Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma


Abstract
:1. Pathological and Genetic Features of Glioblastoma
2. Dysregulated Metabolic Pathways in Glioblastoma
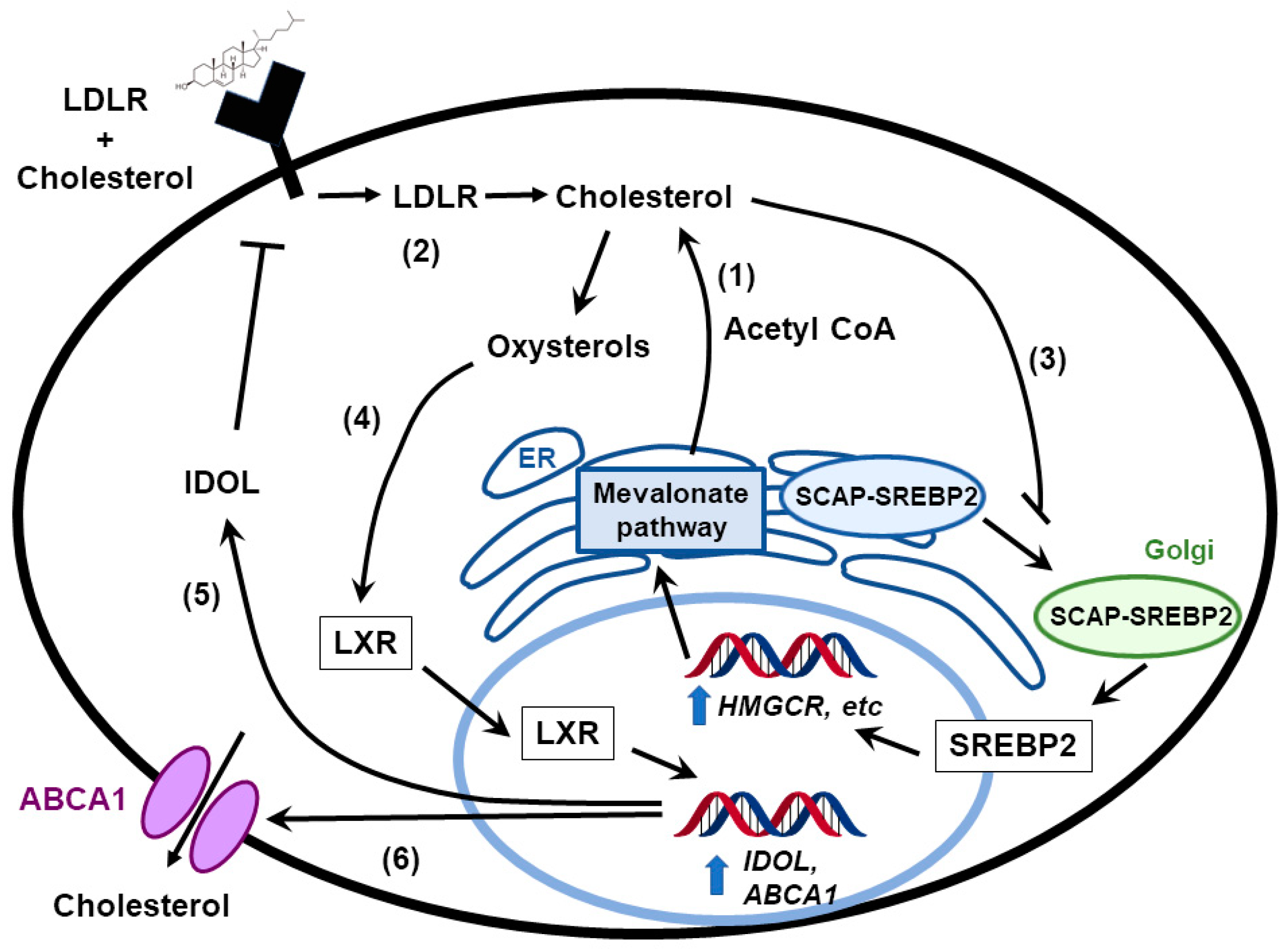
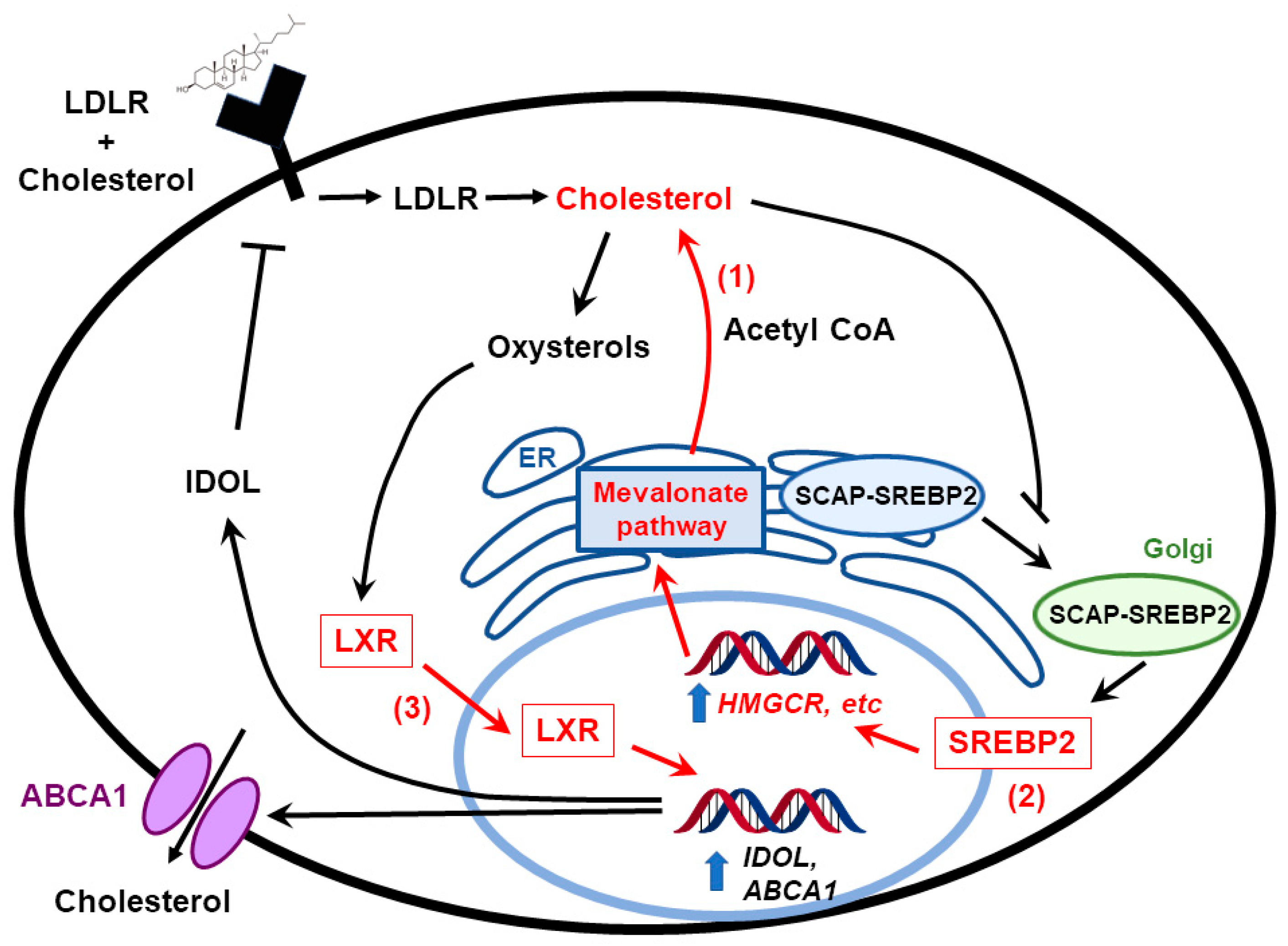
3. Feedback Pathways in Cholesterol Biosynthesis
4. Cholesterol in the Normal Brain
5. Cholesterol Metabolism in Embryonic vs. Adult Brain
6. Cholesterol Excretion from the Brain
7. Cholesterol Metabolism in the Liver vs. Brain
8. Cholesterol Metabolism Pathways Are Altered in Brain Tumors
9. Targeting Cholesterol Metabolism as a Glioblastoma Therapy
10. Cholesterol and its Derivatives as Anti-Glioblastoma Agents
11. Cholesterol-Based Intracerebral Delivery of Chemotherapeutics in Brain Tumors
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed]
- Alphandery, E. Glioblastoma Treatments: An Account of Recent Industrial Developments. Front. Pharmacol. 2018, 9, 879. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Prados, M.D.; Byron, S.A.; Tran, N.L.; Phillips, J.J.; Molinaro, A.M.; Ligon, K.L.; Wen, P.Y.; Kuhn, J.G.; Mellinghoff, I.K.; de Groot, J.F.; et al. Toward precision medicine in glioblastoma: The promise and the challenges. Neuro Oncol. 2015, 17, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Miyai, M.; Tomita, H.; Soeda, A.; Yano, H.; Iwama, T.; Hara, A. Current trends in mouse models of glioblastoma. J. Neurooncol. 2017, 135, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Weissenberger, J.; Steinbach, J.P.; Malin, G.; Spada, S.; Rulicke, T.; Aguzzi, A. Development and malignant progression of astrocytomas in GFAP-v-src transgenic mice. Oncogene 1997, 14, 2005–2013. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Guignard, F.; Zhao, D.; Liu, L.; Burns, D.K.; Mason, R.P.; Messing, A.; Parada, L.F. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell 2005, 8, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Misuraca, K.L.; Hu, G.; Barton, K.L.; Chung, A.; Becher, O.J. A Novel Mouse Model of Diffuse Intrinsic Pontine Glioma Initiated in Pax3-Expressing Cells. Neoplasia 2016, 18, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef]
- Ashizawa, T.; Miyata, H.; Iizuka, A.; Komiyama, M.; Oshita, C.; Kume, A.; Nogami, M.; Yagoto, M.; Ito, I.; Oishi, T.; et al. Effect of the STAT3 inhibitor STX-0119 on the proliferation of cancer stem-like cells derived from recurrent glioblastoma. Int. J. Oncol. 2013, 43, 219–227. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- De Witt Hamer, P.C. Small molecule kinase inhibitors in glioblastoma: A systematic review of clinical studies. Neuro Oncol. 2010, 12, 304–316. [Google Scholar] [CrossRef]
- Thorne, A.H.; Zanca, C.; Furnari, F. Epidermal growth factor receptor targeting and challenges in glioblastoma. Neuro Oncol. 2016, 18, 914–918. [Google Scholar] [CrossRef] [Green Version]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef]
- Lai, A.; Kharbanda, S.; Pope, W.B.; Tran, A.; Solis, O.E.; Peale, F.; Forrest, W.F.; Pujara, K.; Carrillo, J.A.; Pandita, A.; et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J. Clin. Oncol. 2011, 29, 4482–4490. [Google Scholar] [CrossRef]
- Franceschi, S.; Corsinovi, D.; Lessi, F.; Tantillo, E.; Aretini, P.; Menicagli, M.; Scopelliti, C.; Civita, P.; Pasqualetti, F.; Naccarato, A.G.; et al. Mitochondrial enzyme GLUD2 plays a critical role in glioblastoma progression. EBioMedicine 2018, 37, 56–67. [Google Scholar] [CrossRef]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Umetani, M.; Domoto, H.; Gormley, A.K.; Yuhanna, I.S.; Cummins, C.L.; Javitt, N.B.; Korach, K.S.; Shaul, P.W.; Mangelsdorf, D.J. 27-Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen. Nat. Med. 2007, 13, 1185–1192. [Google Scholar] [CrossRef]
- Wei, W.; Schwaid, A.G.; Wang, X.; Wang, X.; Chen, S.; Chu, Q.; Saghatelian, A.; Wan, Y. Ligand Activation of ERRalpha by Cholesterol Mediates Statin and Bisphosphonate Effects. Cell Metab. 2016, 23, 479–491. [Google Scholar] [CrossRef]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef]
- Wang, F.; Beck-Garcia, K.; Zorzin, C.; Schamel, W.W.; Davis, M.M. Inhibition of T cell receptor signaling by cholesterol sulfate, a naturally occurring derivative of membrane cholesterol. Nat. Immunol. 2016, 17, 844–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, J.D. Sterol regulatory element-binding proteins: Transcriptional activators of lipid synthesis. Biochem. Soc. Trans. 2002, 30, 1091–1095. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef]
- Gong, Y.; Lee, J.N.; Lee, P.C.; Goldstein, J.L.; Brown, M.S.; Ye, J. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 2006, 3, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Adams, C.M.; Reitz, J.; De Brabander, J.K.; Feramisco, J.D.; Li, L.; Brown, M.S.; Goldstein, J.L. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J. Biol. Chem. 2004, 279, 52772–52780. [Google Scholar] [CrossRef]
- Sakai, J.; Duncan, E.A.; Rawson, R.B.; Hua, X.; Brown, M.S.; Goldstein, J.L. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 1996, 85, 1037–1046. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Heiniger, H.J.; Marshall, J.D. Cholesterol synthesis in polyclonally activated cytotoxic lymphocytes and its requirement for differentiation and proliferation. Proc. Natl. Acad. Sci. USA 1982, 79, 3823–3827. [Google Scholar] [CrossRef]
- Reboldi, A.; Dang, E.V.; McDonald, J.G.; Liang, G.; Russell, D.W.; Cyster, J.G. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 2014, 345, 679–684. [Google Scholar] [CrossRef]
- Tricarico, P.M.; Gratton, R.; Braga, L.; Celsi, F.; Crovella, S. 25-Hydroxycholesterol and inflammation in Lovastatin-deregulated mevalonate pathway. Int. J. Biochem. Cell Biol. 2017, 92, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Mangelsdorf, D.J. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu. Rev. Cell Dev. Biol. 2000, 16, 459–481. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Hong, C.; Boyadjian, R.; Tontonoz, P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 2009, 325, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Courtney, R.; Landreth, G.E. LXR Regulation of Brain Cholesterol: From Development to Disease. Trends Endocrinol. Metab. 2016, 27, 404–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkhem, I.; Meaney, S. Brain cholesterol: Long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Virkkunen, M.; Penttinen, H. Serum cholesterol in aggressive conduct disorder: A preliminary study. Biol. Psychiatry 1984, 19, 435–439. [Google Scholar] [PubMed]
- Goldstein, M.R. Cholesterol and violence: Is there a connection? Ann. Intern. Med. 1998, 129, 668–669, author reply 669–670. [Google Scholar] [CrossRef]
- Golomb, B.A.; Stattin, H.; Mednick, S. Low cholesterol and violent crime. J. Psychiatry Res. 2000, 34, 301–309. [Google Scholar] [CrossRef]
- Lifshitz, F.; Moses, N. Growth failure. A complication of dietary treatment of hypercholesterolemia. Am. J. Dis. Child. 1989, 143, 537–542. [Google Scholar] [CrossRef]
- Barness, L.A. Nutritional requirements of infants and children with respect to cholesterol and related compounds. Am. J. Med. Genet. 1994, 50, 353–354. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Kita, T.; Suckling, K.E.; Goldstein, J.L.; Brown, M.S. Cholesterol synthesis in vivo and in vitro in the WHHL rabbit, an animal with defective low density lipoprotein receptors. J. Lipid Res. 1983, 24, 469–480. [Google Scholar] [PubMed]
- Osono, Y.; Woollett, L.A.; Herz, J.; Dietschy, J.M. Role of the low density lipoprotein receptor in the flux of cholesterol through the plasma and across the tissues of the mouse. J. Clin. Investig. 1995, 95, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Turley, S.D.; Burns, D.K.; Rosenfeld, C.R.; Dietschy, J.M. Brain does not utilize low density lipoprotein-cholesterol during fetal and neonatal development in the sheep. J. Lipid Res. 1996, 37, 1953–1961. [Google Scholar] [PubMed]
- Quan, G.; Xie, C.; Dietschy, J.M.; Turley, S.D. Ontogenesis and regulation of cholesterol metabolism in the central nervous system of the mouse. Brain Res. Dev. Brain Res. 2003, 146, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Bloch, K. Sterol molecule: Structure, biosynthesis, and function. Steroids 1992, 57, 378–383. [Google Scholar] [CrossRef]
- Geng, F.; Guo, D. Lipid droplets, potential biomarker and metabolic target in glioblastoma. Intern. Med. Rev. 2017, 3. [Google Scholar] [CrossRef] [Green Version]
- Dietschy, J.M.; Turley, S.D. Thematic review series: Brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef] [PubMed]
- Betters, J.L.; Yu, L. NPC1L1 and cholesterol transport. FEBS Lett. 2010, 584, 2740–2747. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.S.; Weickert, C.S.; Garner, B. Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J. Neurochem. 2008, 104, 1145–1166. [Google Scholar] [CrossRef] [Green Version]
- Whitney, K.D.; Watson, M.A.; Collins, J.L.; Benson, W.G.; Stone, T.M.; Numerick, M.J.; Tippin, T.K.; Wilson, J.G.; Winegar, D.A.; Kliewer, S.A. Regulation of cholesterol homeostasis by the liver X receptors in the central nervous system. Mol. Endocrinol. 2002, 16, 1378–1385. [Google Scholar] [CrossRef]
- Davison, A.N.; Wajda, M. Cerebral lipids in multiple sclerosis. J. Neurochem. 1962, 9, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Jurevics, H.; Morell, P. Cholesterol for synthesis of myelin is made locally, not imported into brain. J. Neurochem. 1995, 64, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Cavender, C.P.; Turley, S.D.; Dietschy, J.M. Sterol metabolism in fetal, newborn, and suckled lambs and their response to cholesterol after weaning. Am. J. Physiol. 1995, 269, E331–E340. [Google Scholar] [CrossRef] [PubMed]
- Muse, E.D.; Jurevics, H.; Toews, A.D.; Matsushima, G.K.; Morell, P. Parameters related to lipid metabolism as markers of myelination in mouse brain. J. Neurochem. 2001, 76, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Elmberger, P.G.; Edlund, C.; Kristensson, K.; Dallner, G. Rates of cholesterol, ubiquinone, dolichol and dolichyl-P biosynthesis in rat brain slices. FEBS Lett. 1990, 269, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, H.; Campenot, R.B.; Vance, D.E.; Vance, J.E. Glial lipoproteins stimulate axon growth of central nervous system neurons in compartmented cultures. J. Biol. Chem. 2004, 279, 14009–14015. [Google Scholar] [CrossRef] [PubMed]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef]
- Pfrieger, F.W. Outsourcing in the brain: Do neurons depend on cholesterol delivery by astrocytes? Bioessays 2003, 25, 72–78. [Google Scholar] [CrossRef]
- Lutjohann, D.; Breuer, O.; Ahlborg, G.; Nennesmo, I.; Siden, A.; Diczfalusy, U.; Bjorkhem, I. Cholesterol homeostasis in human brain: Evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc. Natl. Acad. Sci. USA 1996, 93, 9799–9804. [Google Scholar] [CrossRef]
- Dash, S.; Xiao, C.; Morgantini, C.; Lewis, G.F. New Insights into the Regulation of Chylomicron Production. Annu. Rev. Nutr. 2015, 35, 265–294. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D.; Spady, D.K. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J. Lipid Res. 1993, 34, 1637–1659. [Google Scholar]
- Doonan, L.M.; Fisher, E.A.; Brodsky, J.L. Can modulators of apolipoproteinB biogenesis serve as an alternate target for cholesterol-lowering drugs? Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2018, 1863, 762–771. [Google Scholar] [CrossRef]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [PubMed]
- Midzak, A.; Papadopoulos, V. Binding domain-driven intracellular trafficking of sterols for synthesis of steroid hormones, bile acids and oxysterols. Traffic 2014, 15, 895–914. [Google Scholar] [CrossRef] [PubMed]
- Oude Elferink, R.P.; Groen, A.K. Mechanisms of biliary lipid secretion and their role in lipid homeostasis. Semin. Liver Dis. 2000, 20, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; La Vecchia, C.; de Groh, M.; Negri, E.; Morrison, H.; Mery, L.; Canadian Cancer Registries Epidemiology Research Group. Dietary cholesterol intake and cancer. Ann. Oncol. 2012, 23, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Kandutsch, A.A.; Russell, A.E. Preputial gland tumor sterols. 3. A metabolic pathway from lanosterol to cholesterol. J. Biol. Chem. 1960, 235, 2256–2261. [Google Scholar]
- Bloch, K. The biological synthesis of cholesterol. Science 1965, 150, 19–28. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef]
- Celiku, O.; Johnson, S.; Zhao, S.; Camphausen, K.; Shankavaram, U. Visualizing molecular profiles of glioblastoma with GBM-BioDP. PLoS ONE 2014, 9, e101239. [Google Scholar] [CrossRef]
- Kambach, D.M.; Halim, A.S.; Cauer, A.G.; Sun, Q.; Tristan, C.A.; Celiku, O.; Kesarwala, A.H.; Shankavaram, U.; Batchelor, E.; Stommel, J.M. Disabled cell density sensing leads to dysregulated cholesterol synthesis in glioblastoma. Oncotarget 2017, 8, 14860–14875. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Kim, D.K.; Bae, S.H.; Gwak, H.; Jeon, J.H.; Kim, J.K.; Lee, B.I.; You, H.J.; Shin, D.H.; Kim, Y.H.; et al. Farnesyl diphosphate synthase is important for the maintenance of glioblastoma stemness. Exp. Mol. Med. 2018, 50, 137. [Google Scholar] [CrossRef] [PubMed]
- Abate, M.; Laezza, C.; Pisanti, S.; Torelli, G.; Seneca, V.; Catapano, G.; Montella, F.; Ranieri, R.; Notarnicola, M.; Gazzerro, P.; et al. Deregulated expression and activity of Farnesyl Diphosphate Synthase (FDPS) in Glioblastoma. Sci. Rep. 2017, 7, 14123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, G.R.; Hulce, J.J.; Zanca, C.; Bi, J.; Ikegami, S.; Cahill, G.L.; Gu, Y.; Lum, K.M.; Masui, K.; Yang, H.; et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell 2016, 30, 683–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef] [Green Version]
- Buchwald, H. Cholesterol inhibition, cancer, and chemotherapy. Lancet 1992, 339, 1154–1156. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef]
- Ginestier, C.; Monville, F.; Wicinski, J.; Cabaud, O.; Cervera, N.; Josselin, E.; Finetti, P.; Guille, A.; Larderet, G.; Viens, P.; et al. Mevalonate metabolism regulates Basal breast cancer stem cells and is a potential therapeutic target. Stem Cells 2012, 30, 1327–1337. [Google Scholar] [CrossRef]
- Cruz, P.M.; Mo, H.; McConathy, W.J.; Sabnis, N.; Lacko, A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: A review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 2013, 4, 119. [Google Scholar] [CrossRef]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [Green Version]
- Repa, J.J.; Turley, S.D.; Lobaccaro, J.A.; Medina, J.; Li, L.; Lustig, K.; Shan, B.; Heyman, R.A.; Dietschy, J.M.; Mangelsdorf, D.J. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 2000, 289, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- DiBlasio-Smith, E.A.; Arai, M.; Quinet, E.M.; Evans, M.J.; Kornaga, T.; Basso, M.D.; Chen, L.; Feingold, I.; Halpern, A.R.; Liu, Q.Y.; et al. Discovery and implementation of transcriptional biomarkers of synthetic LXR agonists in peripheral blood cells. J. Transl. Med. 2008, 6, 59. [Google Scholar] [CrossRef] [Green Version]
- Warita, K.; Warita, T.; Beckwitt, C.H.; Schurdak, M.E.; Vazquez, A.; Wells, A.; Oltvai, Z.N. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci. Rep. 2014, 4, 7593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeder, S.L.; University of Iowa. A Study of the Proper Dosage of Lovastatin and Docetaxel for Patients with Cancer. Available online: https://ClinicalTrials.gov/show/NCT00584012 (accessed on 2 January 2008).
- Claes, A.; Wesseling, P.; Jeuken, J.; Maass, C.; Heerschap, A.; Leenders, W.P. Antiangiogenic compounds interfere with chemotherapy of brain tumors due to vessel normalization. Mol. Cancer Ther. 2008, 7, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Center, L.U.M.; Institute, N.C. Simvastatin and Panitumumab in Treating Patients with Advanced or Metastatic Colorectal Cancer. Available online: https://ClinicalTrials.gov/show/NCT01110785 (accessed on 27 April 2010).
- Center, C.-S.M.; Institute, R.P.C. Cellular Effect of Cholesterol-Lowering Prior to Prostate Removal. Available online: https://ClinicalTrials.gov/show/NCT02534376 (accessed on 27 August 2015).
- Centre, L.H.S. Metformin and Simvastatin Use in Bladder Cancer. Available online: https://ClinicalTrials.gov/show/NCT02360618 (accessed on 10 February 2015).
- Higgins, M.J.; Prowell, T.M.; Blackford, A.L.; Byrne, C.; Khouri, N.F.; Slater, S.A.; Jeter, S.C.; Armstrong, D.K.; Davidson, N.E.; Emens, L.A.; et al. A short-term biomarker modulation study of simvastatin in women at increased risk of a new breast cancer. Breast Cancer Res. Treat. 2012, 131, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.; Elson, C.E. Studies of the isoprenoid-mediated inhibition of mevalonate synthesis applied to cancer chemotherapy and chemoprevention. Exp. Biol. Med. 2004, 229, 567–585. [Google Scholar] [CrossRef]
- Zhou, X.; Qian, J.; Hua, L.; Shi, Q.; Liu, Z.; Xu, Y.; Sang, B.; Mo, J.; Yu, R. Geranylgeranyltransferase I promotes human glioma cell growth through Rac1 membrane association and activation. J. Mol. Neurosci. 2013, 49, 130–139. [Google Scholar] [CrossRef]
- Ciaglia, E.; Grimaldi, M.; Abate, M.; Scrima, M.; Rodriquez, M.; Laezza, C.; Ranieri, R.; Pisanti, S.; Ciuffreda, P.; Manera, C.; et al. The isoprenoid derivative N(6)-benzyladenosine CM223 exerts antitumor effects in glioma patient-derived primary cells through the mevalonate pathway. Br. J. Pharmacol. 2017, 174, 2287–2301. [Google Scholar] [CrossRef]
- Ciaglia, E.; Abate, M.; Laezza, C.; Pisanti, S.; Vitale, M.; Seneca, V.; Torelli, G.; Franceschelli, S.; Catapano, G.; Gazzerro, P.; et al. Antiglioma effects of N6-isopentenyladenosine, an endogenous isoprenoid end product, through the downregulation of epidermal growth factor receptor. Int. J. Cancer 2017, 140, 959–972. [Google Scholar] [CrossRef]
- Awad, A.B.; Fink, C.S. Phytosterols as anticancer dietary components: Evidence and mechanism of action. J. Nutr. 2000, 130, 2127–2130. [Google Scholar] [CrossRef] [PubMed]
- Putz, M.V.; Lazea, M.; Sandjo, L.P. Quantitative Structure Inter-Activity Relationship (QSInAR). Cytotoxicity Study of Some Hemisynthetic and Isolated Natural Steroids and Precursors on Human Fibrosarcoma Cells HT1080. Molecules 2011, 16, 6603–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, F.; Patrick, S.; Sheikh, T.; Sharma, V.; Pathak, P.; Malgulwar, P.B.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Telomerase reverse transcriptase (TERT)—Enhancer of zeste homolog 2 (EZH2) network regulates lipid metabolism and DNA damage responses in glioblastoma. J. Neurochem. 2017, 143, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, C.; Yonezawa, Y.; Kuriyama, I.; Nishida, M.; Yoshida, H.; Mizushina, Y. Inhibitory effects of cholesterol derivatives on DNA polymerase and topoisomerase activities, and human cancer cell growth. Lipids 2008, 43, 373–382. [Google Scholar] [CrossRef]
- Peacock, G.; Sidwell, R.; Pan, G.; Ole, S.; Lu, D.R. In vitro uptake of a new cholesteryl carborane ester compound by human glioma cell lines. J. Pharm. Sci. 2004, 93, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Zee-Cheng, R.K.; Cheng, C.C. Delivery of anticancer drugs. Methods Find. Exp. Clin. Pharmacol. 1989, 11, 439–529. [Google Scholar] [PubMed]
- Lu, D.R.; Mehta, S.C.; Chen, W. Selective boron drug delivery to brain tumors for boron neutron capture therapy. Adv. Drug Deliv. Rev. 1997, 26, 231–247. [Google Scholar] [PubMed]
- Ahmad, F.; Dixit, D.; Sharma, V.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Nrf2-driven TERT regulates pentose phosphate pathway in glioblastoma. Cell Death Dis. 2016, 7, e2213. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [Green Version]
- Murai, T. Cholesterol lowering: Role in cancer prevention and treatment. Biol. Chem. 2015, 396, 1–11. [Google Scholar] [CrossRef]
- Llaverias, G.; Danilo, C.; Mercier, I.; Daumer, K.; Capozza, F.; Williams, T.M.; Sotgia, F.; Lisanti, M.P.; Frank, P.G. Role of cholesterol in the development and progression of breast cancer. Am. J. Pathol. 2011, 178, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Land, H. Anticancer activity of the cholesterol exporter ABCA1 gene. Cell Rep. 2012, 2, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Rama, A.R.; Alvarez, P.J.; Madeddu, R.; Aranega, A. ABC transporters as differentiation markers in glioblastoma cells. Mol. Biol. Rep. 2014, 41, 4847–4851. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug | Mechanism |
|---|---|
| Ciprofibrate | PPARα agonist |
| Clofibrates | PPARα agonist |
| Fenofibrate | PPARα agonist |
| Gemifibrozil | PPARα agonist |
| Anacetrapib | CETP inhibitor |
| Avasimibe | ACAT inhibitor |
| Berberine | Increases LDLR expression |
| Lapaquistat acetate | FDFT1 inhibitor |
| Ezetimibe | NPC1L1 inhibitor |
| Atorvastatin | HMGCR inhibitor |
| Fluvastatin | HMGCR inhibitor |
| Pitavastatin | HMGCR inhibitor |
| Rosuvastatin | HMGCR inhibitor |
| Simvastatin | HMGCR inhibitor |
| Pitavastatin | HMGCR inhibitor |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, F.; Sun, Q.; Patel, D.; Stommel, J.M. Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma. Cancers 2019, 11, 146. https://doi.org/10.3390/cancers11020146
Ahmad F, Sun Q, Patel D, Stommel JM. Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma. Cancers. 2019; 11(2):146. https://doi.org/10.3390/cancers11020146
Chicago/Turabian StyleAhmad, Fahim, Qian Sun, Deven Patel, and Jayne M. Stommel. 2019. "Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma" Cancers 11, no. 2: 146. https://doi.org/10.3390/cancers11020146