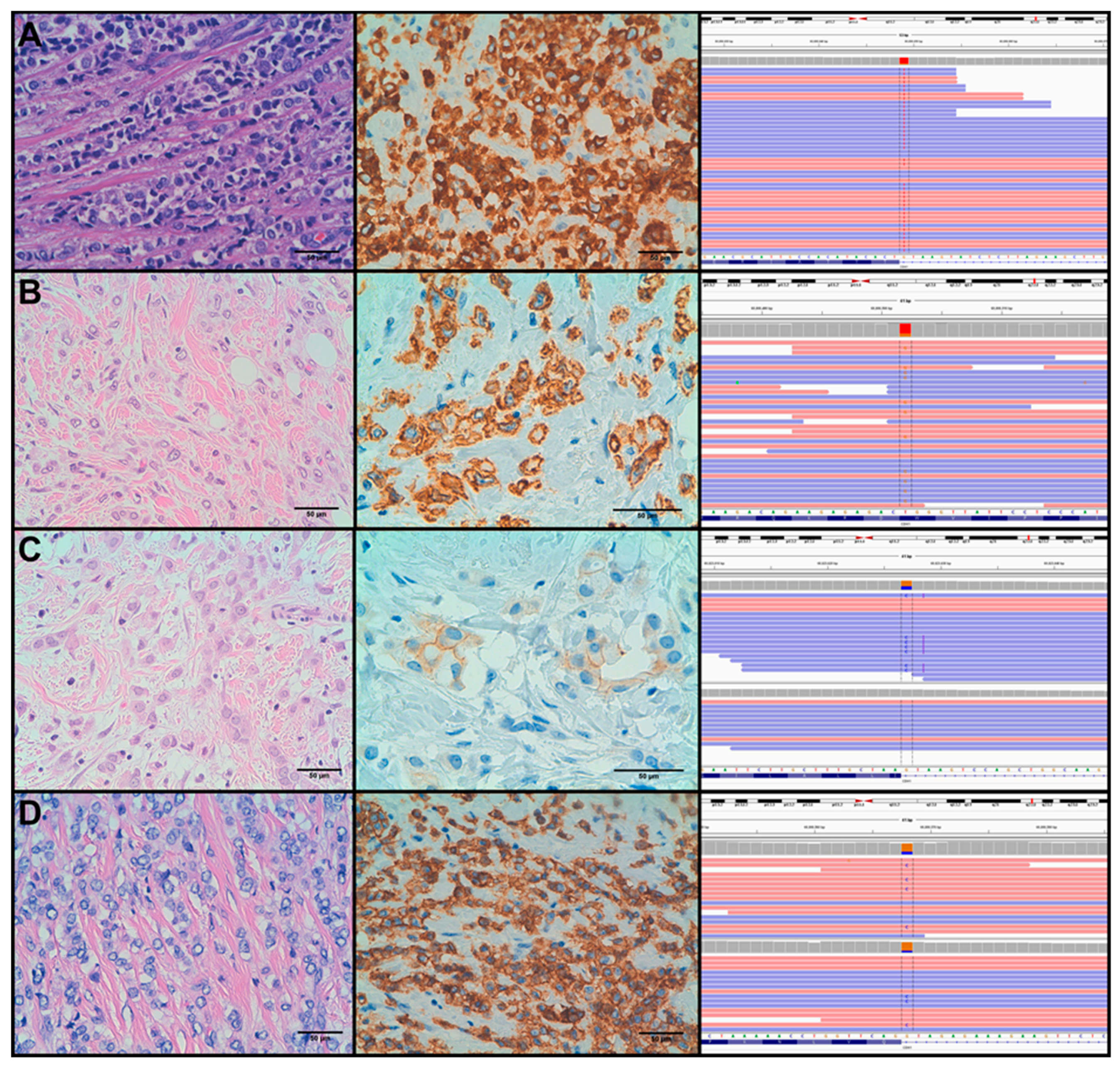
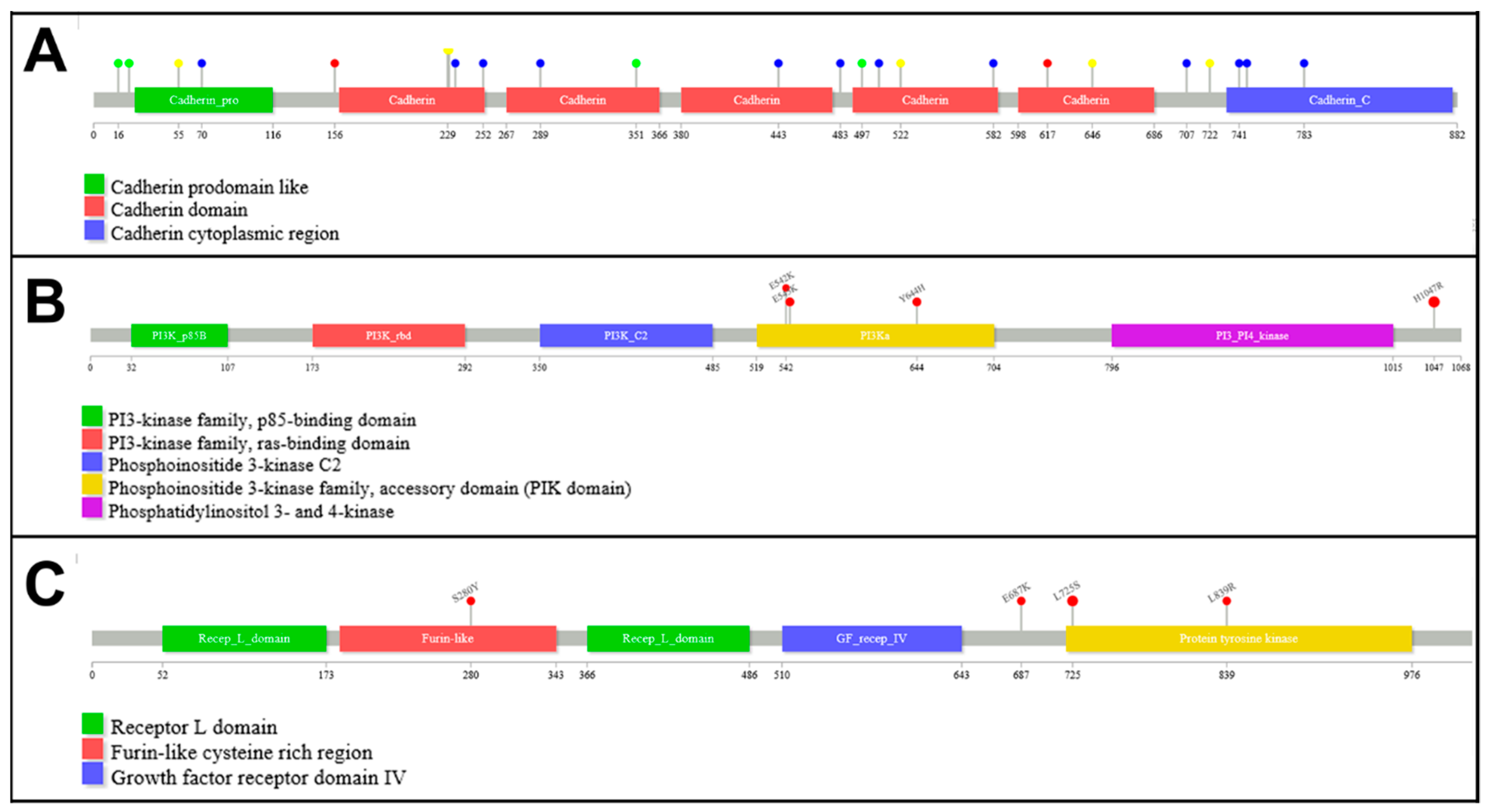
3.2. Mutation Analysis
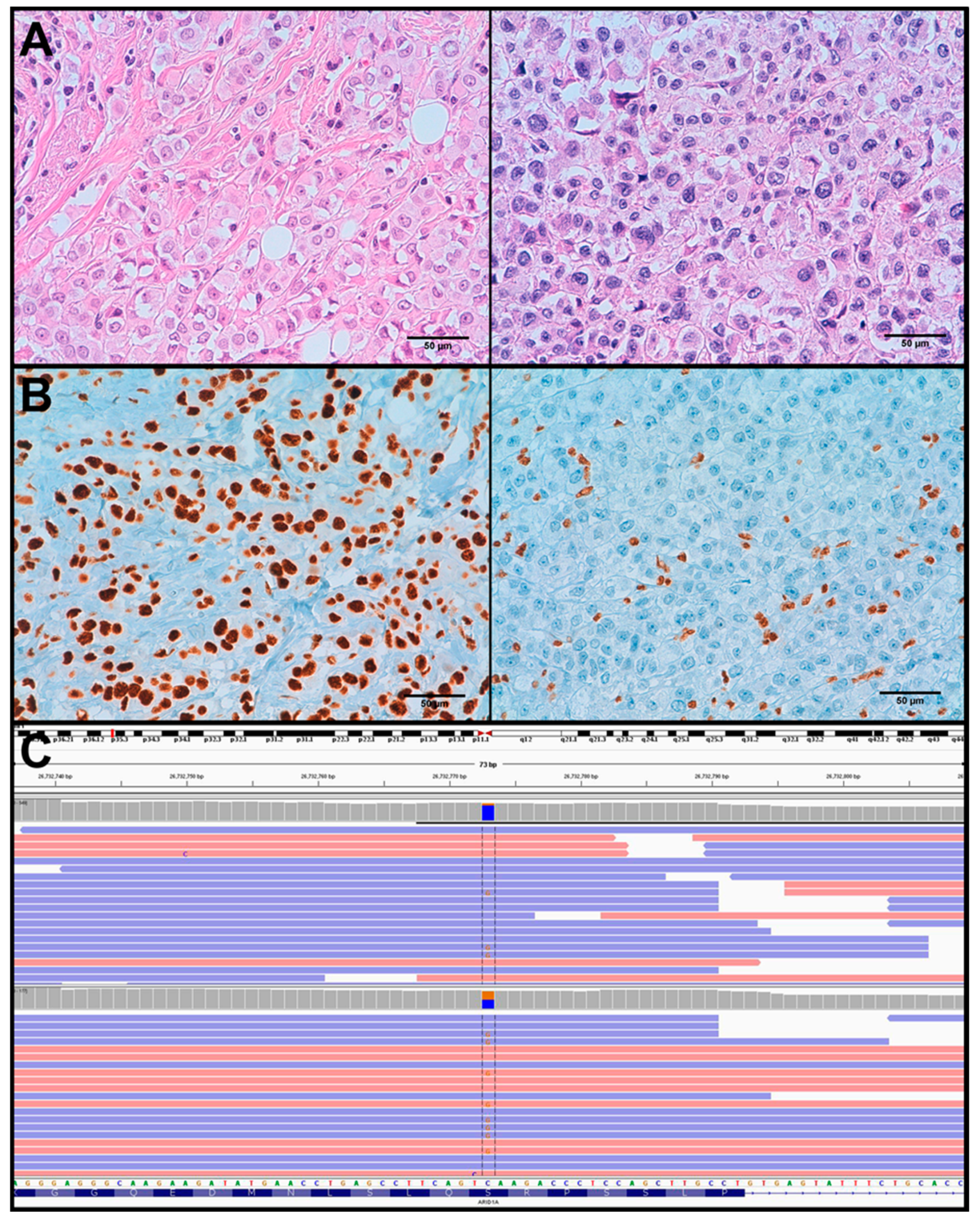
3.3. Molecular Alterations and Histopathological/Immunohistochemical Features
3.4. Molecular Alterations Related to Tumour Progression and Relapse
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakhani, S.R.; Ellis, I.O.; Schnitt, S.J.; Tan, P.H.; van der Vijver, M.J. WHO Classification of Tumours of the Breast, 4th ed.; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- Dixon, J.M.; Anderson, T.J.; Page, D.L.; Lee, D.; Duffy, S.W.; Stewart, H.J. Infiltrating lobular carcinoma of the breast: An evaluation of the incidence and consequence of bilateral disease. Br. J. Surg. 1983, 70, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.; Rosa, M. Pleomorphic lobular carcinoma of the breast: A morphologically and clinically distinct variant of lobular carcinoma. Arch. Pathol. Lab. Med. 2013, 137, 1688–1692. [Google Scholar] [CrossRef] [PubMed]
- Gamallo, C.; Palacios, J.; Suarez, A.; Pizarro, A.; Navarro, P.; Quintanilla, M.; Cano, A. Correlation of E-cadherin expression with differentiation grade and histological type in breast carcinoma. Am. J. Pathol. 1993, 142, 987–993. [Google Scholar]
- Dabbs, D.J.; Schnitt, S.J.; Geyer, F.C.; Weigelt, B.; Baehner, F.L.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Lakhani, S.R.; et al. Lobular neoplasia of the breast revisited with emphasis on the role of E-cadherin immunohistochemistry. Am. J. Surg. Pathol. 2013, 37, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Sarrio, D.; Moreno-Bueno, G.; Hardisson, D.; Sanchez-Estevez, C.; Guo, M.; Herman, J.G.; Gamallo, C.; Esteller, M.; Palacios, J. Epigenetic and genetic alterations of APC and CDH1 genes in lobular breast cancer: Relationships with abnormal E-cadherin and catenin expression and microsatellite instability. Int. J. Cancer 2003, 106, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Desmedt, C.; Zoppoli, G.; Gundem, G.; Pruneri, G.; Larsimont, D.; Fornili, M.; Fumagalli, D.; Brown, D.; Rothe, F.; Vincent, D.; et al. Genomic Characterization of Primary Invasive Lobular Breast Cancer. J. Clin. Oncol. 2016, 34, 1872–1881. [Google Scholar] [CrossRef]
- Sarrio, D.; Moreno-Bueno, G.; Sanchez-Estevez, C.; Banon-Rodriguez, I.; Hernandez-Cortes, G.; Hardisson, D.; Palacios, J. Expression of cadherins and catenins correlates with distinct histologic types of ovarian carcinomas. Hum. Pathol. 2006, 37, 1042–1049. [Google Scholar] [CrossRef]
- Lien, H.C.; Chen, Y.L.; Juang, Y.L.; Jeng, Y.M. Frequent alterations of HER2 through mutation, amplification, or overexpression in pleomorphic lobular carcinoma of the breast. Breast Cancer Res. Treat. 2015, 150, 447–455. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Simpson, P.T.; Jones, C.; Steele, D.; Mackay, A.; Iravani, M.; Fenwick, K.; Valgeirsson, H.; Lambros, M.; Ashworth, A.; et al. Pleomorphic lobular carcinoma of the breast: Role of comprehensive molecular pathology in characterization of an entity. J. Pathol. 2005, 207, 1–13. [Google Scholar] [CrossRef]
- Simpson, P.T.; Reis-Filho, J.S.; Lambros, M.B.; Jones, C.; Steele, D.; Mackay, A.; Iravani, M.; Fenwick, K.; Dexter, T.; Jones, A.; et al. Molecular profiling pleomorphic lobular carcinomas of the breast: Evidence for a common molecular genetic pathway with classic lobular carcinomas. J. Pathol. 2008, 215, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Dieci, M.V.; Smutna, V.; Scott, V.; Yin, G.; Xu, R.; Vielh, P.; Mathieu, M.C.; Vicier, C.; Laporte, M.; Drusch, F.; et al. Whole exome sequencing of rare aggressive breast cancer histologies. Breast Cancer Res. Treat. 2016, 156, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Ward, B.M.; Yu, J.; Matthew-Onabanjo, A.N.; Janusis, J.; Hsieh, C.C.; Tomaszewicz, K.; Hutchinson, L.; Zhu, L.J.; Kandil, D.; et al. IRS2 mutations linked to invasion in pleomorphic invasive lobular carcinoma. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, A.C.; Hammond, M.E.H.; Allison, K.H.; Harvey, B.E.; Mangu, P.B.; Bartlett, J.M.S.; Bilous, M.; Ellis, I.O.; Fitzgibbons, P.; Hanna, W.; et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J. Clin. Oncol. 2018, 36, 2105–2122. [Google Scholar] [CrossRef] [PubMed]
- Sarrio, D.; Perez-Mies, B.; Hardisson, D.; Moreno-Bueno, G.; Suarez, A.; Cano, A.; Martin-Perez, J.; Gamallo, C.; Palacios, J. Cytoplasmic localization of p120ctn and E-cadherin loss characterize lobular breast carcinoma from preinvasive to metastatic lesions. Oncogene 2004, 23, 3272–3283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curigliano, G.; Burstein, H.J.; Winer, E.P.; Gnant, M.; Dubsky, P.; Loibl, S.; Colleoni, M.; Regan, M.M.; Piccart-Gebhart, M.; Senn, H.J.; et al. De-escalating and escalating treatments for early-stage breast cancer: The St. Gallen International Expert Consensus Conference on the Primary Therapy of Early Breast Cancer 2017. Ann. Oncol. 2017, 28, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, P.; Disalvatore, D.; Rotmensz, N.; Curigliano, G.; Colleoni, M.; Dellapasqua, S.; Pruneri, G.; Mastropasqua, M.G.; Luini, A.; Bassi, F.; et al. Proposed new clinicopathological surrogate definitions of luminal A and luminal B (HER2-negative) intrinsic breast cancer subtypes. Breast Cancer Res. 2014, 16, R65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa-Rosa, J.M.; Caniego-Casas, T.; Leskela, S.; Munoz, G.; Del Castillo, F.; Garrido, P.; Palacios, J. Modified SureSelect(QXT) Target Enrichment Protocol for Illumina Multiplexed Sequencing of FFPE Samples. Biol. Proced. Online 2018, 20, 19. [Google Scholar] [CrossRef]
- Shigemizu, D.; Momozawa, Y.; Abe, T.; Morizono, T.; Boroevich, K.A.; Takata, S.; Ashikawa, K.; Kubo, M.; Tsunoda, T. Performance comparison of four commercial human whole-exome capture platforms. Sci. Rep. 2015, 5, 12742. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lupat, R.; Amarasinghe, K.C.; Thompson, E.R.; Doyle, M.A.; Ryland, G.L.; Tothill, R.W.; Halgamuge, S.K.; Campbell, I.G.; Gorringe, K.L. CONTRA: Copy number analysis for targeted resequencing. Bioinformatics 2012, 28, 1307–1313. [Google Scholar] [CrossRef]
- Pena-Jaimes, L.; Gonzalez-Garcia, I.; Reguero-Callejas, M.E.; Pinilla-Pagnon, I.; Perez-Mies, B.; Albarran-Artahona, V.; Martinez-Janez, N.; Rosa-Rosa, J.M.; Palacios, J. Pleomorphic lobular carcinoma of the breast with osteoclast-like giant cells: A case report and review of the literature. Diagn. Pathol. 2018, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Michaut, M.; Chin, S.F.; Majewski, I.; Severson, T.M.; Bismeijer, T.; de Koning, L.; Peeters, J.K.; Schouten, P.C.; Rueda, O.M.; Bosma, A.J.; et al. Integration of genomic, transcriptomic and proteomic data identifies two biologically distinct subtypes of invasive lobular breast cancer. Sci. Rep. 2016, 6, 18517. [Google Scholar] [CrossRef] [Green Version]
- Palacios, J.; Sarrio, D.; Garcia-Macias, M.C.; Bryant, B.; Sobel, M.E.; Merino, M.J. Frequent E-cadherin gene inactivation by loss of heterozygosity in pleomorphic lobular carcinoma of the breast. Mod. Pathol. 2003, 16, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Patel, A.; Powe, D.G.; Benhasouna, A.; Green, A.R.; Lambros, M.B.; Reis-Filho, J.S.; Ellis, I.O. Clinical and biological significance of E-cadherin protein expression in invasive lobular carcinoma of the breast. Am. J. Surg. Pathol. 2010, 34, 1472–1479. [Google Scholar] [CrossRef]
- Deniziaut, G.; Tille, J.C.; Bidard, F.C.; Vacher, S.; Schnitzler, A.; Chemlali, W.; Tremoulet, L.; Fuhrmann, L.; Cottu, P.; Rouzier, R.; et al. ERBB2 mutations associated with solid variant of high-grade invasive lobular breast carcinomas. Oncotarget 2016, 7, 73337–73346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidard, F.C.; Ng, C.K.; Cottu, P.; Piscuoglio, S.; Escalup, L.; Sakr, R.A.; Reyal, F.; Mariani, P.; Lim, R.; Wang, L.; et al. Response to dual HER2 blockade in a patient with HER3-mutant metastatic breast cancer. Ann. Oncol. 2015, 26, 1704–1709. [Google Scholar] [CrossRef]
- Li, G.; Wang, X.; Hibshoosh, H.; Jin, C.; Halmos, B. Modulation of ErbB2 blockade in ErbB2-positive cancers: The role of ErbB2 Mutations and PHLDA1. PLoS ONE 2014, 9, e106349. [Google Scholar] [CrossRef]
- Bleeker, F.E.; Felicioni, L.; Buttitta, F.; Lamba, S.; Cardone, L.; Rodolfo, M.; Scarpa, A.; Leenstra, S.; Frattini, M.; Barbareschi, M.; et al. AKT1(E17K) in human solid tumours. Oncogene 2008, 27, 5648–5650. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Christin, J.R.; Wang, C.; Ge, K.; Oktay, M.H.; Guo, W. Mammary-Stem-Cell-Based Somatic Mouse Models Reveal Breast Cancer Drivers Causing Cell Fate Dysregulation. Cell Rep. 2016, 16, 3146–3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa-Rosa, J.M.; Leskela, S.; Cristobal-Lana, E.; Santon, A.; Lopez-Garcia, M.A.; Munoz, G.; Perez-Mies, B.; Biscuola, M.; Prat, J.; Esther, O.E.; et al. Molecular genetic heterogeneity in undifferentiated endometrial carcinomas. Mod. Pathol. 2016, 29, 1594. [Google Scholar] [CrossRef] [PubMed]
- Ahlin, C.; Lundgren, C.; Embretsen-Varro, E.; Jirstrom, K.; Blomqvist, C.; Fjallskog, M. High expression of cyclin D1 is associated to high proliferation rate and increased risk of mortality in women with ER-positive but not in ER-negative breast cancers. Breast Cancer Res. Treat. 2017, 164, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Dossus, L.; Benusiglio, P.R. Lobular breast cancer: Incidence and genetic and non-genetic risk factors. Breast Cancer Res. 2015, 17, 37. [Google Scholar] [CrossRef]
- Armes, J.E.; Egan, A.J.; Southey, M.C.; Dite, G.S.; McCredie, M.R.; Giles, G.G.; Hopper, J.L.; Venter, D.J. The histologic phenotypes of breast carcinoma occurring before age 40 years in women with and without BRCA1 or BRCA2 germline mutations: A population-based study. Cancer 1998, 83, 2335–2345. [Google Scholar] [CrossRef]
- Hernandez, L.; Wilkerson, P.M.; Lambros, M.B.; Campion-Flora, A.; Rodrigues, D.N.; Gauthier, A.; Cabral, C.; Pawar, V.; Mackay, A.; A’Hern, R.; et al. Genomic and mutational profiling of ductal carcinomas in situ and matched adjacent invasive breast cancers reveals intra-tumour genetic heterogeneity and clonal selection. J. Pathol. 2012, 227, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonleben, F.; Qiu, W.; Ciau, N.T.; Ho, D.J.; Li, X.; Allendorf, J.D.; Remotti, H.E.; Su, G.H. PIK3CA mutations in intraductal papillary mucinous neoplasm/carcinoma of the pancreas. Clin. Cancer Res. 2006, 12, 3851–3855. [Google Scholar] [CrossRef]
- Bellanger, A.; Donini, C.F.; Vendrell, J.A.; Lavaud, J.; Machuca-Gayet, I.; Ruel, M.; Vollaire, J.; Grisard, E.; Gyorffy, B.; Bieche, I.; et al. The critical role of the ZNF217 oncogene in promoting breast cancer metastasis to the bone. J. Pathol. 2017, 242, 73–89. [Google Scholar] [CrossRef]
- Maatta, K.; Rantapero, T.; Lindstrom, A.; Nykter, M.; Kankuri-Tammilehto, M.; Laasanen, S.L.; Schleutker, J. Whole-exome sequencing of Finnish hereditary breast cancer families. Eur. J. Hum. Genet. 2016, 25, 85–93. [Google Scholar] [CrossRef]
- Xie, Y.; Li, G.; Chen, M.; Guo, X.; Tang, L.; Luo, X.; Wang, S.; Yi, W.; Dai, L.; Wang, J. Mutation screening of 10 cancer susceptibility genes in unselected breast cancer patients. Clin. Genet. 2017. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age at Diagnosis (Years) | #Cases | Receptor Status | #Cases |
|---|---|---|---|
| <35 | 0 | ER+ | 20 |
| 35–49 | 4 | ER− | 7 |
| 50–69 | 13 | ||
| 70+ | 10 | PgR+ | 16 |
| Tumour size | PgR− | 11 | |
| N/A | 2 | ||
| <1 cm | 1 | AR+ | 22 |
| 1–2 cm | 7 | AR− | 5 |
| 2–5 cm | 15 | Ki67 | |
| >5 cm | 2 | <14 | 20 |
| Lymph node status | 14–30 | 6 | |
| N0 | 11 | 30+ | 1 |
| N1 | 11 | HER2 | |
| N3 | 4 | 0/+/++ | 26 |
| NX | 1 | +++ | 1 |
| Tumour grade | E-cadherin status | ||
| G2 | 17 | - | 23 |
| G3 | 10 | aberrant | 4 |
| Subtype | |||
| Luminal A | 17 | ||
| Luminal B | 5 | ||
| Triple Negative | 4 | ||
| HER2 enriched | 1 |
| Gene | Number of Mutations Found in Current Study | %Cases Mutated | ||||
|---|---|---|---|---|---|---|
| Current Study (PLC, n = 27) | Zhu et al. [14] (PLC, n = 17) | Desmedt et al. [8] (ILC, n = 413) | Michaut et al. [24] (ILC, n = 144) | Ciriello et al. [1] (ILC, n = 127) | ||
| CDH1 | 25 | 89% | 59% | 65% | 43% | 65% |
| PIK3CA | 12 | 33% | 53% | 43% | 35% | 48% |
| ERBB2 | 7 | 26% | 18% | 5% | 4% | 4% |
| ARID1B | 6 | 22% | - | 0% | 5% | 6% |
| KMT2C | 5 | 19% | 35% | 8% | 10% | 7% |
| MAP3K1 | 5 | 19% | 35% | 5% | 5% | 6% |
| TP53 | 5 | 19% | 12% | 7% | 4% | 8% |
| ARID1A | 4 | 15% | 6% | 6% | 7% | 17% |
| CCND1_amp | 3 | 11% | 12% | 38% | 15% | 17% |
| AKT1 | 2 | 7% | 6% | 4% | 5% | 2% |
| FGFR1_amp | 2 | 7% | - | 25% | 8% | 9% |
| GATA3 | 2 | 7% | 6% | 7% | 5% | 5% |
| NF1 | 2 | 7% | 23% | 0% | 4% | 4% |
| TBX3 | 2 | 7% | 23% | 13% | 8% | 9% |
| ERBB2_amp | 1 | 4% | 6% | 0% | 4% | 7% |
| PIK3CA_amp | 1 | 4% | - | 0% | 1% | 2% |
| BRCA2 | 1 | 4% | 0% | 2% | 4% | - |
| CASP8 | 1 | 4% | - | 0% | 1% | 1% |
| NCOR1 | 1 | 4% | 23% | 0% | 7% | 6% |
| PGAP3 | 1 | 4% | 12% | 0% | 0% | - |
| RB1 | 1 | 4% | 0% | 0% | 3% | 6% |
| Gene | Mutation | Present Study | Available Studies | |||
|---|---|---|---|---|---|---|
| Cases | Frequency | Cases | Frequency | References | ||
| ERBB2 | p.L755S | 4 | 57.1% | 24 | 33.3% | [1,8,24,27] |
| p.L869R | 1 | 14.3% | 4 | 5.6% | [8] | |
| p.S310Y | 1 | 14.3% | 2 | 2.8% | [27] | |
| p.E717K | 1 | 14.3% | - | - | - | |
| p.V777L | 10 | 13.9% | [1,8,14,24] | |||
| p.D769Y | 5 | 6.9% | [8,24] | |||
| p.S310F | 4 | 5.6% | [8,11,24] | |||
| p.A775_G776insYVMA | 3 | 4.2% | [24] | |||
| p.L755_T759del | 3 | 4.2% | [10,24] | |||
| p.I767M | 2 | 2.8% | [24,27] | |||
| p.R678Q | 2 | 2.8% | [1,24] | |||
| c.1647-2A > G | 1 | 1.4% | [24] | |||
| c.2923_2923delG | 1 | 1.4% | [24] | |||
| p.A771V | 1 | 1.4% | [10] | |||
| p.A775V | 1 | 1.4% | [10] | |||
| p.E1021K | 1 | 1.4% | [8] | |||
| p.L755M | 1 | 1.4% | [1] | |||
| p.L755W | 1 | 1.4% | [1] | |||
| p.R434Q | 1 | 1.4% | [14] | |||
| p.R978C | 1 | 1.4% | [8] | |||
| p.S305C | 1 | 1.4% | [1] | |||
| p.T791I | 1 | 1.4% | [10] | |||
| p.V697L | 1 | 1.4% | [8] | |||
| p.V842I | 1 | 1.4% | [24] | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosa-Rosa, J.M.; Caniego-Casas, T.; Leskela, S.; Cristobal, E.; González-Martínez, S.; Moreno-Moreno, E.; López-Miranda, E.; Holgado, E.; Pérez-Mies, B.; Garrido, P.; et al. High Frequency of ERBB2 Activating Mutations in Invasive Lobular Breast Carcinoma with Pleomorphic Features. Cancers 2019, 11, 74. https://doi.org/10.3390/cancers11010074
Rosa-Rosa JM, Caniego-Casas T, Leskela S, Cristobal E, González-Martínez S, Moreno-Moreno E, López-Miranda E, Holgado E, Pérez-Mies B, Garrido P, et al. High Frequency of ERBB2 Activating Mutations in Invasive Lobular Breast Carcinoma with Pleomorphic Features. Cancers. 2019; 11(1):74. https://doi.org/10.3390/cancers11010074
Chicago/Turabian StyleRosa-Rosa, Juan Manuel, Tamara Caniego-Casas, Susanna Leskela, Eva Cristobal, Silvia González-Martínez, Esther Moreno-Moreno, Elena López-Miranda, Esther Holgado, Belén Pérez-Mies, Pilar Garrido, and et al. 2019. "High Frequency of ERBB2 Activating Mutations in Invasive Lobular Breast Carcinoma with Pleomorphic Features" Cancers 11, no. 1: 74. https://doi.org/10.3390/cancers11010074
APA StyleRosa-Rosa, J. M., Caniego-Casas, T., Leskela, S., Cristobal, E., González-Martínez, S., Moreno-Moreno, E., López-Miranda, E., Holgado, E., Pérez-Mies, B., Garrido, P., & Palacios, J. (2019). High Frequency of ERBB2 Activating Mutations in Invasive Lobular Breast Carcinoma with Pleomorphic Features. Cancers, 11(1), 74. https://doi.org/10.3390/cancers11010074