The Volume-Regulated Anion Channel in Glioblastoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Glioblastoma
2. The Swelling Activated Chloride Current
2.1. General
2.2. Basic Biophysical Properties of VRAC
2.3. Pharmacology of VRAC
2.4. Activation of VRAC
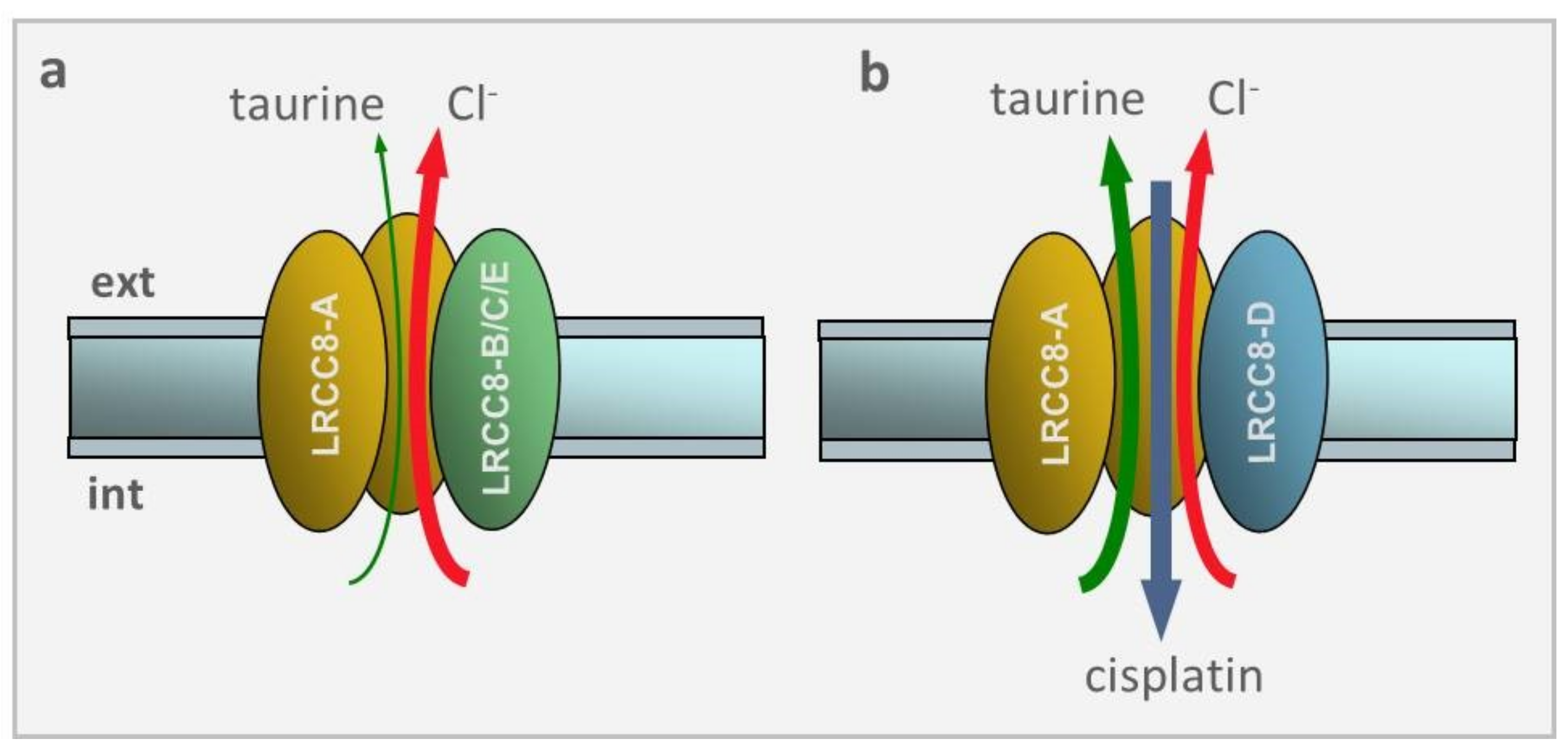
2.5. Molecular Identity of VRAC
2.6. VRAC Classical Role: Control of Cell Volume
2.7. VRAC in Drug Transport and Drug Resistance
3. Activation of VRAC by Pathologically Relevant Conditions
3.1. Blood Serum
3.2. Hypoxia
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Holland, E.C. Gliomagenesis: Genetic alterations and mouse models. Nat. Rev. Genet. 2001, 2, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.A.; Furnari, F.B.; Bachoo, R.M.; Rowitch, D.H.; Louis, D.N.; Cavenee, W.K.; DePinho, R.A. Malignant glioma: Genetics and biology of a grave matter. Genes Dev. 2001, 15, 1311–1333. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Pavlidis, N.; Jelic, S. ESMO Guidelines Task Force. ESMO Minimum Clinical Recommendations for diagnosis, treatment and follow-up of malignant glioma. Ann. Oncol. 2005, 16 (Suppl. 1), i64–i65. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy with Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Nakada, M.; Nakada, S.; Demuth, T.; Tran, N.L.; Hoelzinger, D.B.; Berens, M.E. Molecular targets of glioma invasion. Cell. Mol. Life Sci. 2007, 64, 458–478. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed]
- Sforna, L.; Cenciarini, M.; Belia, S.; D’Adamo, M.C.; Pessia, M.; Franciolini, F.; Catacuzzeno, L. The role of ion channels in the hypoxia-induced aggressiveness of glioblastoma. Front. Cell Neurosci. 2015, 15, 467. [Google Scholar] [CrossRef] [PubMed]
- Zinnhardt, B.; Pigeon, H.; Thézé, B.; Viel, T.; Wachsmuth, L.; Fricke, I.B.; Schelhaas, S.; Honold, L.; Schwegmann, K.; Wagner, S.; et al. Combined PET Imaging of the Inflammatory Tumor Microenvironment Identifies Margins of Unique Radiotracer Uptake. Cancer Res. 2017, 77, 1831–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisapia, D.J. The Updated World Health Organization Glioma Classification: Cellular and Molecular Origins of Adult Infiltrating Gliomas. Arch. Pathol. Lab. Med. 2017, 141, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuddapah, V.A.; Sontheimer, H. Ion channels and transporters [corrected] in cancer. 2. Ion channels and the control of cancer cell migration. Am. J. Physiol. Cell Physiol. 2011, 301, C541–C549. [Google Scholar] [CrossRef] [PubMed]
- Vehlow, A.; Cordes, N. Invasion as target for therapy of glioblastoma multiforme. Biochim. Biophys. Acta 2013, 1836, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A.M. Cytoskeletal interactions with the outside world. Dev. Cell 2005, 9, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Usatyuk, P.V.; Fu, P.; Mohan, V.; Epshtein, Y.; Jacobson, J.R.; Gomez-Cambronero, J.; Wary, K.K.; Bindokas, V.; Dudek, S.M.; Salgia, R.; et al. Role of c-Met/phosphatidylinositol 3-kinase (PI3k)/Akt signaling in hepatocyte growth factor (HGF)-mediated lamellipodia formation, reactive oxygen species (ROS) generation, and motility of lung endothelial cells. J. Biol. Chem. 2014, 289, 13476–13491. [Google Scholar] [CrossRef] [PubMed]
- Chandrika, G.; Natesh, K.; Ranade, D.; Chugh, A.; Shastry, P. Suppression of the invasive potential of Glioblastoma cells by mTOR inhibitors involves modulation of NFκB and PKC-α signaling. Sci. Rep. 2016, 6, 22455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallia, G.L.; Rand, V.; Siu, I.M.; Eberhart, C.G.; James, C.D.; Marie, S.K.; Oba-Shinjo, S.M.; Carlotti, C.G.; Caballero, O.L.; Simpson, A.J.; et al. PIK3CA gene mutations in pediatric and adult glioblastoma multiforme. Mol. Cancer Res. 2006, 4, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, A.S.; Fattet, S.; Fischer, B.; Shalaby, T.; Jackson, S.P.; Schoenwaelder, S.M.; Grotzer, M.A.; Delattre, O.; Arcaro, A. Targeting the PI3K p110alpha isoform inhibit medulloblastoma proliferation, chemoresistance, and migration. Clin. Cancer Res. 2008, 14, 6761–6769. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.L.; Parat, M.O.; Binder, Z.A.; Gallia, G.L.; Riggins, G.J. Abrogation of PIK3CA or PIK3R1 reduces proliferation, migration, and invasion in glioblastoma multiforme cells. Oncotarget 2011, 2, 833–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Ding, X.; Ye, H.; Wang, J.; Shao, J.; Huang, T. Hypoxia enhances the migration and invasion of human glioblastoma U87 cells through PI3K/Akt/mTOR/HIF-1α pathway. Neuroreport 2018, 29, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Domin, J.; Harper, L.; Aubyn, D.; Wheeler, M.; Florey, O.; Haskard, D.; Yuan, M.; Zicha, D. The class II phosphoinositide 3-kinase PI3K-C2beta regulates cell migration by a PtdIns3P dependent mechanism. J. Cell. Physiol. 2005, 205, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Cain, R.J.; Ridley, A.J. Phosphoinositide 3-kinases in cell migration. Biol. Cell 2009, 101, 13–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefranc, F.; Brotchi, J.; Kiss, R. Possible future issues in the treatment of glioblastomas: Special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J. Clin. Oncol. 2005, 23, 2411–2422. [Google Scholar] [CrossRef] [PubMed]
- Lino, M.M.; Merlo, A. PI3Kinase signaling in glioblastoma. J. Neurooncol. 2011, 103, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Galietta, L.J. Chloride channels as drug targets. Nat. Rev. Drug Discov. 2009, 8, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Strange, K.; Emma, F.; Jackson, P.S. Cellular and molecular physiology of volume-sensitive anion channels. Am. J. Physiol. 1996, 270 Pt 1, C711–C730. [Google Scholar] [CrossRef]
- Strange, K.; Yamada, T.; Denton, J.S. A 30-year journey from volume-regulated anion currents to molecular structure of the LRRC8 channel. J. Gen. Physiol. 2019, 151, 100–117. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Eggermont, J.; Voets, T.; Buyse, G.; Manolopoulos, V.; Droogmans, G. Properties of volume-regulated anion channels in mammalian cells. Prog. Biophys. Mol. Biol. 1997, 68, 69–119. [Google Scholar] [CrossRef]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of cell volume regulation in vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Klausen, T.K.; Nilius, B. The identification of a volume-regulated anion channel: An amazing Odyssey. Acta Physiol. 2015, 213, 868–881. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Okada, Y.; Nilius, B. Biophysics and Physiology of the Volume-Regulated Anion Channel (VRAC)/Volume-Sensitive Outwardly Rectifying Anion Channel (VSOR). Pflug. Arch. 2016, 468, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, T.J.; Lutter, D.; Planells-Cases, R.; Ullrich, F.; Voss, F.K. VRAC: Molecular identification as LRRC8 heteromers with differential functions. Pflug. Arch. 2016, 468, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Fioretti, B.; Castigli, E.; Calzuola, I.; Harper, A.A.; Franciolini, F.; Catacuzzeno, L. NPPB block of the intermediate-conductance Ca2+-activated K+ channel. Eur. J. Pharmacol. 2004, 497, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Catacuzzeno, L.; Michelucci, A.; Sforna, L.; Aiello, F.; Sciaccaluga, M.; Fioretti, B.; Castigli, E.; Franciolini, F. Identification of key signaling molecules involved in the activation of the swelling-activated chloride current in human glioblastoma cells. J. Membr. Biol. 2014, 247, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Sforna, L.; Cenciarini, M.; Belia, S.; Michelucci, A.; Pessia, M.; Franciolini, F.; Catacuzzeno, L. Hypoxia Modulates the Swelling-Activated Cl Current in Human Glioblastoma Cells: Role in Volume Regulation and Cell Survival. J. Cell. Physiol. 2017, 232, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Formaggio, F.; Saracino, E.; Mola, M.G.; Rao, S.B.; Amiry-Moghaddam, M.; Muccini, M.; Zamboni, R.; Nicchia, G.P.; Caprini, M.; Benfenati, V. LRRC8A is essential for swelling-activated chloride current and for regulatory volume decrease in astrocytes. FASEB J. 2019, 33, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.; Chen, W.; Zhong, X.; Rutka, J.T.; Feng, Z.P.; Sun, H.S. Swelling-induced chloride current in glioblastoma proliferation, migration, and invasion. J. Cell. Physiol. 2018, 233, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Rubino, S.; Bach, M.D.; Schober, A.L.; Lambert, I.H.; Mongin, A.A. Downregulation of Leucine-Rich Repeat-Containing 8A Limits Proliferation and Increases Sensitivity of Glioblastoma to Temozolomide and Carmustine. Front. Oncol. 2018, 8, 142. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Kim, W.J.; Park, J.A.; Choi, Y.K.; Kwon, Y.W.; Kim, K.W. Blood-brain barrier interfaces and brain tumors. Arch. Pharm. Res. 2006, 29, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Sehrer, J.; Viana, F.; De Greef, C.; Raeymaekers, L.; Eggermont, J.; Droogmans, G. Volume-activated Cl− currents in different mammalian non-excitable cell types. Pflug. Arch. 1994, 428, 364–371. [Google Scholar] [CrossRef]
- Voets, T.; Droogmans, G.; Nilius, B. Modulation of voltage-dependent properties of a swelling-activated Cl− current. J. Gen. Physiol. 1997, 110, 313–325. [Google Scholar] [PubMed]
- Nilius, B.; Droogmans, G. Amazing chloride channels: An overview. Acta Physiol. Scand. 2003, 177, 119–147. [Google Scholar] [CrossRef] [PubMed]
- Kimelberg, H.K.; Goderie, S.K.; Higman, S.; Pang, S.; Waniewski, R.A. Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J. Neurosci. 1990, 10, 1583–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banderali, U.; Roy, G. Activation of K+ and Cl− channels in MDCK cells during volume regulation in hypotonic media. J. Membr. Biol. 1992, 126, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.S.; Strange, K. Volume-sensitive anion channels mediate swelling-activated inositol and taurine efflux. Am. J. Physiol. 1993, 265 Pt 1, C1489–C1500. [Google Scholar] [CrossRef]
- Droogmans, G.; Prenen, J.; Eggermont, J.; Voets, T.; Nilius, B. Voltage-dependent block of endothelial volume-regulated anion channels by calix[4]arenes. Am. J. Physiol. 1998, 275 Pt 1, C646–C652. [Google Scholar] [CrossRef]
- Ternovsky, V.I.; Okada, Y.; Sabirov, R.Z. Sizing the pore of the volume-sensitive anion channel by differential polymer partitioning. FEBS Lett. 2004, 576, 433–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planells-Cases, R.; Lutter, D.; Guyader, C.; Gerhards, N.M.; Ullrich, F.; Elger, D.A.; Kucukosmanoglu, A.; Xu, G.; Voss, F.K.; Reincke, S.M.; et al. Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J. 2015, 34, 2993–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deneka, D.; Sawicka, M.; Lam, A.K.M.; Paulino, C.; Dutzler, R. Structure of a volume-regulated anion channel of the LRRC8 family. Nature 2018, 558, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Kefauver, J.M.; Saotome, K.; Dubin, A.E.; Pallesen, J.; Cottrell, C.A.; Cahalan, S.M.; Qiu, Z.; Hong, G.; Crowley, C.S.; Whitwam, T.; et al. Structure of the human volume regulated anion channel. eLife 2018, 7, e38461. [Google Scholar] [CrossRef] [PubMed]
- Voets, T.; Nilius, B.; Vennekens, R. VRACs swallow platinum drugs. EMBO J. 2015, 34, 2985–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Kubo, M.; Oiki, S.; Petersen, C.C.; Tominaga, M.; Hazama, A.; Morishima, S. Properties of volume-sensitive Cl− channels in a human epithelial cell line. Jpn. J. Physiol. 1994, 44 (Suppl. 2), S31–S35. [Google Scholar] [PubMed]
- Jackson, P.S.; Strange, K. Single-channel properties of a volume-sensitive anion conductance. Current activation occurs by abrupt switching of closed channels to an open state. J. Gen. Physiol. 1995, 105, 643–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syeda, R.; Qiu, Z.; Dubin, A.E.; Murthy, S.E.; Florendo, M.N.; Mason, D.E.; Mathur, J.; Cahalan, S.M.; Peters, E.C.; Montal, M.; et al. LRRC8 Proteins Form Volume-Regulated Anion Channels that Sense Ionic Strength. Cell 2016, 164, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Decher, N.; Lang, H.J.; Nilius, B.; Brüggemann, A.; Busch, A.E.; Steinmeyer, K. DCPIB is a novel selective blocker of I(Cl,swell) and prevents swelling-induced shortening of guinea-pig atrial action potential duration. Br. J. Pharmacol. 2001, 134, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Abdullaev, I.F.; Rudkouskaya, A.; Schools, G.P.; Kimelberg, H.K.; Mongin, A.A. Pharmacological comparison of swelling-activated excitatory amino acid release and Cl− currents in cultured rat astrocytes. J. Physiol. 2006, 572 Pt 3, 677–689. [Google Scholar] [CrossRef]
- Min, X.J.; Li, H.; Hou, S.C.; He, W.; Liu, J.; Hu, B.; Wang, J. Dysfunction of volume-sensitive chloride channels contributes to cisplatin resistance in human lung adenocarcinoma cells. Exp. Biol. Med. 2011, 236, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Minieri, L.; Pivonkova, H.; Caprini, M.; Harantova, L.; Anderova, M.; Ferroni, S. The inhibitor of volume-regulated anion channels DCPIB activates TREK potassium channels in cultured astrocytes. Br. J. Pharmacol. 2013, 168, 1240–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Mahajan, R.; Baumgarten, C.M.; Logothetis, D.E. The ICl,swell inhibitor DCPIB blocks Kir channels that possess weak affinity for PIP2. Pflug. Arch. 2016, 468, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.M.; Oh, S.; Hite, R.K.; Brohawn, S.G. Cryo-EM structures of the DCPIB-inhibited volume-regulated anion channel LRRC8A in lipid nanodiscs. BioRxiv 2018. [Google Scholar] [CrossRef] [PubMed]
- Feustel, P.J.; Jin, Y.; Kimelberg, H.K. Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke 2004, 35, 1164–1168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H.; Feustel, P.J.; Kimelberg, H.K. DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Exp. Neurol. 2008, 210, 514–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef]
- Bowens, N.H.; Dohare, P.; Kuo, Y.H.; Mongin, A.A. DCPIB, the proposed selective blocker of volume-regulated anion channels, inhibits several glutamate transport pathways in glial cells. Mol. Pharmacol. 2013, 83, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Benfenati, V.; Caprini, M.; Nicchia, G.P.; Rossi, A.; Dovizio, M.; Cervetto, C.; Nobile, M.; Ferroni, S. Carbenoxolone inhibits volume-regulated anion conductance in cultured rat cortical astroglia. Channels 2009, 3, 323–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilius, B.; Prenen, J.; Voets, T.; Eggermont, J.; Droogmans, G. Activation of volume-regulated chloride currents by reduction of intracellular ionic strength in bovine endothelial cells. J. Physiol. 1998, 506 Pt 2, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Voets, T.; Droogmans, G.; Raskin, G.; Eggermont, J.; Nilius, B. Reduced intracellular ionic strength as the initial trigger for activation of endothelial volume-regulated anion channels. Proc. Natl. Acad. Sci. USA 1999, 96, 5298–5303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabirov, R.Z.; Prenen, J.; Tomita, T.; Droogmans, G.; Nilius, B. Reduction of ionic strength activates single volume-regulated anion channels (VRAC) in endothelial cells. Pflug. Arch. 2000, 439, 315–320. [Google Scholar] [CrossRef]
- Akita, T.; Okada, Y. Characteristics and roles of the volume-sensitive outwardly rectifying (VSOR) anion channel in the central nervous system. Neuroscience 2014, 275, 211–231. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.M.; Oh, R.K.H.; Brohawn, S.G. Cryo-EM structures of the DCPIB-inhibited volume-regulated anion channel LRRC8A in lipid nanodiscs. bioRxiv 2018. [Google Scholar] [CrossRef]
- Abramovici, H.; Mojtabaie, P.; Parks, R.J.; Zhong, X.P.; Koretzky, G.A.; Topham, M.K.; Gee, S.H. Diacylglycerol kinase zeta regulates actin cytoskeleton reorganization through dissociation of Rac1 from RhoGDI. Mol. Biol. Cell. 2009, 20, 2049–2059. [Google Scholar] [CrossRef] [PubMed]
- Chianale, F.; Cutrupi, S.; Rainero, E.; Baldanzi, G.; Porporato, P.E.; Traini, S.; Filigheddu, N.; Gnocchi, V.F.; Santoro, M.M.; Parolini, O.; et al. Diacylglycerol kinase-alpha mediates hepatocyte growth factor-induced epithelial cell scatter by regulating Rac activation and membrane ruffling. Mol. Biol. Cell. 2007, 18, 4859–4871. [Google Scholar] [CrossRef] [PubMed]
- Chianale, F.; Rainero, E.; Cianflone, C.; Bettio, V.; Pighini, A.; Porporato, P.E.; Filigheddu, N.; Serini, G.; Sinigaglia, F.; Baldanzi, G.; et al. Diacylglycerol kinase alpha mediates HGF-induced Rac activation and membrane ruffling by regulating atypical PKC and RhoGDI. Proc. Natl. Acad. Sci. USA 2010, 107, 4182–4187. [Google Scholar] [CrossRef] [PubMed]
- Tolias, K.F.; Couvillon, A.D.; Cantley, L.C.; Carpenter, C.L. Characterization of a Rac1- and RhoGDI-associated lipid kinase signaling complex. Mol. Cell Biol. 1998, 18, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Dubin, A.E.; Mathur, J.; Tu, B.; Reddy, K.; Miraglia, L.J.; Reinhardt, J.; Orth, A.P.; Patapoutian, A. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 2014, 157, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Voss, F.K.; Ullrich, F.; Münch, J.; Lazarow, K.; Lutter, D.; Mah, N.; Andrade-Navarro, M.A.; von Kries, J.P.; Stauber, T.; Jentsch, T.J. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 2014, 344, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R. LRRC8 proteins share a common ancestor with pannexins, and may form hexameric channels involved in cell-cell communication. Bioessays 2012, 34, 551–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Lu, Y.; Gunasekar, S.; Zhang, Y.; Benson, C.J.; Chapleau, M.W.; Sah, R.; Abboud, F.M. The volume-regulated anion channel (LRRC8) in nodose neurons is sensitive to acidic pH. JCI Insight. 2017, 2, e90632. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, A.; Brandl, C.; Milenkovic, V.M.; Jendryke, T.; Sirianant, L.; Wanitchakool, P.; Zimmermann, S.; Reiff, C.M.; Horling, F.; Schrewe, H.; et al. Bestrophin 1 is indispensable for volume regulation in human retinal pigment epithelium cells. Proc. Natl. Acad. Sci. USA 2015, 112, E2630–E2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirianant, L.; Wanitchakool, P.; Ousingsawat, J.; Benedetto, R.; Zormpa, A.; Cabrita, I.; Schreiber, R.; Kunzelmann, K. Non-essential contribution of LRRC8A to volume regulation. Pflug. Arch. 2016, 468, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Shumilina, E.; Ritter, M.; Gulbins, E.; Vereninov, A.; Huber, S.M. Ion channels and cell volume in regulation of cell proliferation and apoptotic cell death. Contrib. Nephrol. 2006, 152, 142–160. [Google Scholar] [PubMed]
- Anbari, K.; Schultz, R.M. Effect of sodium and betaine in culture media on development and relative rates of protein synthesis in preimplantation mouse embryos in vitro. Mol. Reprod. Dev. 1993, 35, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Dubois, J.M.; Rouzaire-Dubois, B. The influence of cell volume changes on tumour cell proliferation. Eur. Biophys. J. 2004, 33, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Rouzaire-Dubois, B.; O’Regan, S.; Dubois, J.M. Cell size-dependent and independent proliferation of rodent neuroblastoma x glioma cells. J. Cell. Physiol. 2005, 203, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Maeno, E.; Ishizaki, Y.; Kanaseki, T.; Hazama, A.; Okada, Y. Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9487–9492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, A.; Stock, C. Ion channels and transporters in tumour cell migration and invasion. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130102. [Google Scholar] [CrossRef] [PubMed]
- Lui, V.C.; Lung, S.S.; Pu, J.K.; Hung, K.N.; Leung, G.K. Invasion of human glioma cells is regulated by multiple chloride channels including ClC-3. Anticancer Res. 2010, 30, 4515–4524. [Google Scholar] [PubMed]
- Habela, C.W.; Ernest, N.J.; Swindall, A.F.; Sontheimer, H. Chloride accumulation drives volume dynamics underlying cell proliferation and migration. J. Neurophysiol. 2009, 101, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Ransom, C.B.; O’Neal, J.T.; Sontheimer, H. Volume-activated chloride currents contribute to the resting conductance and invasive migration of human glioma cells. J. Neurosci. 2001, 21, 7674–7683. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Manning, T.J., Jr.; Sontheimer, H. Modulation of glioma cell migration and invasion using Cl(−) and K(+) ion channel blockers. J. Neurosci. 1999, 19, 5942–5954. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, B.B.; Mahesparan, R. Biological mechanisms of glioma invasion and potential therapeutic targets. J. Neurooncol. 2001, 53, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Catacuzzeno, L.; Aiello, F.; Fioretti, B.; Sforna, L.; Castigli, E.; Ruggieri, P.; Tata, A.M.; Calogero, A.; Franciolini, F. Serum-activated K and Cl currents underlay U87-MG glioblastoma cell migration. J. Cell. Physiol. 2011, 226, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Ise, T.; Shimizu, T.; Lee, E.L.; Inoue, H.; Kohno, K.; Okada, Y. Roles of volume-sensitive Cl− channel in cisplatin-induced apoptosis in human epidermoid cancer cells. J. Membr. Biol. 2005, 205, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Heimlich, G.; Cidlowski, J.A. Selective role of intracellular chloride in the regulation of the intrinsic but not extrinsic pathway of apoptosis in Jurkat T-cells. J. Biol. Chem. 2006, 281, 2232–2241. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.L.; Shimizu, T.; Ise, T.; Numata, T.; Kohno, K.; Okada, Y. Impaired activity of volume-sensitive Cl− channel is involved in cisplatin resistance of cancer cells. J. Cell. Physiol. 2007, 211, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, K.A.; Andersen, E.C.; Hansen, C.F.; Klausen, T.K.; Hougaard, C.; Lambert, I.H.; Hoffmann, E.K. Deregulation of apoptotic volume decrease and ionic movements in multidrug-resistant tumor cells: Role of chloride channels. Am. J. Physiol. Cell Physiol. 2010, 298, C14–C25. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, B.H.; Dam, C.S.; Stürup, S.; Lambert, I.H. Dual role of LRRC8A-containing transporters on cisplatin resistance in human ovarian cancer cells. J. Inorg. Biochem. 2016, 160, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, B.H.; Nielsen, D.; Thorsteinsdottir, U.A.; Hoffmann, E.K.; Lambert, I.H. Downregulation of LRRC8A protects human ovarian and alveolar carcinoma cells against Cisplatin-induced expression of p53, MDM2, p21Waf1/Cip1, and Caspase-9/-3 activation. Am. J. Physiol. Cell Physiol. 2016, 310, C857–C873. [Google Scholar] [CrossRef] [PubMed]
- Bicker, J.; Alves, G.; Fortuna, A.; Falcão, A. Blood-brain barrier models and their relevance for a successful development of CNS drug delivery systems: A review. Eur. J. Pharm. Biopharm. 2014, 87, 409–432. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.V.; van Roosmalen, I.A.; Busschers, E.; Tomar, T.; Conroy, S.; Eggens-Meijer, E.; Peñaranda Fajardo, N.; Pore, M.M.; Balasubramanyian, V.; Wagemakers, M.; et al. Serum-Induced Differentiation of Glioblastoma Neurospheres Leads to Enhanced Migration/Invasion Capacity That Is Associated with Increased MMP9. PLoS ONE 2015, 10, e0145393. [Google Scholar] [CrossRef] [PubMed]
- Vescovi, A.L.; Galli, R.; Reynolds, B.A. Brain tumour stem cells. Nat. Rev. Cancer 2006, 6, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Van Strien, M.E.; van den Berge, S.A.; Hol, E.M. Migrating neuroblasts in the adult human brain: A stream reduced to a trickle. Cell Res. 2011, 21, 1523–1525. [Google Scholar] [CrossRef] [PubMed]
- Rondé, P.; Giannone, G.; Gerasymova, I.; Stoeckel, H.; Takeda, K.; Haiech, J. Mechanism of calcium oscillations in migrating human astrocytoma cells. Biochim. Biophys. Acta 2000, 1498, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Bordey, A.; Sontheimer, H.; Trouslard, J. Muscarinic activation of BK channels induces membrane oscillations in glioma cells and leads to inhibition of cell migration. J. Membr. Biol. 2000, 176, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Manning, T.J., Jr.; Parker, J.C.; Sontheimer, H. Role of lysophosphatidic acid and rho in glioma cell motility. Cell Motil. Cytoskeleton. 2000, 45, 185–199. [Google Scholar] [CrossRef]
- Mariggio, M.A.; Mazzoleni, G.; Pietrangelo, T.; Guarnieri, S.; Morabito, C.; Steimberg, N.; Fano, G. Calcium-mediated transductive systems and functionally active gap junctions in astrocyte-like GL15 cells. BMC Physiol. 2001, 1, 4. [Google Scholar] [CrossRef]
- Rolfe, D.F.; Brand, M.D. Contribution of mitochondrial proton leak to skeletal muscle respiration and to standard metabolic rate. Am. J. Physiol. 1996, 271 Pt 1, C1380–C1389. [Google Scholar] [CrossRef]
- Priebe, L.; Friedrich, M.; Benndorf, K. Functional interaction between K(ATP) channels and the Na(+)-K(+) pump in metabolically inhibited heart cells of the guinea-pig. J. Physiol. 1996, 492 Pt 2, 405–417. [Google Scholar] [CrossRef]
- Barros, L.F.; Hermosilla, T.; Castro, J. Necrotic volume increase and the early physiology of necrosis. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2001, 130, 401–409. [Google Scholar] [CrossRef]
- Nabekura, T.; Morishima, S.; Cover, T.L.; Mori, S.; Kannan, H.; Komune, S.; Okada, Y. Recovery from lactacidosis-induced glial cell swelling with the aid of exogenous anion channels. Glia 2003, 41, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Maeno, E.; Shimizu, T.; Manabe, K.; Mori, S.; Nabekura, T. Dual roles of plasmalemmal chloride channels in induction of cell death. Pflug. Arch. 2004, 448, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Hochachka, P.W. Defense strategies against hypoxia and hypothermia. Science 1986, 231, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, J.A.; Chan, P.H.; Chan, T.Y.; Gregory, G.A. Modification of hypoxia-induced injury in cultured rat astrocytes by high levels of glucose. Stroke 1993, 24, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, J.P.; Wolburg, H.; Klumpp, A.; Probst, H.; Weller, M. Hypoxia-induced cell death in human malignant glioma cells: Energy deprivation promotes decoupling of mitochondrial cytochrome c release from caspase processing and necrotic cell death. Cell Death Differ. 2003, 10, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. ‘Pseudopalisading’ necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Amberger-Murphy, V. Hypoxia helps glioma to fight therapy. Curr. Cancer Drug Targets 2009, 9, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, C.; Wang, L.; Guo, H.; Wang, X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp. Cell Res. 2012, 318, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Oike, M.; Droogmans, G.; Nilius, B. The volume-activated chloride current in human endothelial cells depends on intracellular ATP. Pflug. Arch. 1994, 427, 184–186. [Google Scholar] [CrossRef]
- Okada, Y.; Oiki, S.; Tominaga, M.; Kubo, M.; Miwa, A.; Tominaga, T.; Tsumura, T.; Ueda, K. Volume-sensitive Cl− channel in human epithelial cells: Regulation by ATP and relation to P-glycoprotein. Jpn. J. Physiol. 1997, 47 (Suppl. 1), S19–S20. [Google Scholar] [PubMed]
- Crépel, V.; Panenka, W.; Kelly, M.E.; MacVicar, B.A. Mitogen-activated protein and tyrosine kinases in the activation of astrocyte volume-activated chloride current. J. Neurosci. 1998, 18, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.I.; Lee, Y.W.; Kim, Y.K. Chemical hypoxia-induced cell death in human glioma cells: Role of reactive oxygen species, ATP depletion, mitochondrial damage and Ca2+. Neurochem. Res. 2003, 28, 1201–1211. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caramia, M.; Sforna, L.; Franciolini, F.; Catacuzzeno, L. The Volume-Regulated Anion Channel in Glioblastoma. Cancers 2019, 11, 307. https://doi.org/10.3390/cancers11030307
Caramia M, Sforna L, Franciolini F, Catacuzzeno L. The Volume-Regulated Anion Channel in Glioblastoma. Cancers. 2019; 11(3):307. https://doi.org/10.3390/cancers11030307
Chicago/Turabian StyleCaramia, Martino, Luigi Sforna, Fabio Franciolini, and Luigi Catacuzzeno. 2019. "The Volume-Regulated Anion Channel in Glioblastoma" Cancers 11, no. 3: 307. https://doi.org/10.3390/cancers11030307
APA StyleCaramia, M., Sforna, L., Franciolini, F., & Catacuzzeno, L. (2019). The Volume-Regulated Anion Channel in Glioblastoma. Cancers, 11(3), 307. https://doi.org/10.3390/cancers11030307